In Silico Analysis of the Age-Dependent Evolution of the Transcriptome of Mouse Skin Stem Cells



Abstract
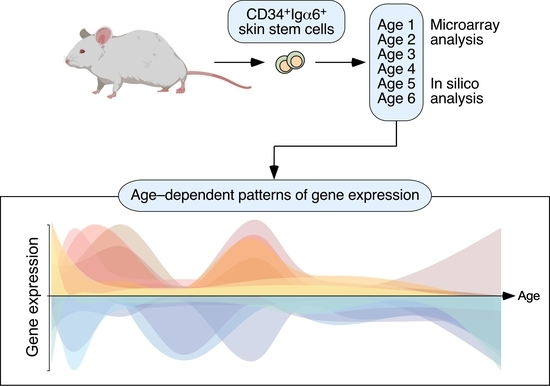
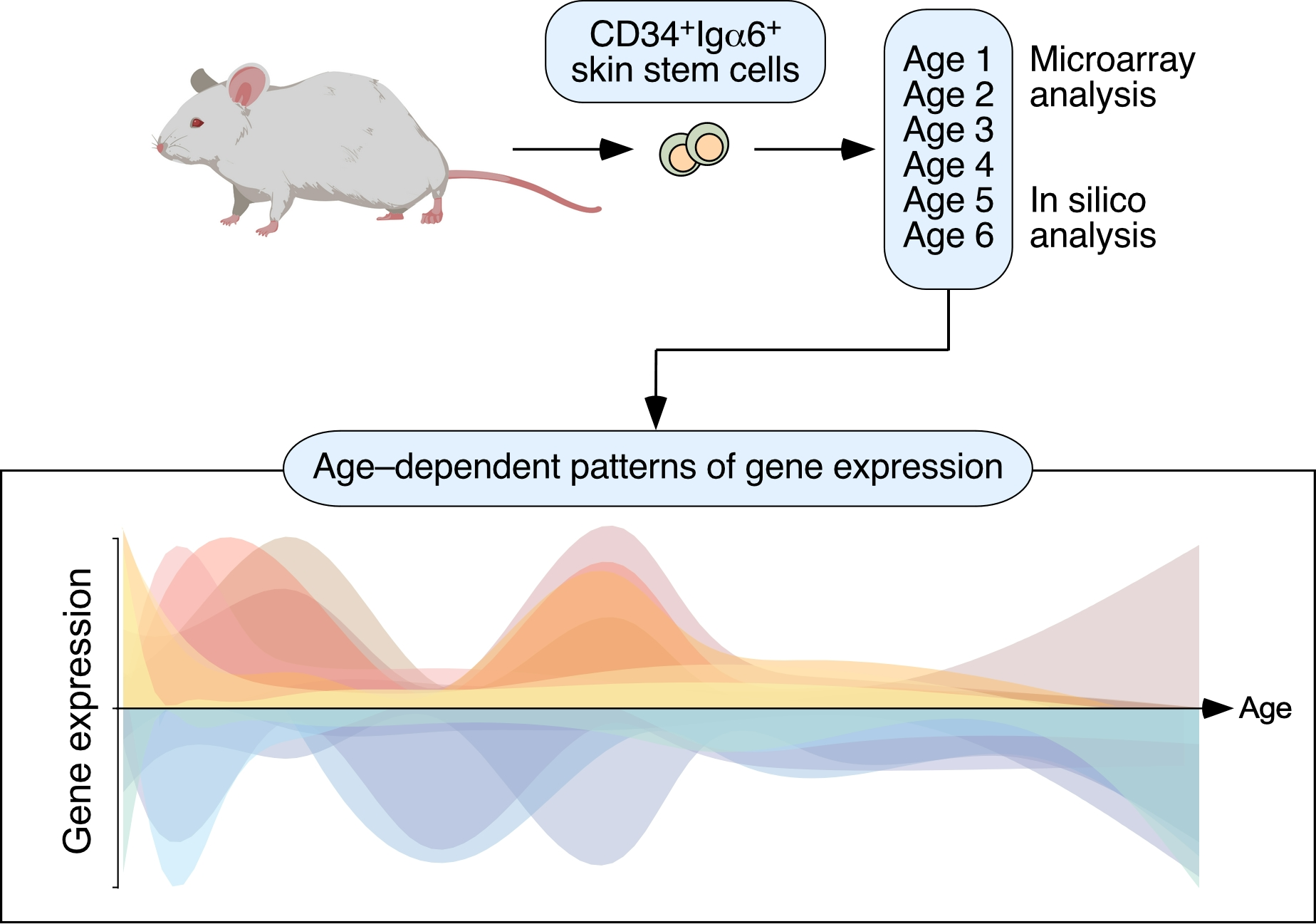{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Animal Studies
2.2. Isolation of CD34+ Itgα6+ SSCs
2.3. RNA Extraction and Transcriptome Profiling
2.4. Establishment of Gene Expression Patterns
2.5. Gene Set Annotation and Enrichment
2.6. Weighted Correlation Network Analyses
2.7. Data Availability
3. Results
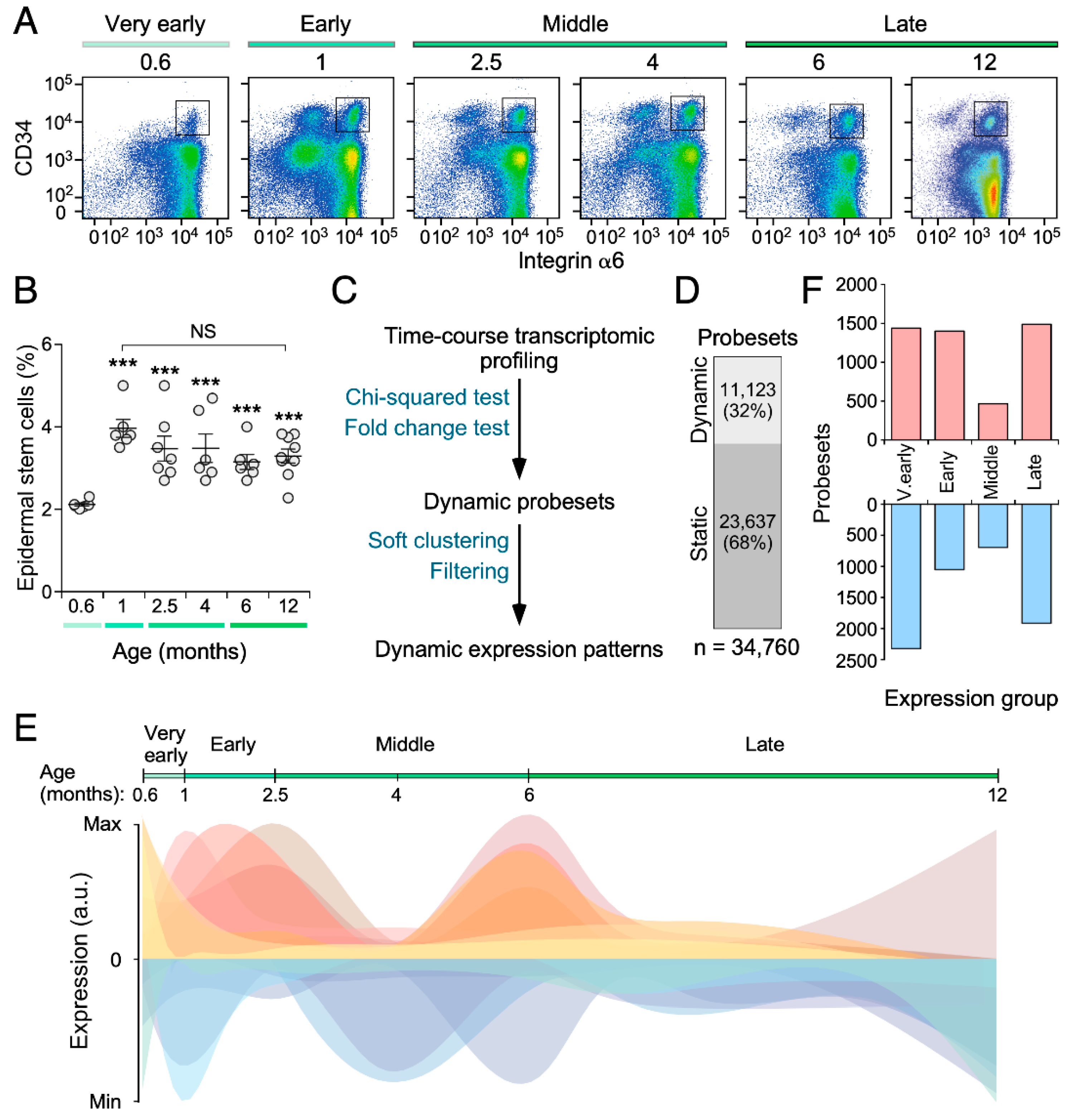
3.1. The SSC Transcriptome Fluctuates in an Age-Dependent Manner
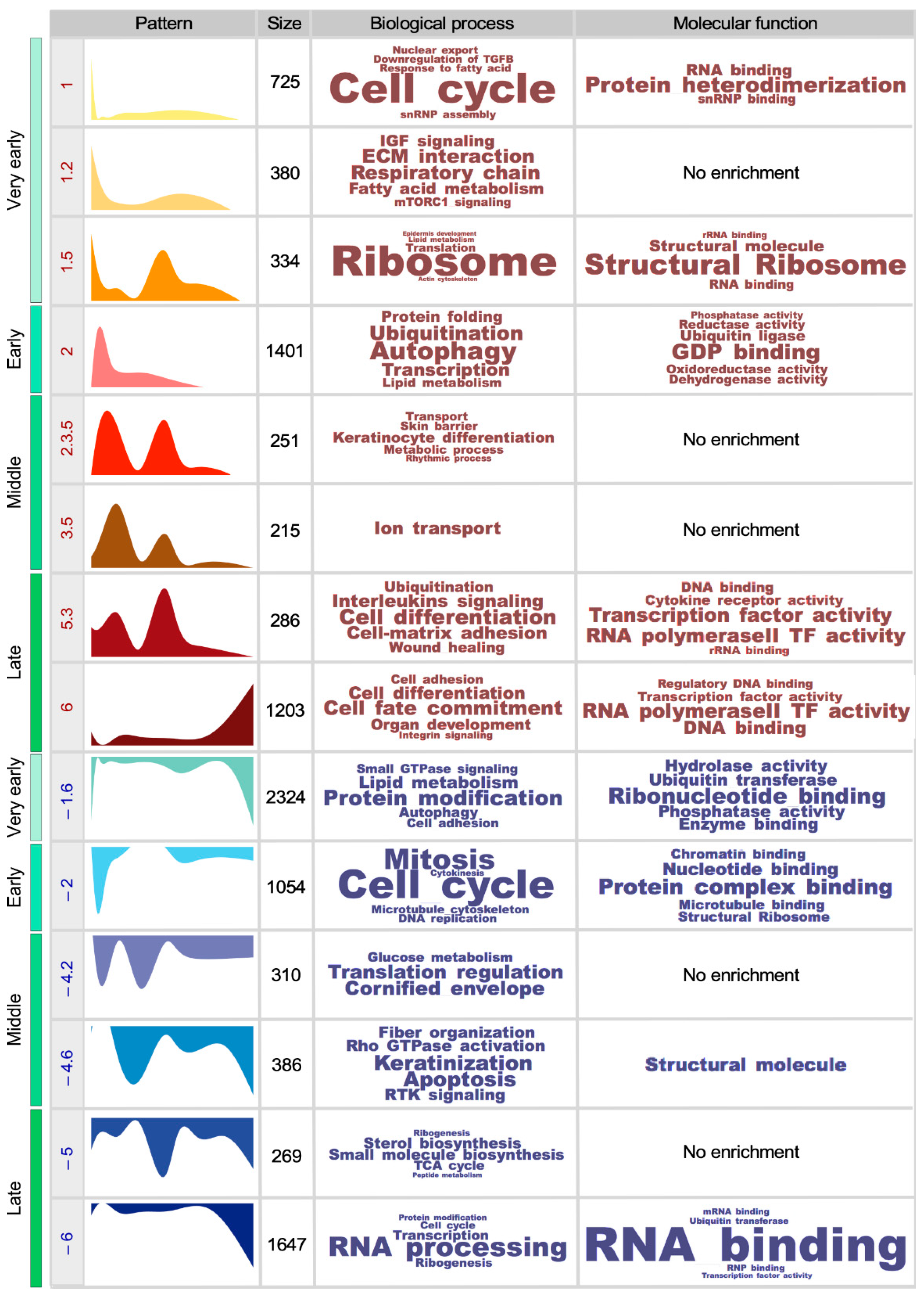
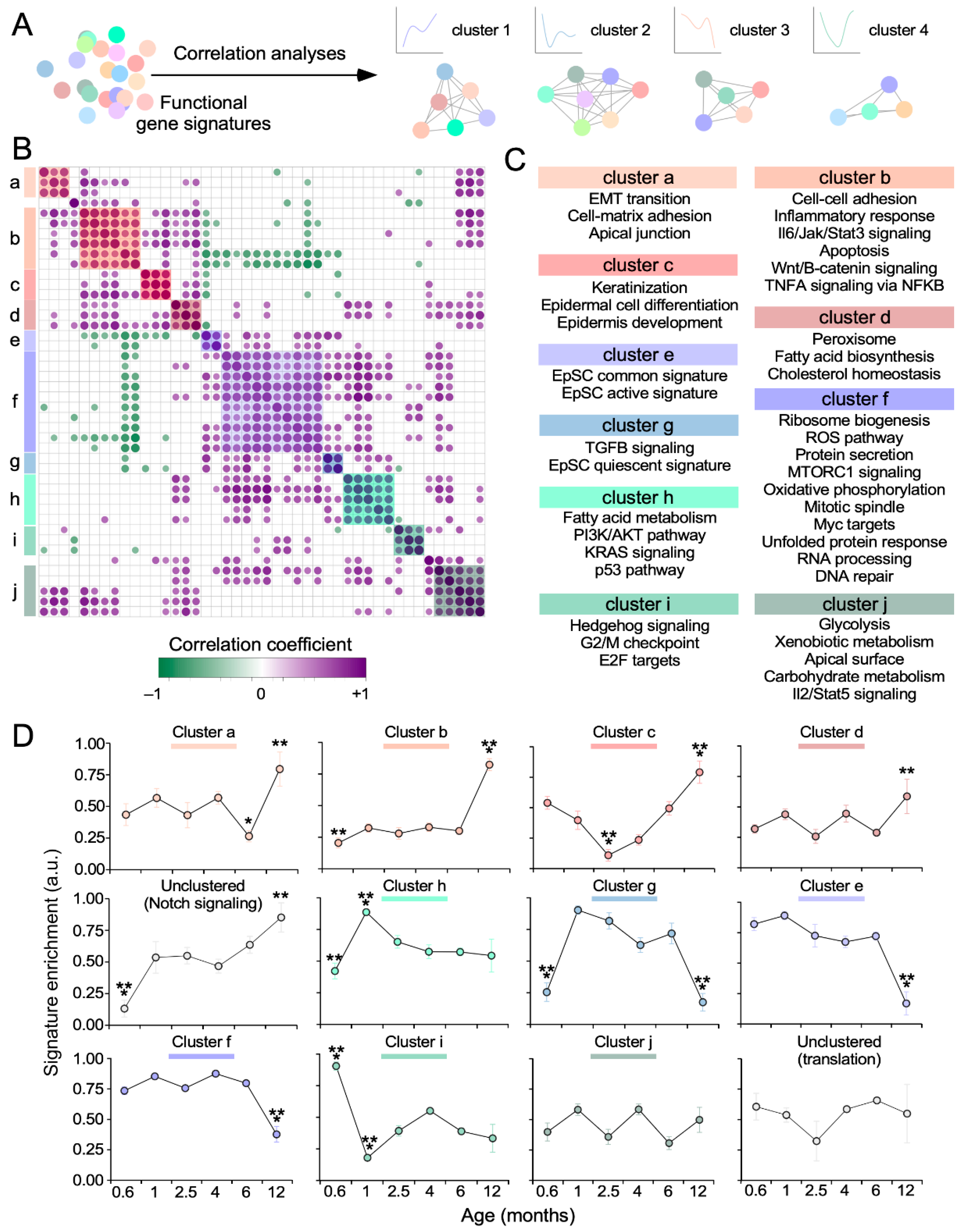
3.2. The Functionality of the SSC Transcriptome Drifts over Time
3.3. Key SSC-Related Signaling Pathways are also Age-Sensitive
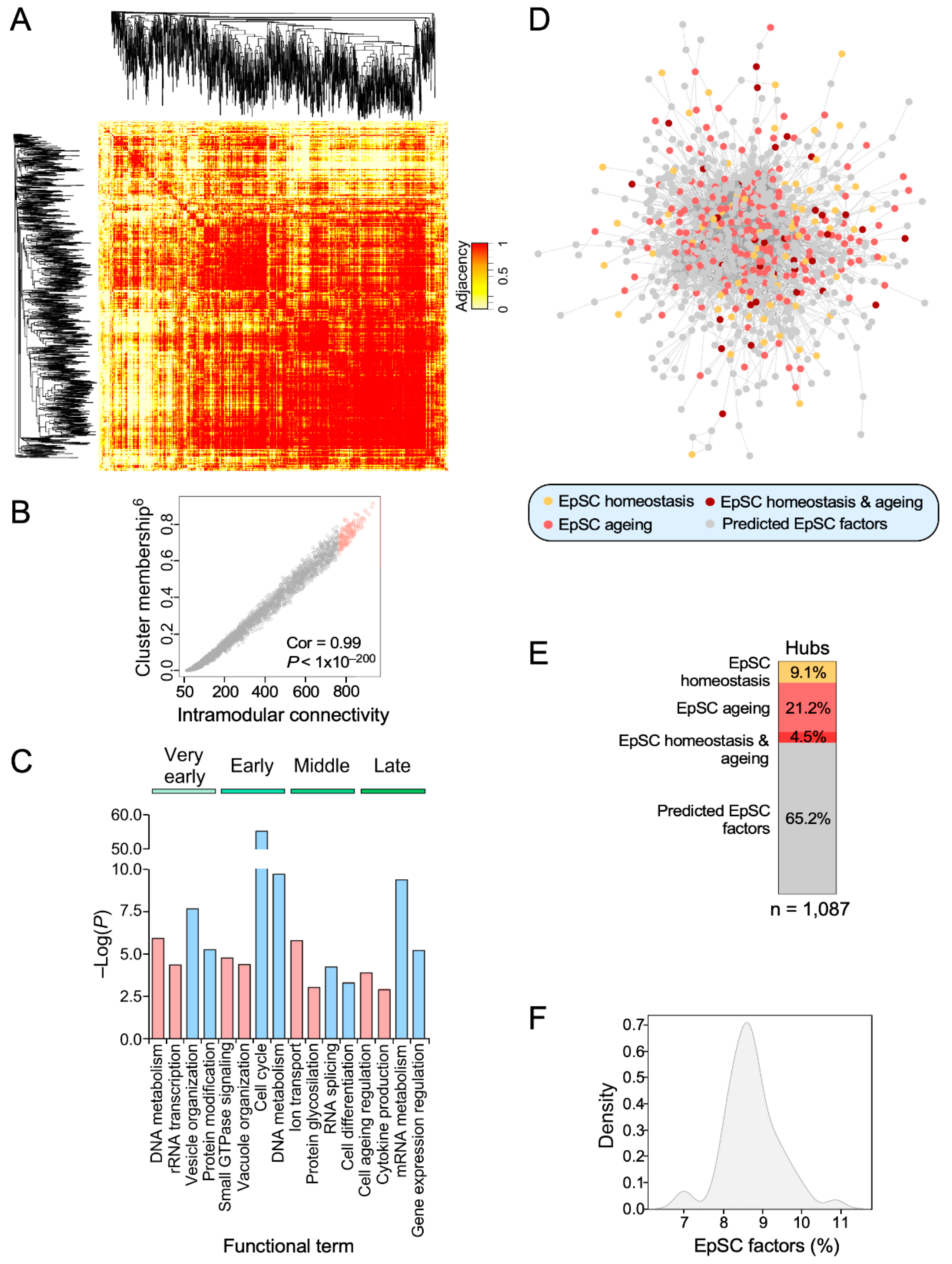
3.4. Identification of Age-Dependent Transcriptional Hubs in SSCs
