Pathogens MenTORing Macrophages and Dendritic Cells: Manipulation of mTOR and Cellular Metabolism to Promote Immune Escape

Abstract
1. Introduction
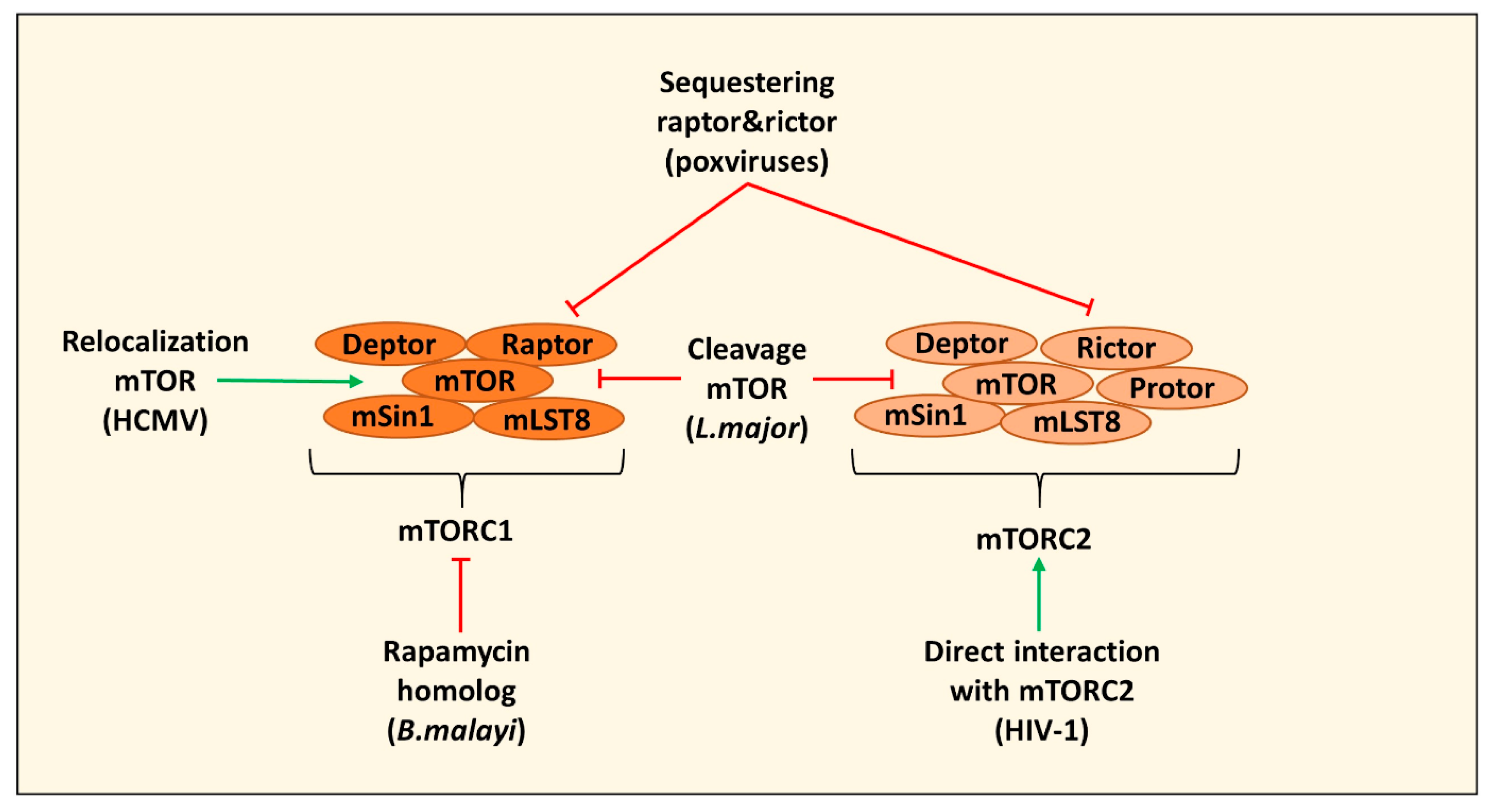
2. Direct Targeting of mTOR Complexes by Pathogens
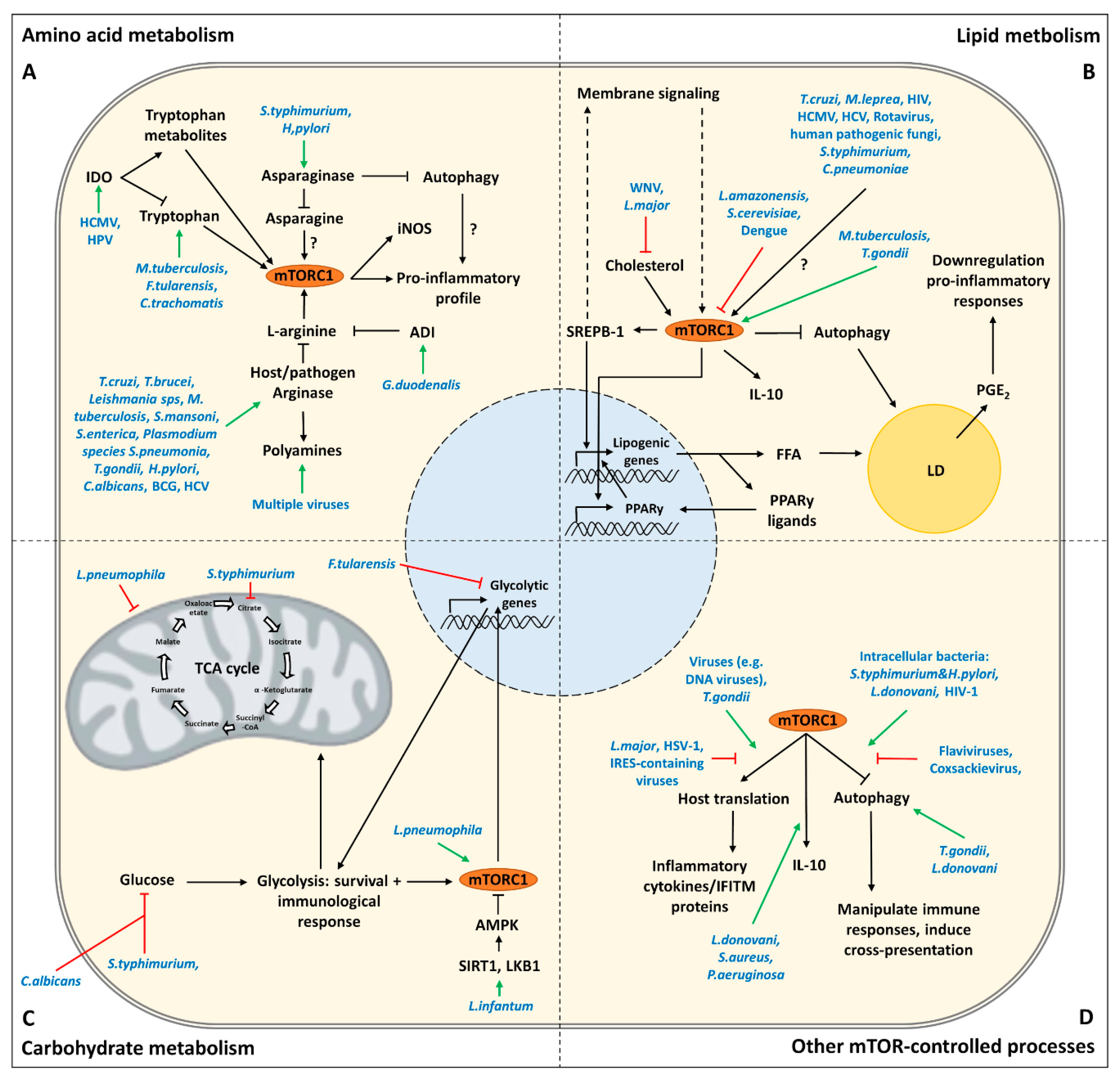
3. Targeting of Amino Acid Metabolism by Pathogens
3.1. Tryptophan
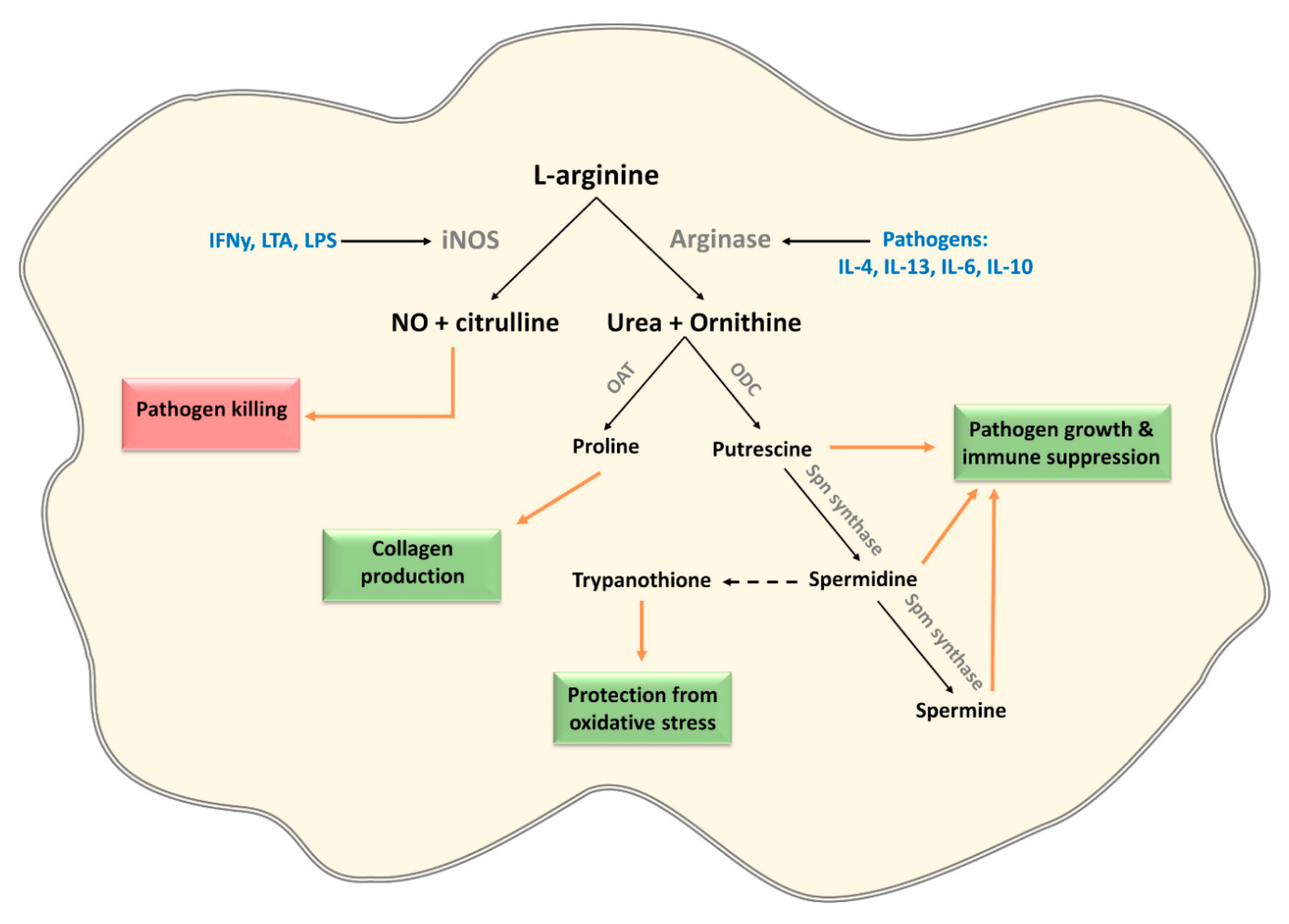
3.2. L-Arginine
3.3. Asparagine
4. Targeting of Lipid Metabolism by Pathogens
4.1. Lipid Droplets
4.2. Membranes
5. Targeting of Carbohydrate Metabolism by Pathogens
6. Targeting of Other mTOR-Controlled Processes by Pathogens
6.1. Autophagy
6.2. Host Translation
6.3. Innate Cytokines
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.M.; Lawless, S.J.; Pearce, E.J. Metabolism and acetylation in innate immune cell function and fate. Sem. Immunol. 2016, 28, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef]
- Collette, J.R.; Lorenz, M.C. Mechanisms of immune evasion in fungal pathogens. Curr. Opin. Microbiol. 2011, 14, 668–675. [Google Scholar] [CrossRef]
- Schmid-Hempel, P. Immune defence, parasite evasion strategies and their relevance for “macroscopic phenomena” such as virulence. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 85–98. [Google Scholar] [CrossRef]
- Freyberg, Z.; Harvill, E.T. Pathogen manipulation of host metabolism: A common strategy for immune evasion. PLoS Pathog. 2017, 13, e1006669. [Google Scholar] [CrossRef]
- Olive, A.J.; Sassetti, C.M. Metabolic crosstalk between host and pathogen: Sensing, adapting and competing. Nat. Rev. Microbiol. 2016, 14, 221–234. [Google Scholar] [CrossRef]
- Moreno-Altamirano, M.M.B.; Kolstoe, S.E.; Sánchez-García, F.J. Virus Control of Cell Metabolism for Replication and Evasion of Host Immune Responses. Front. Cell Infect. Microbiol. 2019, 9, 95. [Google Scholar] [CrossRef]
- Martin, S.; Saha, B.; Riley, J.L. The battle over mTOR: An emerging theatre in host-pathogen immunity. PLoS Pathog. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed]
- Brunton, J.; Steele, S.; Ziehr, B.; Moorman, N.; Kawula, T. Feeding uninvited guests: mTOR and AMPK set the table for intracellular pathogens. PLoS Pathog. 2013, 9, e1003552. [Google Scholar] [CrossRef] [PubMed]
