Study of Melatonin as Preventive Agent of Gastrointestinal Damage Induced by Sodium Diclofenac

 ,
,  , and
, and 
Abstract
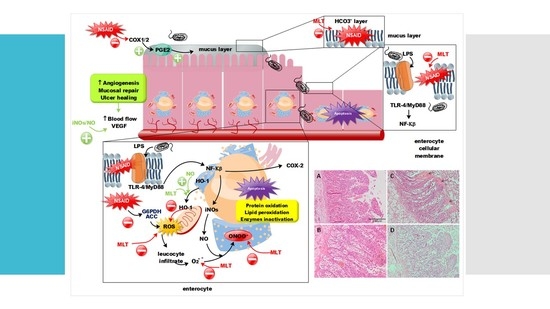
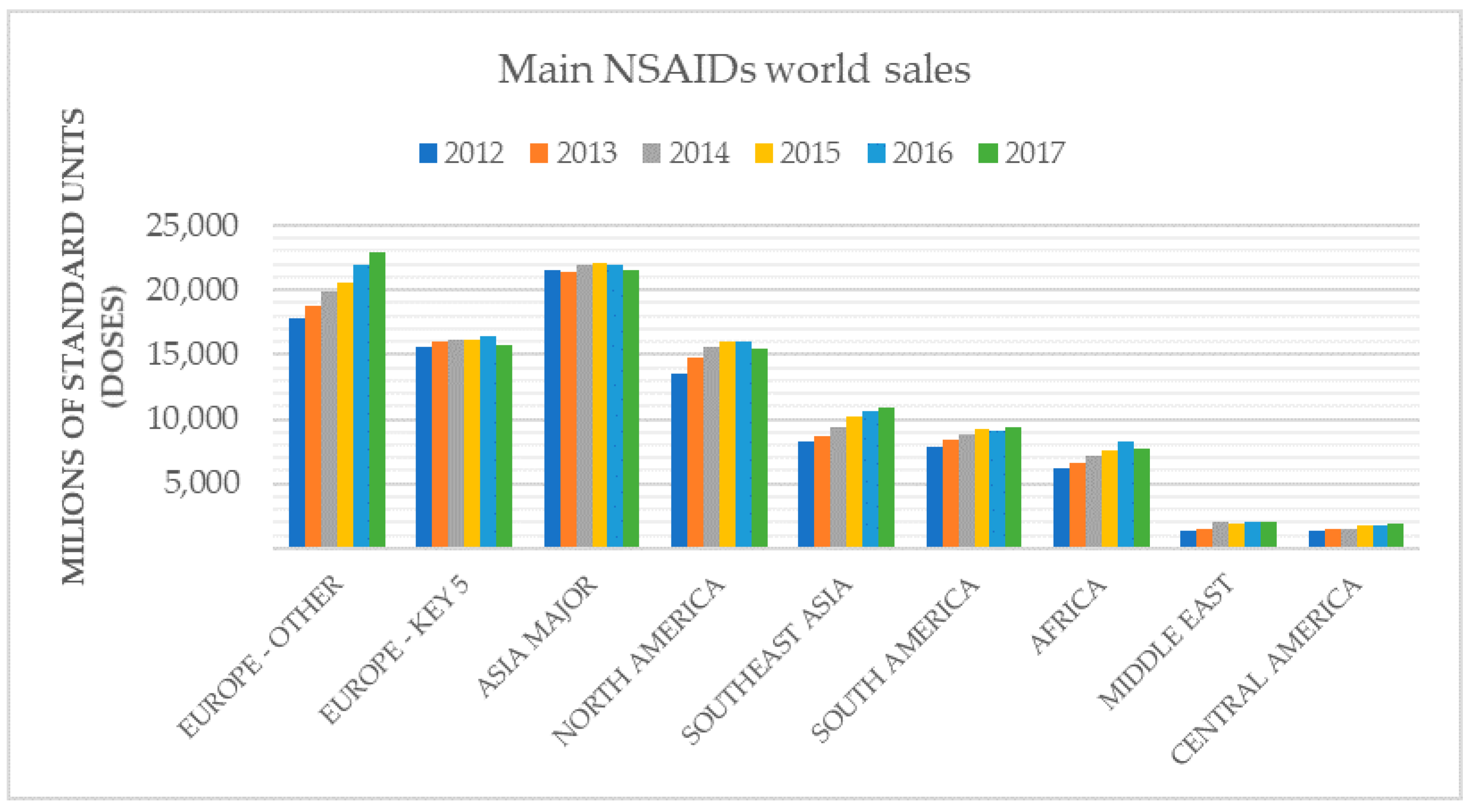
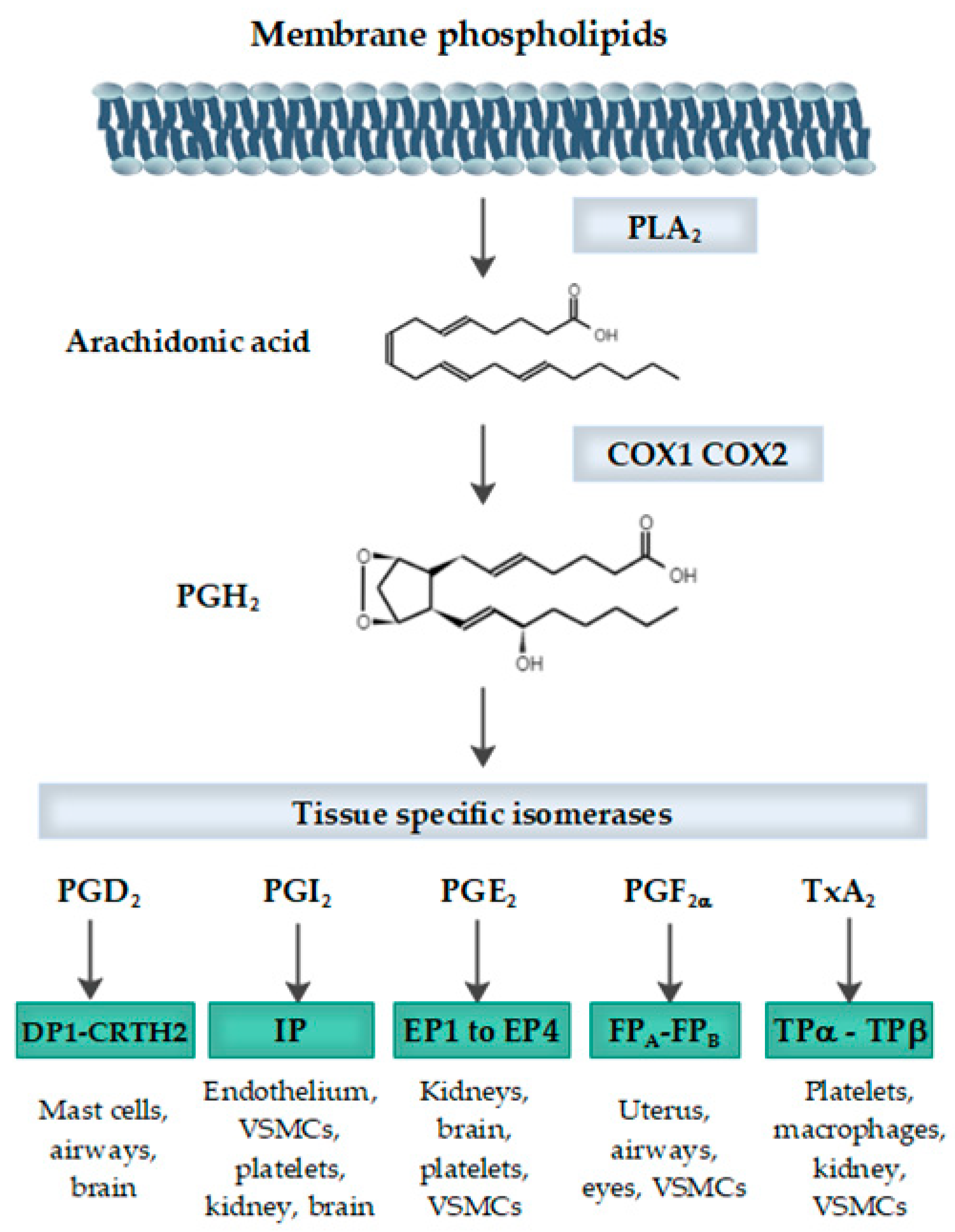
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Ex Vivo NSAID/ML Administration in Pig Intestine
2.2.1. HPLC-UV Procedure and Instrumentation
2.2.2. Validation and Verification of Analytical Methods
2.2.3. Sample Analysis
2.2.4. Data Treatment and Statistical Analysis
2.3. In Vivo Studies: Oral Administration of NSAID and MLT in Mice
2.4. Histological Analysis
2.5. COX-2 and iNOS Determination
2.5.1. RNA Isolation
2.5.2. RT-qPCR
2.5.3. Data Treatment and Statistical Analysis
3. Results
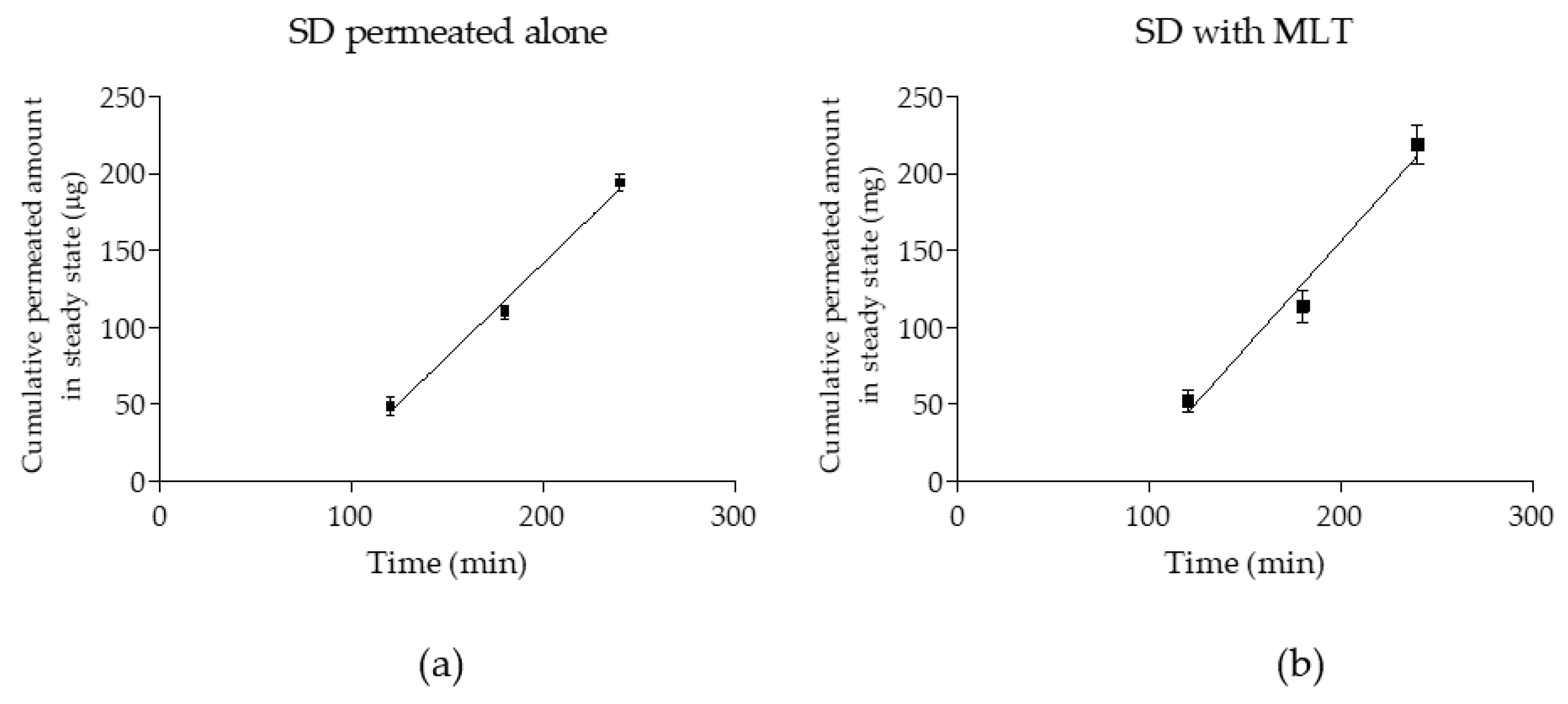
3.1. Obtained Kinetics Parameters, Papp Calculation, and Statistical Analysis
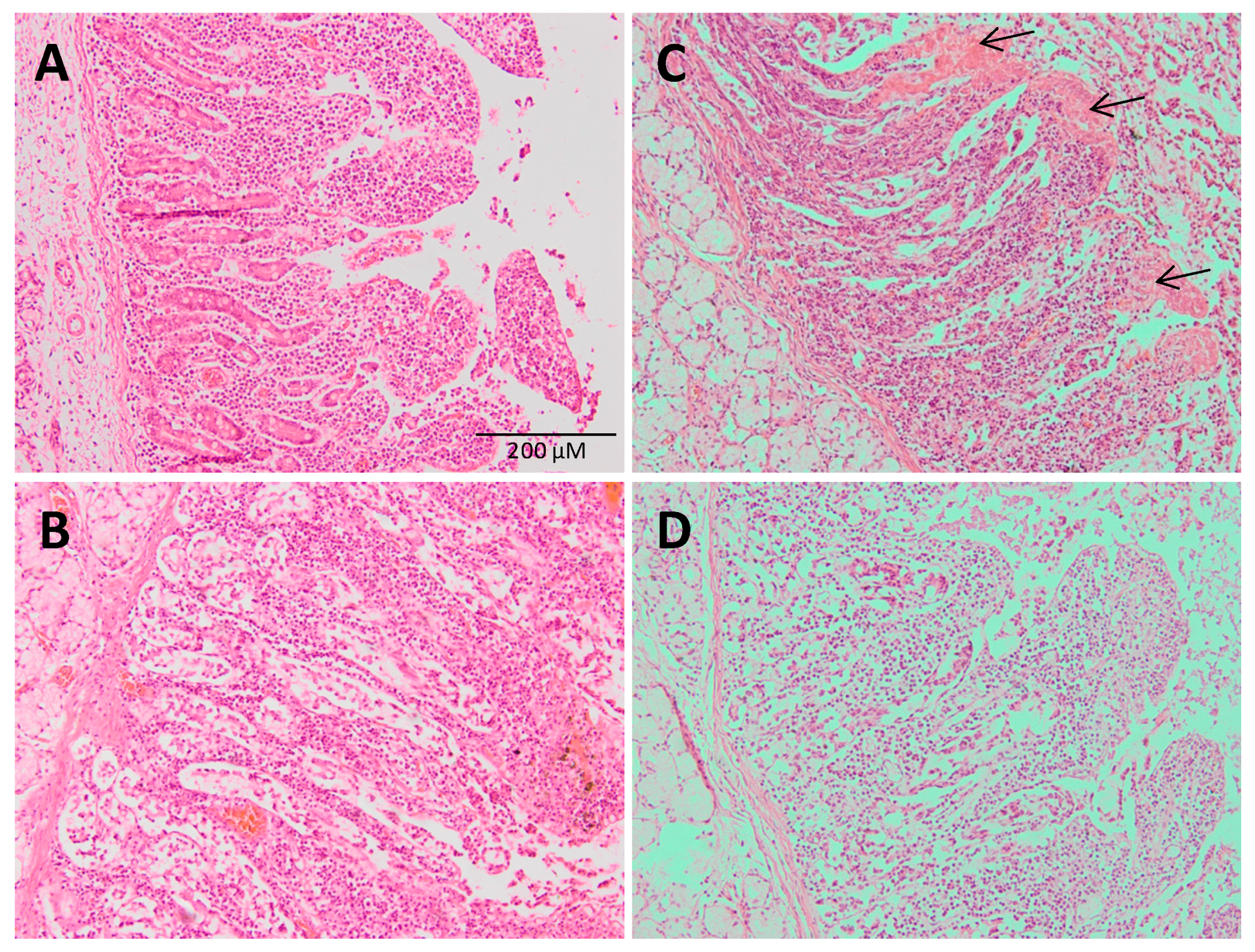
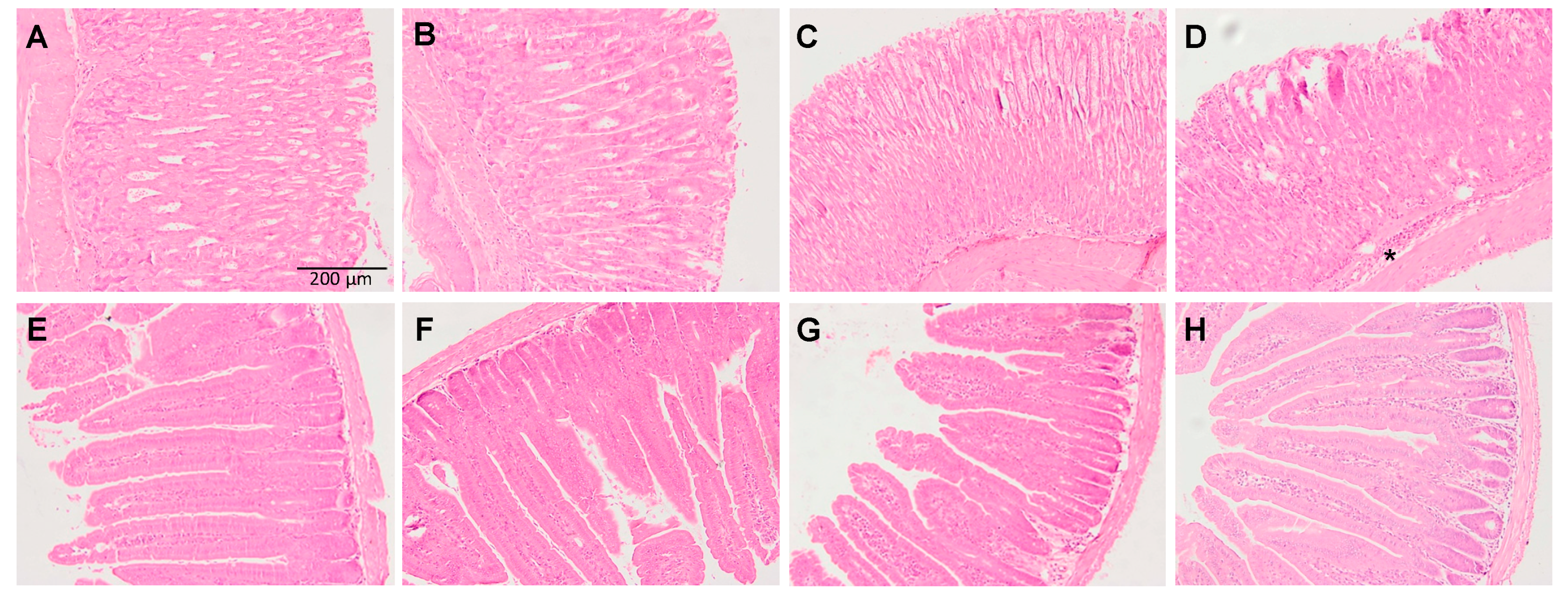
3.2. Histological Results: Pig Intestine and Mice Stomach and Intestine
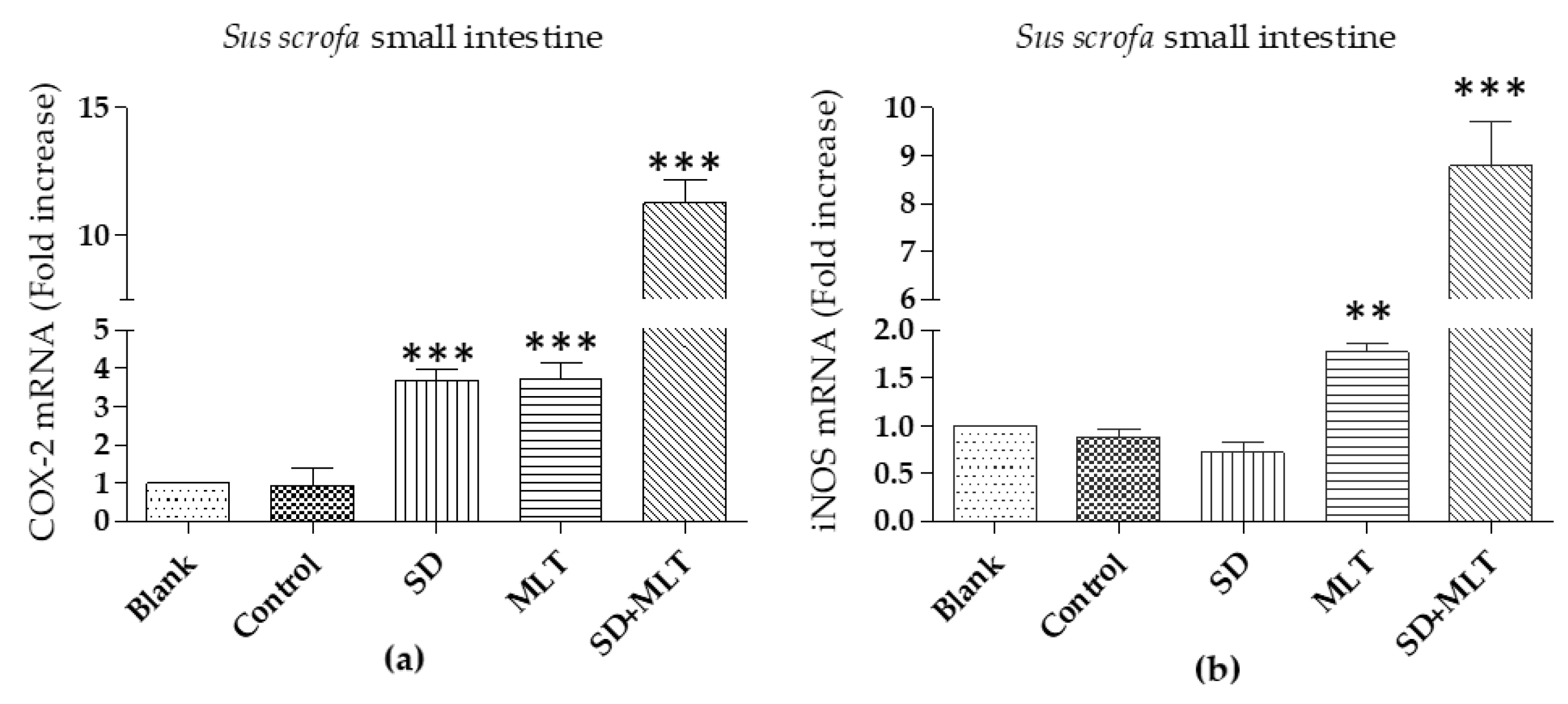
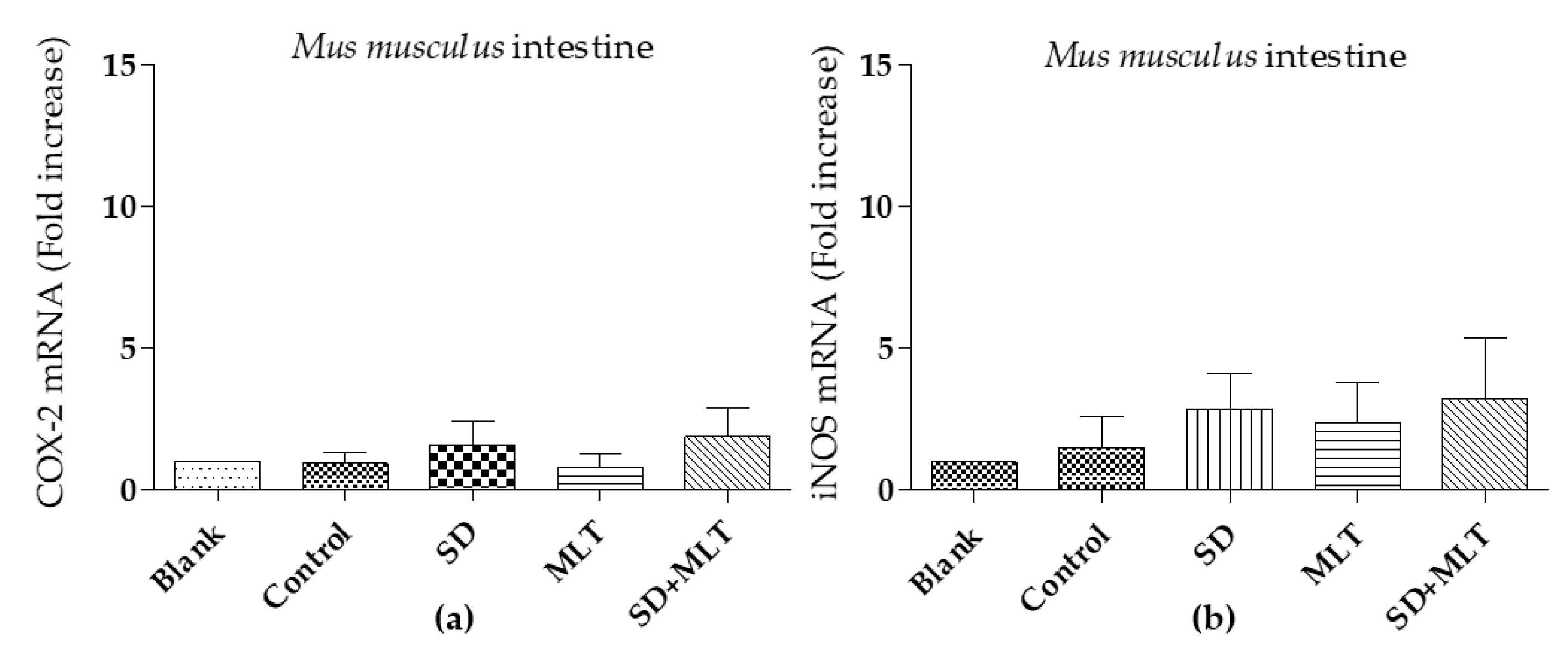
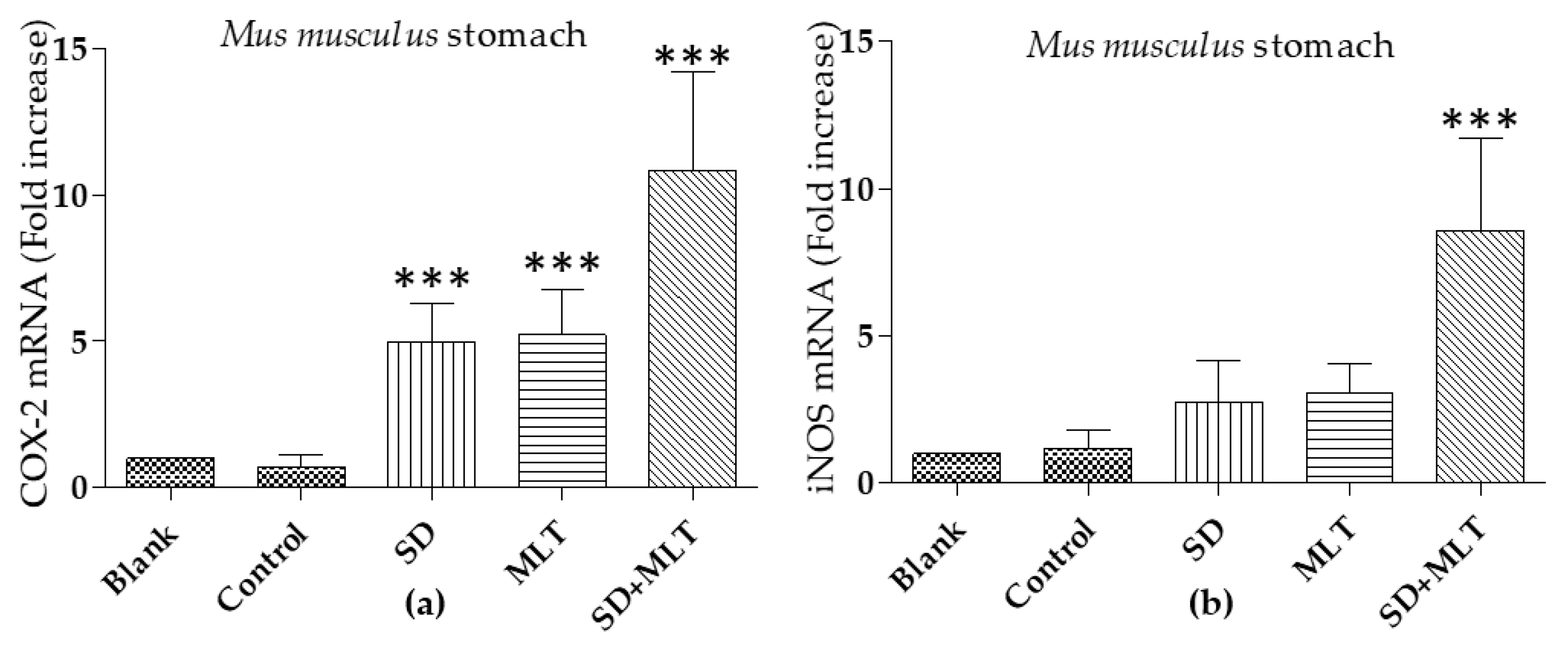
3.3. COX-2 and iNOs Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kargman, S.; Charleson, S.; Cartwright, M.; Frank, J.; Riendeau, D.; Mancini, J.; O’Neill, G. Characterization of prostaglandin G/H synthase 1 and 2 in rat, dog, monkey, and human gastrointestinal tracts. Gastroenterology 1996, 111, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, F.A. Cyclooxygenase enzymes: Regulation and function. Curr. Pharm. Des. 2004, 10, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Singer, I.I.; Kawka, D.W.; Schloemann, S.; Tessner, T.; Riehl, T.; Stenson, W.F. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 1998, 115, 297–306. [Google Scholar] [CrossRef]
- Xie, W.L.; Chipman, J.G.; Robertson, D.L.; Erikson, R.L.; Simmons, D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. PNAS 1991, 88, 2692–2696. [Google Scholar] [CrossRef]
- Robert, A.; Nezamis, J.E.; Lancaster, C.; Hanchar, A.J. Cytoprotection by prostaglandins in rats. Prevention of gastric necrosis produced by alcohol, HCl, NaOH, hypertonic NaCl, and thermal injury. Gastroenterology 1979, 77, 433–443. [Google Scholar] [CrossRef]