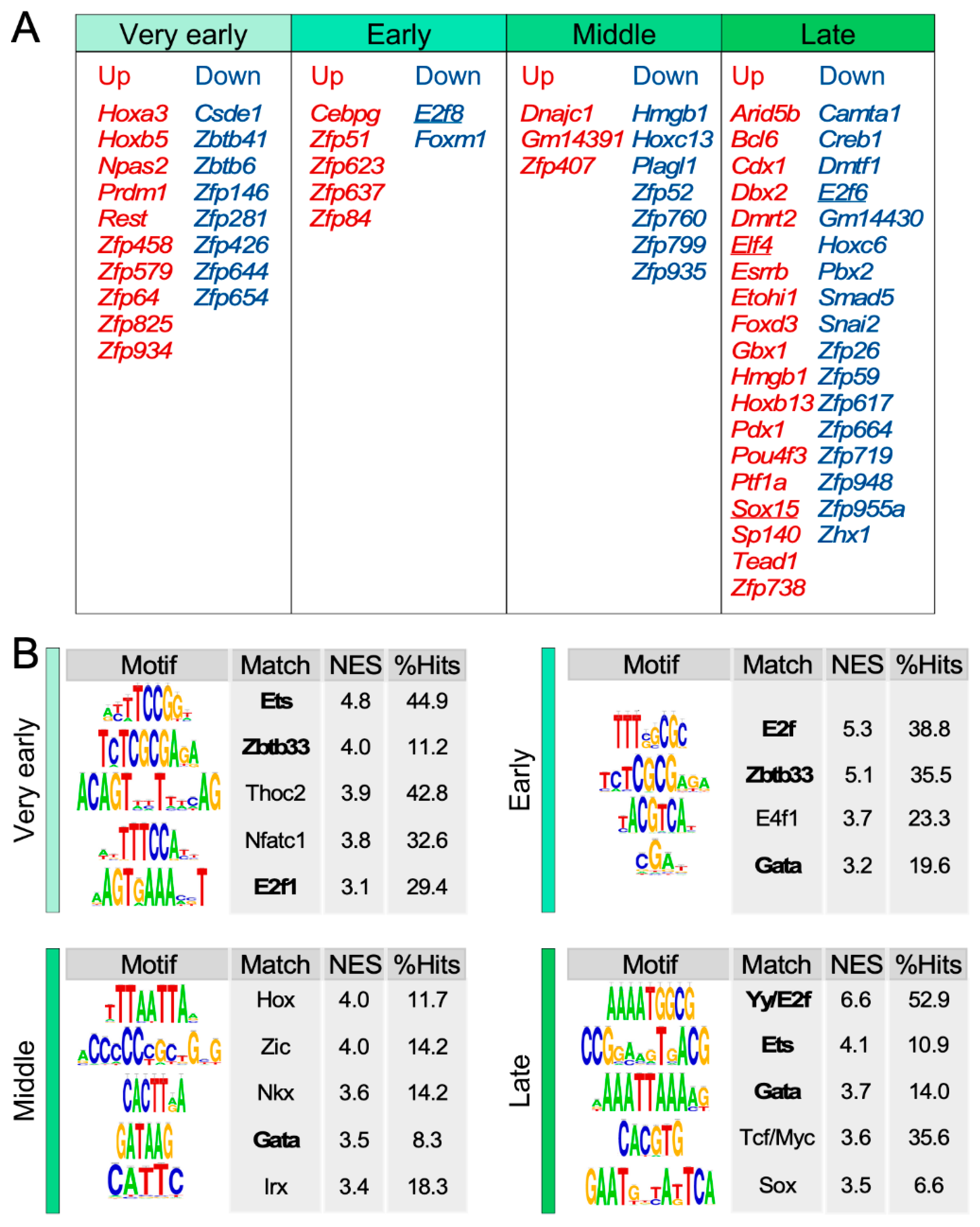
3.5. Identification of Transcriptional Factors Potentially Involved in the SSC Transcriptome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blanpain, C.; Fuchs, E. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 2006, 22, 339–373. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Skin stem cells: Rising to the surface. J. Cell Biol. 2008, 180, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Racila, D.; Bickenbach, J.R. Are epidermal stem cells unique with respect to aging? Aging 2009, 1, 746–750. [Google Scholar] [CrossRef]
- Giangreco, A.; Qin, M.; Pintar, J.E.; Watt, F.M. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 2008, 7, 250–259. [Google Scholar] [CrossRef]
- Doles, J.; Storer, M.; Cozzuto, L.; Roma, G.; Keyes, W.M. Age-associated inflammation inhibits epidermal stem cell function. Genes Dev. 2012, 26, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 2004, 118, 635–648. [Google Scholar] [CrossRef]