- Osada-Oka, M.; Goda, N.; Saiga, H.; Yamamoto, M.; Takeda, K.; Ozeki, Y.; Yamaguchi, T.; Soga, T.; Tateishi, Y.; Miura, K.; et al. Metabolic adaptation to glycolysis is a basic defense mechanism of macrophages for Mycobacterium tuberculosis infection. Int. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Heesemann, J.; Rudel, T.; Goebel, W. Metabolic host responses to infection by intracellular bacterial pathogens. Front. Cell Infect. Microbiol. 2013, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Shea-Donohue, T.; Qin, B.; Smith, A. Parasites, nutrition, immune responses and biology of metabolic tissues. Parasite Immunol. 2017, 39, e12422. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.-T.; Liu, X.-G. mTOR Signaling in T Cell Immunity and Autoimmunity. Int. Rev. Immunol. 2015, 34, 50–66. [Google Scholar] [CrossRef]
- Keating, R.; McGargill, M.A. mTOR Regulation of Lymphoid Cells in Immunity to Pathogens. Front. Immunol. 2016, 7, 180. [Google Scholar] [CrossRef]
- Guri, Y.; Nordmann, T.M.; Roszik, J. mTOR at the Transmitting and Receiving Ends in Tumor Immunity. Front. Immunol. 2018, 9, 578. [Google Scholar] [CrossRef]
- Schroeder, J.H.; McCarthy, D.; Szestak, T.; Cook, D.A.; Taylor, M.J.; Craig, A.G.; Lawson, C.; Lawrence, R.A. Brugia malayi microfilariae adhere to human vascular endothelial cells in a C3-dependent manner. PLoS Negl. Trop. Dis. 2017, 11, e0005592. [Google Scholar] [CrossRef]
- Narasimhan, P.B.; Bennuru, S.; Meng, Z.; Cotton, R.N.; Elliott, K.R.; Ganesan, S.; McDonald-Fleming, R.; Veenstra, T.D.; Nutman, T.B.; Semnani, R.T. Microfilariae of Brugia malayi Inhibit the mTOR Pathway and Induce Autophagy in Human Dendritic Cells. Infect. Immun. 2016, 84, 2463–2472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jaramillo, M.; Gomez, M.A.; Larsson, O.; Shio, M.T.; Topisirovic, I.; Contreras, I.; Luxenburg, R.; Rosenfeld, A.; Colina, R.; McMaster, R.W.; et al. Leishmania repression of host translation through mTOR cleavage is required for parasite survival and infection. Cell Host Microbe 2011, 9, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J. Virol. 2011, 85, 9369–9376. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Alwine, J.C. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes Dev. 2012, 26, 2015–2026. [Google Scholar] [CrossRef]