- Wilson, D.E.; Phillips, C.; Levine, R.A. Inhibition of gastric secretion in man by prostaglandin A1. Gastroenterology 1971, 61, 201–206. [Google Scholar] [CrossRef]
- Main, I.H.M.; Whittle, B.J.R. The effects of E and A prostaglandins on gastric mucosal blood flow and acid secretion in the rat. Br. J. Pharmacol. 1973, 49, 428–436. [Google Scholar] [CrossRef]
- Jackson, L.M.; Wu, K.; Mahida, Y.R.; Jenkins, D.; Donnelly, M.T.; Hawkey, L.C.J. Cox-1 expression in human gastric mucosa infected with helicobacter pylori: Constitutive or induced? Gut 2000, 47, 762–770. [Google Scholar] [CrossRef]
- Whittle, B.; Boughton-Smith, N.K.; Moncada, S.; Vane, J.R. Actions of prostacyclin (PGI2) and its product 6-oxo-PGF1 on the rat gastric mucosa in vivo and vitro. Prostaglandins 1978, 15, 955–967. [Google Scholar] [CrossRef]
- Masferrer, J.L.; Isakson, P.C.; Seibert, K. Cyclooxygenase-2 inhibitors: A new class of anti-inflammatory agents that spare the gastrointestinal tract. Gastroenterol. Clin. N. Am. 1996, 25, 363–372. [Google Scholar] [CrossRef]
- Kearney, P.M.; Baigent, C.; Godwin, J. Do selective cyclo-oxygenase-2 inhibitors and traditional nonsteroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 2006, 332, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.; Henry, D. Cardiovascular risk and inhibition of cyclooxygenase: A systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA 2006, 296, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Majed, B.H.; Khalil, R.A. Molecular Mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol. Rev. 2012, 64, 540–582. [Google Scholar] [CrossRef]
- Langenbach, R.; Morham, S.G.; Tiano, H.F.; Loftin, C.D.; Ghanayem, B.I.; Chulada, P.C.; Smithies, O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 1995, 83, 483–492. [Google Scholar] [CrossRef]
- Langenbach, R.; Loftin, C.; Lee, C.; Tiano, H. Cyclooxygenase knockout mice. Biochem. Pharmacol. 2002, 58, 1237–1246. [Google Scholar] [CrossRef]
- Karrasch, T.; Steinbrecher, K.A.; Allard, B.; Baldwin, A.S.; Jobin, C. Wound-induced p38MAPK-dependent histone H3 phosphorylation correlates with increased COX- 2 expression in enterocytes. J. Cell Physiol. 2006, 207, 809–815. [Google Scholar] [CrossRef]