- Joost, S.; Zeisel, A.; Jacob, T.; Sun, X.; La Manno, G.; Lonnerberg, P.; Linnarsson, S.; Kasper, M. Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst. 2016, 3, 221–237. [Google Scholar] [CrossRef]
- Zhang, Y.V.; Cheong, J.; Ciapurin, N.; McDermitt, D.J.; Tumbar, T. Distinct self-renewal and differentiation phases in the niche of infrequently dividing hair follicle stem cells. Cell Stem Cell 2009, 5, 267–278. [Google Scholar] [CrossRef]
- Lien, W.H.; Guo, X.; Polak, L.; Lawton, L.N.; Young, R.A.; Zheng, D.; Fuchs, E. Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 2011, 9, 219–232. [Google Scholar] [CrossRef]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar] [CrossRef] [PubMed]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 2017, 20, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Janich, P.; Pascual, G.; Merlos-Suarez, A.; Batlle, E.; Ripperger, J.; Albrecht, U.; Cheng, H.Y.; Obrietan, K.; Di Croce, L.; Benitah, S.A. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature 2011, 480, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Menacho-Marquez, M.; Garcia-Escudero, R.; Ojeda, V.; Abad, A.; Delgado, P.; Costa, C.; Ruiz, S.; Alarcon, B.; Paramio, J.M.; Bustelo, X.R. The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol. 2013, 11, e1001615. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Toufighi, K.; Yang, J.S.; Luis, N.M.; Aznar Benitah, S.; Lehner, B.; Serrano, L.; Kiel, C. Dissecting the calcium-induced differentiation of human primary keratinocytes stem cells by integrative and structural network analyses. PLoS Comput. Biol. 2015, 11, e1004256. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Tripathi, S.; Pohl, M.O.; Zhou, Y.; Rodriguez-Frandsen, A.; Wang, G.; Stein, D.A.; Moulton, H.M.; DeJesus, P.; Che, J.; Mulder, L.C.; et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 2015, 18, 723–735. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Kumar, V.; Geyfman, M.; Chudova, D.; Ihler, A.T.; Smyth, P.; Paus, R.; Takahashi, J.S.; Andersen, B. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 2009, 5, e1000573. [Google Scholar] [CrossRef] [PubMed]
- Janky, R.; Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Naval Sanchez, M.; Potier, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Han, J.D.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef]
- Akagi, T.; Kuure, S.; Uranishi, K.; Koide, H.; Costantini, F.; Yokota, T. ETS-related transcription factors ETV4 and ETV5 are involved in proliferation and induction of differentiation-associated genes in embryonic stem (ES) cells. J. Biol. Chem. 2015, 290, 22460–22473. [Google Scholar] [CrossRef]
- Wang, L.; Miao, Y.L.; Zheng, X.; Lackford, B.; Zhou, B.; Han, L.; Yao, C.; Ward, J.M.; Burkholder, A.; Lipchina, I.; et al. The THO complex regulates pluripotency gene mRNA export and controls embryonic stem cell self-renewal and somatic cell reprogramming. Cell Stem Cell 2013, 13, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Kim, S.W.; Lyons, J.P.; Ji, H.; Nguyen, T.T.; Cho, K.; Barton, M.C.; Deroo, T.; Vleminckx, K.; Moon, R.T.; et al. Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev. Cell 2005, 8, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Horsley, V.; Aliprantis, A.O.; Polak, L.; Glimcher, L.H.; Fuchs, E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 2008, 132, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Caramel, J.; Goguet-Rubio, P.; Linares, L.K.; Estrach, S.; Hatchi, E.; Rodier, G.; Lledo, G.; De Bettignies, C.; Thepot, A.; et al. Transcription factor E4F1 is essential for epidermal stem cell maintenance and skin homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 21076–21081. [Google Scholar] [CrossRef]