- Bayer, C.; Varani, S.; Wang, L.; Walther, P.; Zhou, S.; Straschewski, S.; Bachem, M.; Söderberg-Naucler, C.; Mertens, T.; Frascaroli, G. Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J. Virol. 2013, 87, 67–79. [Google Scholar] [CrossRef]
- Gredmark-Russ, S.; Söderberg-Nauclér, C. Dendritic cell biology in human cytomegalovirus infection and the clinical consequences for host immunity and pathology. Virulence 2012, 3, 621–634. [Google Scholar] [CrossRef]
- Meade, N.; King, M.; Munger, J.; Walsh, D. mTOR Dysregulation by Vaccinia Virus F17 Controls Multiple Processes with Varying Roles in Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143–1157. [Google Scholar] [CrossRef]
- Vekariya, U.; Saxena, R.; Singh, P.; Rawat, K.; Kumar, B.; Kumari, S.; Agnihotri, S.K.; Kaur, S.; Sachan, R.; Nazir, A.; et al. HIV-1 Nef-POTEE; A novel interaction modulates macrophage dissemination via mTORC2 signaling pathway. Life Sci. 2018, 214, 158–166. [Google Scholar] [CrossRef]
- Yoon, B.R.; Oh, Y.-J.; Kang, S.W.; Lee, E.B.; Lee, W.-W. Role of SLC7A5 in Metabolic Reprogramming of Human Monocyte/Macrophage Immune Responses. Front. Immunol. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Nada, S.; Takegahara, N.; Okuno, T.; Nojima, S.; Kang, S.; Ito, D.; Morimoto, K.; Hosokawa, T.; Hayama, Y.; et al. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat. Commun. 2016, 7, 13130. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- McGaha, T.L.; Huang, L.; Lemos, H.; Metz, R.; Mautino, M.; Prendergast, G.C.; Mellor, A.L. Amino acid catabolism: A pivotal regulator of innate and adaptive immunity. Immunol. Rev. 2012, 249, 135. [Google Scholar] [CrossRef]
- Zelante, T.; Fallarino, F.; Bistoni, F.; Puccetti, P.; Romani, L. Indoleamine 2,3-dioxygenase in infection: The paradox of an evasive strategy that benefits the host. Microbes Infect. 2009, 11, 133–141. [Google Scholar] [CrossRef]
- Friedman, M. Analysis, Nutrition, and Health Benefits of Tryptophan. Int. J. Tryptophan Res. 2018, 11, 1178646918802282. [Google Scholar] [CrossRef]
- Passalacqua, K.D.; Charbonneau, M.-E.; O’Riordan, M.X.D. Bacterial Metabolism Shapes the Host-Pathogen Interface. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Qualls, J.E.; Murray, P.J. Immunometabolism within the tuberculosis granuloma: Amino acids, hypoxia, and cellular respiration. Semin. Immunopathol. 2016, 38, 139–152. [Google Scholar] [CrossRef]