- Kato, S.; Tanaka, A.; Kunikata, T.; Umeda, M.; Takeuchi, K. Protective effect of lafutidine against indomethacin-induced intestinal ulceration in rats: Relation to capsaicin-sensitive sensory neurons. Digestion 2000, 61, 39–46. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Wang, Z.M.; Romero, J.J.; Ulloa, C.; Perez, J.C.; Giraud, M.N.; Barreto, J.C. Nonsteroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat. Med. 1995, 1, 154–158. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Zhou, Y.; Jayaraman, V.; Doyen, J.R.; O’Neil, R.G.; Dial, E.J.; Volk, D.E.; Gorenstein, D.G.; Boggara, M.B.; Krishnamoorti, R. Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: Characterization of interaction of NSAIDs with phosphatidylcholine. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2012, 1821, 994–1002. [Google Scholar] [CrossRef]
- Bernhard, W.; Postle, A.D.; Linck, M.; Sewing, K.F. Composition of phospholipid classes and phosphatidylcholine molecular species of gastric mucosa and mucus. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 1995, 1255, 99–104. [Google Scholar] [CrossRef]
- Scheiman, J.M.; Hindley, C.E. Strategies to optimize treatment with NSAIDs in patients at risk for gastrointestinal and cardiovascular adverse events. Clin. Ther. 2010, 32, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Higuchi, K.; Kobata, A.; Nishio, H.; Tanigawa, T.; Shiba, M.; Arakawa, T. Intestinal damage is Toll-like receptor 4 dependent. Gut 2008, 57, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, L.; Zhong, C.; Geng, J. NF-KB activation for constitutive expression of VCAM-1 and ICAM-1 on B lymphocytes and plasma cells. Biochem. Biophys. Res. Commun. 2001, 289, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Konaka, A.; Kato, S.; Tanaka, A.; Kunikata, T.; Korolkiewicz, R.; Takeuchi, K. Roles of enterobacteria, nitric oxide and neutrophil in pathogenesis of indomethacin-induced small intestinal lesions in rats. Pharmacol. Res. 1999, 40, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, O.; Miwa, H.; Watanabe, S.; Tsujii, M.; Dubois, R.N.; Sato, N.; Dubois, R.N. Cyclooxygenase-2 downregulates inducible nitric oxide synthase in rat intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Asensio, C.; Levoin, N.; Guillaume, C.; Guerquin, M.J.; Rouguieg, K.; Chrétien, F.; Lapicque, F. Irreversible inhibition of glucose-6-phosphate dehydrogenase by the coenzyme A conjugate of ketoprofen: A key to oxidative stress induced by non-steroidal anti-inflammatory drugs? Biochem. Pharmacol. 2007, 73, 405–416. [Google Scholar] [CrossRef]