- Kaufman, C.K.; Zhou, P.; Pasolli, H.A.; Rendl, M.; Bolotin, D.; Lim, K.C.; Dai, X.; Alegre, M.L.; Fuchs, E. GATA-3: An unexpected regulator of cell lineage determination in skin. Genes Dev. 2003, 17, 2108–2122. [Google Scholar] [CrossRef]
- Wang, A.B.; Zhang, Y.V.; Tumbar, T. Gata6 promotes hair follicle progenitor cell renewal by genome maintenance during proliferation. EMBO J. 2017, 36, 61–78. [Google Scholar] [CrossRef]
- Kadaja, M.; Keyes, B.E.; Lin, M.; Pasolli, H.A.; Genander, M.; Polak, L.; Stokes, N.; Zheng, D.; Fuchs, E. SOX9: A stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev. 2014, 28, 328–341. [Google Scholar] [CrossRef]
- Foronda, M.; Martinez, P.; Schoeftner, S.; Gomez-Lopez, G.; Schneider, R.; Flores, J.M.; Pisano, D.G.; Blasco, M.A. Sox4 links tumor suppression to accelerated aging in mice by modulating stem cell activation. Cell Rep. 2014, 8, 487–500. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzo-Martín, L.F.; Bustelo, X.R. In Silico Analysis of the Age-Dependent Evolution of the Transcriptome of Mouse Skin Stem Cells. Cells 2020, 9, 165. https://doi.org/10.3390/cells9010165
Lorenzo-Martín LF, Bustelo XR. In Silico Analysis of the Age-Dependent Evolution of the Transcriptome of Mouse Skin Stem Cells. Cells. 2020; 9(1):165. https://doi.org/10.3390/cells9010165
Chicago/Turabian StyleLorenzo-Martín, L. Francisco, and Xosé R. Bustelo. 2020. "In Silico Analysis of the Age-Dependent Evolution of the Transcriptome of Mouse Skin Stem Cells" Cells 9, no. 1: 165. https://doi.org/10.3390/cells9010165
APA StyleLorenzo-Martín, L. F., & Bustelo, X. R. (2020). In Silico Analysis of the Age-Dependent Evolution of the Transcriptome of Mouse Skin Stem Cells. Cells, 9(1), 165. https://doi.org/10.3390/cells9010165