- Majumdar, T.; Sharma, S.; Kumar, M.; Hussain, M.A.; Chauhan, N.; Kalia, I.; Sahu, A.K.; Rana, V.S.; Bharti, R.; Haldar, A.K.; et al. Tryptophan-kynurenine pathway attenuates β-catenin-dependent pro-parasitic role of STING-TICAM2-IRF3-IDO1 signalosome in Toxoplasma gondii infection. Cell Death Dis. 2019, 10, 161. [Google Scholar] [CrossRef]
- Nino-Castro, A.; Abdullah, Z.; Popov, A.; Thabet, Y.; Beyer, M.; Knolle, P.; Domann, E.; Chakraborty, T.; Schmidt, S.V.; Schultze, J.L. The IDO1-induced kynurenines play a major role in the antimicrobial effect of human myeloid cells against Listeria monocytogenes. Innate Immune 2014, 20, 401–411. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Reddy, M.C.; Ioerger, T.R.; Rothchild, A.C.; Dartois, V.; Schuster, B.M.; Trauner, A.; Wallis, D.; Galaviz, S.; Huttenhower, C.; et al. Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 2013, 155, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. Ido expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Wang, X.F.; Wang, H.S.; Wang, H.; Zhang, F.; Wang, K.F.; Guo, Q.; Zhang, G.; Cai, S.H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Poormasjedi-Meibod, M.-S.; Jalili, R.B.; Hosseini-Tabatabaei, A.; Hartwell, R.; Ghahary, A. Immuno-regulatory function of indoleamine 2,3 dioxygenase through modulation of innate immune responses. PLoS ONE 2013, 8, e71044. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Avdic, S.; McSharry, B.P.; Slobedman, B. Modulation of dendritic cell functions by viral IL-10 encoded by human cytomegalovirus. Front. Microbiol. 2014, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Raftery, M.J.; Wieland, D.; Gronewald, S.; Kraus, A.A.; Giese, T.; Schönrich, G. Shaping Phenotype, Function, and Survival of Dendritic Cells by Cytomegalovirus-Encoded IL-10. J. Immunol. 2004, 173, 3383–3391. [Google Scholar] [CrossRef]
- Mittal, D.; Kassianos, A.J.; Tran, L.S.; Bergot, A.S.; Gosmann, C.; Hofmann, J.; Blumenthal, A.; Leggatt, G.R.; Frazer, I.H. Indoleamine 2,3-dioxygenase activity contributes to local immune suppression in the skin expressing human papillomavirus oncoprotein e7. J. Investig Dermatol. 2013, 133, 2686–2694. [Google Scholar] [CrossRef]