- Macpherson, A.; Rafi, S.; Mahmod, T.; Hayllar, J.; Jacob, M.; Scott, D.; Wrigglesworth, J.M. Mitochondrial damage: A possible mechanism of the “topical” phase of NSAID induced injury to the rat intestine. Gut 2010, 41, 344–353. [Google Scholar]
- Basivireddy, J.; Vasudevan, A.; Jacob, M.; Balasubramanian, K.A. Indomethacin-induced mitochondrial dysfunction and oxidative stress in villus enterocytes. Biochem. Pharmacol. 2002, 64, 339–349. [Google Scholar] [CrossRef]
- Mahmud, T.; Rafi, S.S.; Scott, D.L.; Wrigglesworth, J.M.; Bjarnason, I. Nonsteroidal antiinflammatory drugs and uncoupling of mitochondrial oxidative phosphorylation. Arthritis Rheum. 1996, 39, 1998–2003. [Google Scholar] [CrossRef]
- Maharaj, A.S.; D’Amore, P.A. Roles for VEGF in the adult. Microvasc. Res. 2007, 74, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, F.; Whittle, B.J.R. Role of nitric oxide and platelet-activating factor in the initiation of indomethacin-provoked intestinal inflammation in rats. Eur. J. Pharmacol. 1998, 344, 191–195. [Google Scholar] [CrossRef]
- Khattab, M.M. Protective role of nitric oxide in indomethacin-induced gastric ulceration by a mechanism independent of gastric acid secretion. Pharmacol. Res. 2001, 43, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kitamura, M.; Korolkiewicz, R.P.; Takeuchi, K. Role of nitric oxide in regulation of gastric acid secretion in rats: Effects of NO donors and NO synthase inhibitor. Br. J. Pharmacol. 1998, 839–846. [Google Scholar] [CrossRef]
- Lee, M.; Kallal, S.M.; Feldman, M. Omeprazole prevents indomethacin-induced gastric ulcers in rabbits. Aliment. Pharmacol. Ther. 1996, 10, 571–576. [Google Scholar] [CrossRef]
- Fornai, M.; Natale, G.; Colucci, R.; Tuccori, M.; Carazzina, G.; Antonioli, L.; Del Tacca, M. Mechanisms of protection by pantoprazole against NSAID-induced gastric mucosal damage. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2005, 372, 79–87. [Google Scholar] [CrossRef]