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef]
- Battu, S.; Minhas, G.; Mishra, A.; Khan, N. Amino Acid Sensing via General Control Nonderepressible-2 Kinase and Immunological Programming. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Castilho, B.A.; Shanmugam, R.; Silva, R.C.; Ramesh, R.; Himme, B.M.; Sattlegger, E. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim. Biophys. Acta 2014, 1843, 1948–1968. [Google Scholar] [CrossRef]
- Tattoli, I.; Sorbara, M.T.; Vuckovic, D.; Ling, A.; Soares, F.; Carneiro, L.A.; Yang, C.; Emili, A.; Philpott, D.J.; Girardin, S.E. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 2012, 11, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Ravishankar, B.; Liu, H.; Shinde, R.; Chaudhary, K.; Xiao, W.; Bradley, J.; Koritzinsky, M.; Madaio, M.P.; McGaha, T.L. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc. Natl. Acad. Sci. USA 2015, 112, 10774–10779. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.; Hanczko, R.; Lai, Z.W.; Oaks, Z.; Kelly, R.; Borsuk, R.; Asara, J.M.; Phillips, P.E. Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: Implications for activation of the mechanistic target of rapamycin. Metabolomics 2015, 11, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Kanamori, H.; Seki, E.; Hoshi, M.; Ohtaki, H.; Yasuda, Y.; Ito, H.; Suetsugu, A.; Nagaki, M.; Moriwaki, H.; et al. L-tryptophan-mediated enhancement of susceptibility to nonalcoholic fatty liver disease is dependent on the mammalian target of rapamycin. J. Biol. Chem. 2011, 286, 34800–34808. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Sorgdrager, F.J.H.; Naudé, P.J.W.; Kema, I.P.; Nollen, E.A.; Deyn, P.P.D. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Front. Immunol. 2019, 10, 2565. [Google Scholar] [CrossRef]
- Uchiya, K.-I.; Groisman, E.A.; Nikai, T. Involvement of Salmonella pathogenicity island 2 in the up-regulation of interleukin-10 expression in macrophages: Role of protein kinase A signal pathway. Infect. Immun. 2004, 72, 1964–1973. [Google Scholar] [CrossRef]
- Tsalikis, J.; Croitoru, D.O.; Philpott, D.J.; Girardin, S.E. Nutrient sensing and metabolic stress pathways in innate immunity. Cell Microbiol. 2013, 15, 1632–1641. [Google Scholar] [CrossRef]