- Fornai, M.; Colucci, R.; Antonioli, L.; Awwad, O.; Ugolini, C.; Tuccori, M.; Blandizzi, C. Effects of esomeprazole on healing of nonsteroidal anti-inflammatory drug (NSAID)-induced gastric ulcers in the presence of a continued NSAID treatment: Characterization of molecular mechanisms. Pharmacol. Res. 2011, 63, 59–67. [Google Scholar] [CrossRef]
- Rodriguez-Stanley, S.; Redinger, N.; Miner, P.B. Effect of naproxen on gastric acid secretion and gastric pH. Aliment. Pharmacol. Ther. 2006, 23, 1719–1724. [Google Scholar] [CrossRef]
- Pearson, S.P.; Kelberman, I. Gastrointestinal effects of NSAIDs and coxibs. Postgrad. Med. 1996, 100, 131–143. [Google Scholar] [CrossRef]
- Li, Y.; Soendergaard, C.; Bergenheim, F.H.; Aronoff, D.M.; Milne, G.; Riis, L.B.; Nielsen, O.H. COX-2–PGE 2 signaling impairs intestinal epithelial regeneration and associates with TNF inhibitor responsiveness in ulcerative colitis. EBioMedicine 2018, 36, 497–507. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Vacca, M.; Moschetta, A.; Cinzia Sasso, R.; Palasciano, G.; van Erpecum, K.J.; Portincasa, P. Intestinal mucosal damage caused by non-steroidal anti-inflammatory drugs: Role of bile salts. Clin. Biochem. 2007, 40, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Poplawski, C.; Sosnowski, D.; Szaflarska-Popławska, A.; Sarosiek, J.; McCallum, R.; Bartuzi, Z. Role of bile acids, prostaglandins and COX inhibitors in chronic esophagitis in a mouse model. World J. Gastroenterol. 2006, 12, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Barrios, J.M.; Lichtenberger, L.M. Role of biliary phosphatidylcholine in bile acid protection and NSAIDs injury of the lleal mucosa in rats. Gastroenterology 2000, 118, 1179–1186. [Google Scholar] [CrossRef]
- Savarino, V.; Dulbecco, P.; Savarino, E. Are proton pump inhibitors really so dangerous? Dig. Liver Dis. 2016, 48, 851–859. [Google Scholar] [CrossRef]
- Seekatz, A.M.; Schnizlein, M.K.; Koenigsknecht, M.J.; Baker, J.R.; Bleske, B.E.; Young, V.B. Spatial and temporal analysis of the stomach and small-intestinal microbiota in fasted healthy humans. mSphere 2019, 4, e00126-19. [Google Scholar] [CrossRef]
- Lo, W.K.; Chan, W.W. Proton pump inhibitor use and the risk of small intestinal bacterial overgrowth: A meta-analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 483–490. [Google Scholar] [CrossRef]