- McRae, M.P. Therapeutic Benefits of l-Arginine: An Umbrella Review of Meta-analyses. J. Chiropr. Med. 2016, 15, 184. [Google Scholar] [CrossRef] [PubMed]
- Popovic, P.J.; Zeh, H.J., 3rd; Ochoa, J.B. Arginine and immunity. J Nutr. 2007, 137, 1681S–1686S. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C. Perspectives series: Host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Investig. 1997, 99, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Lahiri, A.; Lahiri, A.; Chakravortty, D. Modulation of the arginase pathway in the context of microbial pathogenesis: A metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 2010, 6, e1000899. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, M.; Datey, A.; Wilson, K.T.; Chakravortty, D. Dual role of arginine metabolism in establishing pathogenesis. Curr. Opin. Microbiol. 2016, 29, 43–48. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Qualls, J.E.; Pesce, J.T.; Smith, A.M.; Thompson, R.W.; Henao-Tamayo, M.; Basaraba, R.J.; König, T.; Schleicher, U.; Koo, M.S.; et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 2008, 9, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Holzmuller, P.; Geiger, A.; Nzoumbou-Boko, R.; Pissarra, J.; Hamrouni, S.; Rodrigues, V.; Dauchy, F.A.; Lemesre, J.L.; Vincendeau, P.; Bras-Gonçalves, R. Trypanosomatid Infections: How Do Parasites and Their Excreted–Secreted Factors Modulate the Inducible Metabolism of l-Arginine in Macrophages? Front. Immunol. 2018, 9, 778. [Google Scholar] [CrossRef]
- Kropf, P.; Fuentes, J.M.; Fähnrich, E.; Arpa, L.; Herath, S.; Weber, V.; Soler, G.; Celada, A.; Modolell, M.; Müller, I. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. 2005, 19, 1000–1002. [Google Scholar] [CrossRef]
- Duque-Correa, M.A.; Kühl, A.A.; Rodriguez, P.C.; Zedler, U.; Schommer-Leitner, S.; Rao, M.; Weiner, J.; Hurwitz, R.; Qualls, J.E.; Kosmiadi, G.A.; et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc. Natl. Acad. Sci. USA 2014, 111, E4024–E4032. [Google Scholar] [CrossRef]
- Badirzadeh, A.; Taheri, T.; Taslimi, Y.; Abdossamadi, Z.; Heidari-Kharaji, M.; Gholami, E.; Sedaghat, B.; Niyyati, M.; Rafati, S. Arginase activity in pathogenic and non-pathogenic species of Leishmania parasites. PLoS Negl. Trop. Dis. 2017, 11, e0005774. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.F.L.; Floeter-Winter, L.M. Arginase in Leishmania. Subcell. Biochem. 2014, 74, 103–117. [Google Scholar] [CrossRef]
- Boitz, J.M.; Gilroy, C.A.; Olenyik, T.D.; Paradis, D.; Perdeh, J.; Dearman, K.; Davis, M.J.; Yates, P.A.; Li, Y.; Riscoe, M.K.; et al. Arginase Is Essential for Survival of Leishmania donovani Promastigotes but Not Intracellular Amastigotes. Infect. Immun. 2017, 85, e00554-16. [Google Scholar] [CrossRef] [PubMed]
- Gobert, A.P.; McGee, D.J.; Akhtar, M.; Mendz, G.L.; Newton, J.C.; Cheng, Y.; Mobley, H.L.; Wilson, K.T. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: A strategy for bacterial survival. Proc. Natl. Acad. Sci. USA 2001, 98, 13844–13849. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.; Asim, M.; Lewis, N.D.; Algood, H.M.; Cover, T.L.; Kim, P.Y.; Wilson, K.T. L-Arginine Availability Regulates Inducible Nitric Oxide Synthase-Dependent Host Defense against Helicobacter pylori. Infect. Immun. 2007, 75, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-López, C.; Collette, J.R.; Brothers, K.M.; Shepardson, K.M.; Cramer, R.A.; Wheeler, R.T.; Lorenz, M.C. Candida albicans induces arginine biosynthetic genes in response to host-derived reactive oxygen species. Eukaryot. Cell 2013, 12, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Buck, M.D.; Geltink, R.I.; Kyle, R.L.; Caputa, G.; O’Sullivan, D.; Cameron, A.M.; Castoldi, A.; Musa, Y.; Kabat, A.M.; et al. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab. 2019, 30, 352–363.e8. [Google Scholar] [CrossRef]
- Raniga, K.; Liang, C. Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses 2018, 10, 36. [Google Scholar] [CrossRef]
- Goh, C.C.; Roggerson, K.M.; Lee, H.-C.; Golden-Mason, L.; Rosen, H.R.; Hahn, Y.S. Hepatitis C Virus-Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-γ Production by Altering Cellular Metabolism via Arginase-1. J. Immunol. 2016, 196, 2283–2292. [Google Scholar] [CrossRef]
- Banik, S.; Renner Viveros, P.; Seeber, F.; Klotz, C.; Ignatius, R.; Aebischer, T. Giardia duodenalis arginine deiminase modulates the phenotype and cytokine secretion of human dendritic cells by depletion of arginine and formation of ammonia. Infect. Immun. 2013, 81, 2309–2317. [Google Scholar] [CrossRef]