- Miazga, A.; Osiński, M.; Cichy, W.; Żaba, R. Advances in medical sciences current views on the etiopathogenesis, clinical manifestation, diagnostics, treatment and correlation with other nosological entities of SIBO. Adv. Med. Sci. 2015, 60, 118–124. [Google Scholar] [CrossRef]
- Daniell, H.W. Acid suppressing therapy as a risk factor for Candida esophagitis. Dis. Esophagus 2016, 29, 479–483. [Google Scholar] [CrossRef]
- Jung, S.B.; Nagaraja, V.; Kapur, A.; Eslick, G.D. Association between vitamin B12 deficiency and long-term use of acid-lowering agents: A systematic review and meta-analysis. Intern. Med. J. 2015, 45, 409–416. [Google Scholar] [CrossRef]
- Sánchez, A.; Calpena, A.C.; Clares, B. Evaluating the Oxidative Stress in Inflammation: Role of Melatonin. Int. J. Mol. Sci. 2015, 16, 16981–17004. [Google Scholar] [CrossRef] [PubMed]
- Adeyeye, C.M.; Li, P.K. Diclofenac sodium. In Analytical Profiles of Drug Substances; Florey, K., Ed.; Academic Press Inc.: San Diego, CA, USA, 1990; Volume 19, pp. 123–144. [Google Scholar]
- Mei, Q.; Diao, L.; Xu, J.; Liu, X.; Jin, J. A protective effect of melatonin on intestinal permeability is induced by diclofenac via regulation of mitochondrial function in mice. Acta Pharmacol. Sin. 2011, 32, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.B.; Calpena, A.C.; Mallandrich, M.; Clares, B. Validation of an ex vivo permeation method for the intestinal permeability of different BCS drugs and its correlation with caco-2 in vitro experiments. Pharmaceutics 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Pineal Melatonin: Cell biology of its synthesis and of its physiological interactions. EndocR. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef]
- Shida, C.S.; Castrucci, A.M.; Lamy-Freund, M.T. High melatonin solubility in aqueous medium. J. Pineal Res. 1994, 16, 198–201. [Google Scholar] [CrossRef]
- Munford, M.L.; Pasa, A. Influence of melatonin on the order of phosphatidylcholine-based membranes. J. Pineal Res. 2010, 49, 169–175. [Google Scholar]
- Dies, H.; Cheung, B.; Tang, J.; Rheinstädter, M.C. The organization of melatonin in lipid membranes. Biochim. Biophys. Acta 2015, 1848, 1032–1040. [Google Scholar] [CrossRef]
- Johns, J. Estimation of melatonin blood brain barrier permeability. J. Bioanaly. Biomed. 2011, 3, 64–69. [Google Scholar] [CrossRef]
- Alluri, H.; Wilson, R.L.; Shaji, C.A.; Wiggins, K. Melatonin preserves blood-brain barrier integrity and permeability via matrix metalloproteinase-9 inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef]
- Reiter, R.J.; Fuentes-broto, L.; Paredes, S.D.; Tan, D.; Garcia, J.J. Melatonin and the pathophysiology of cellular membranes. Marmara Pharm. J. 2010, 14, 1–9. [Google Scholar] [CrossRef]
- Res, P. Melatonin in the duodenal lumen is a potent stimulant of mucosal bicarbonate secretion. J. Pineal Res. 2003, 34, 288–293. [Google Scholar]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. PNAS 2006, 86, 99–103. [Google Scholar] [CrossRef]
- Kim, H.J.; So, H.S.; Lee, J.H.; Lee, J.H.; Park, C.; Park, S.Y.; Park, R. Heme oxygenase-1 attenuates the cisplatin-induced apoptosis of auditory cells via down-regulation of reactive oxygen species generation. Free Radic. Biol. Med. 2006, 40, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Pal, C.; Dey, S.; Goyal, M.; Alam, A.; Iqbal, M.S.; Bandyopadhyay, U. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 2011, 286, 39387–39402. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lu, K.C.; Lin, G.J.; Hsieh, H.Y.; Chu, P.; Lin, S.H.; Sytwu, H.K. Melatonin enhances endogenous heme oxygenase-1 and represses immune responses to ameliorate experimental murine membranous nephropathy. J. Pineal Res. 2012, 52, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Tang, S.; Tseng, H.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef]
- Machowska, A.; Szlachcic, A.; Pawlik, M.; Brzozowski, T.; Konturek, S.J.; Pawlik, W.W. The role of female and male sex hormones in the healing process of preexisting lingual and gastric ulcerations. J. Physiol. Pharmacol. 2004, 55 (Suppl. 2), 91–104. [Google Scholar]
- Sangma, T.K.; Jain, S.; Mediratta, P.K. Effect of ovarian sex hormones on non-steroidal anti-inflammatory drug-induced gastric lesions in female rats. Indian J. Pharmacol. 2014, 46, 113–116. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Number | Permeated Drug | Permeated Solution | Dose |
|---|---|---|---|
| 1 (n = 6) | No drug (Reference group) | - | - |
| 2 (n = 6) | SD | 1 mg/mL in PBS pH 7.4 | 1 mL |
| 3 (n = 6) | MLT | 0.5 mg/mL in PBS pH 7.4 | 1 mL |
| 4 (n = 6) | SD (a) + MLT (b) | 2 mg/mL (a) 1 mg/mL (b) | 0.5 mL + 0.5 mL |
| Group number | Group | Administered Drugs | Dose |
|---|---|---|---|
| 1 (n = 6) | REF | No drug (Reference group) | - |
| 2 (n = 6) | SD | Sodium Diclofenac | 2.5 mg/kg |
| 3 (n = 6) | MLT | Melatonin | 10 mg/kg |
| 4 (n = 6) | SD + MLT | Sodium Diclofenac and Melatonin | 2.5 mg/kg and 10 mg/kg |
| Primer | Animal Model | Sequence (5′ to 3′) | Acc. Number |
|---|---|---|---|
| COX-2 | Sus scrofa | FW: GGAGAGACAGCATAAACTGC | AF207824 |
| RV: GTGTGTTAAACTCAGCAGCA | |||
| Mus musculus | FW: CCACTTCAAGGGAGTCTGGA | NM_011198.4 | |
| RV: AGTCATCTGCTACGGGAGGA | |||
| iNOS | Sus scrofa | FW: CAACAATGGCAACATCAGG | U59390 |
| RV: CATCAGGCATCTGGTAGC | |||
| Mus musculus | FW: GTTCTCAGCCCAACAATACAAGA | NM_010927 | |
| RV: GTGGACGGGTCGATGTCAC | |||
| β-actin | Sus scrofa | FW: GACATCCGCAAGGACCTCTA | DQ845171 |
| RV: ACACGGAGTACTTGCGCTCT | |||
| Mus musculus | FW: GGCCGGGACCTGACAGACTACCTC | NM_007393 | |
| RV: GTCACGCACGATTTCCCTCTCAGC |
| Permeation Parameters | SD Alone | SD+MLT | Unpaired t-Test (p) |
|---|---|---|---|
| Flux (µg/min) | 1.21 ± 0.11 | 1.39 ± 0.20 | - |
| Flux/sup (µg/(cm/min)) | 0.48 ± 0.04 | 0.55 ± 0.10 | - |
| Co (µg/mL) | 1000 | 1000 | - |
| Papp (×10−6) (cm/s) | 7.92 ± 0.73 | 9.12 ± 0.13 | 0.078 (>0.05) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, A.B.; Clares, B.; Rodríguez-Lagunas, M.J.; Fábrega, M.J.; Calpena, A.C. Study of Melatonin as Preventive Agent of Gastrointestinal Damage Induced by Sodium Diclofenac. Cells 2020, 9, 180. https://doi.org/10.3390/cells9010180
Sánchez AB, Clares B, Rodríguez-Lagunas MJ, Fábrega MJ, Calpena AC. Study of Melatonin as Preventive Agent of Gastrointestinal Damage Induced by Sodium Diclofenac. Cells. 2020; 9(1):180. https://doi.org/10.3390/cells9010180
Chicago/Turabian StyleSánchez, Aroha B., Beatriz Clares, María J. Rodríguez-Lagunas, María J. Fábrega, and Ana C. Calpena. 2020. "Study of Melatonin as Preventive Agent of Gastrointestinal Damage Induced by Sodium Diclofenac" Cells 9, no. 1: 180. https://doi.org/10.3390/cells9010180
APA StyleSánchez, A. B., Clares, B., Rodríguez-Lagunas, M. J., Fábrega, M. J., & Calpena, A. C. (2020). Study of Melatonin as Preventive Agent of Gastrointestinal Damage Induced by Sodium Diclofenac. Cells, 9(1), 180. https://doi.org/10.3390/cells9010180