- Ban, H.; Shigemitsu, K.; Yamatsuji, T.; Haisa, M.; Nakajo, T.; Takaoka, M.; Nobuhisa, T.; Gunduz, M.; Tanaka, N.; Naomoto, Y. Arginine and Leucine regulate p70 S6 kinase and 4E-BP1 in intestinal epithelial cells. Int. J. Mol. Med. 2004, 13, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Averous, J.; Lambert-Langlais, S.; Mesclon, F.; Carraro, V.; Parry, L.; Jousse, C.; Bruhat, A.; Maurin, A.C.; Pierre, P.; Proud, C.G.; et al. GCN2 contributes to mTORC1 inhibition by leucine deprivation through an ATF4 independent mechanism. Sci. Rep. 2016, 6, 27698. [Google Scholar] [CrossRef]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine Depletion Blunts Antitumor T-cell Responses by Inducing Myeloid-Derived Suppressor Cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef]
- Shaheen, Z.R.; Naatz, A.; Corbett, J.A. CCR5-Dependent Activation of mTORC1 Regulates Translation of Inducible NO Synthase and COX-2 during Encephalomyocarditis Virus Infection. J. Immunol. 2015, 195, 4406–4414. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Kullas, A.L.; McClelland, M.; Yang, H.J.; Tam, J.W.; Torres, A.; Porwollik, S.; Mena, P.; McPhee, J.B.; Bogomolnaya, L.; Andrews-Polymenis, H.; et al. L-asparaginase II produced by Salmonella typhimurium inhibits T cell responses and mediates virulence. Cell Host Microbe 2012, 12, 791–798. [Google Scholar] [CrossRef]
- Shibayama, K.; Takeuchi, H.; Wachino, J.-I.; Mori, S.; Arakawa, Y. Biochemical and pathophysiological characterization of Helicobacter pylori asparaginase. Microbiol. Immunol. 2011, 55, 408–417. [Google Scholar] [CrossRef]
- Torres, A.; Luke, J.D.; Kullas, A.L.; Kapilashrami, K.; Botbol, Y.; Koller, A.; Tonge, P.J.; Chen, E.I.; Macian, F.; van der Velden, A.W. Asparagine deprivation mediated by Salmonella asparaginase causes suppression of activation-induced T cell metabolic reprogramming. J. Leukoc. Biol. 2016, 99, 387–398. [Google Scholar] [CrossRef]
- Cachumba, J.J.M.; Antunes, F.A.F.; Peres, G.F.D.; Brumano, L.P.; Santos, J.C.D.; Da Silva, S.S. Current applications and different approaches for microbial l-asparaginase production. Braz. J. Microbiol. 2016, 47, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Hofreuter, D.; Novik, V.; Galán, J.E. Metabolic diversity in Campylobacter jejuni enhances specific tissue colonization. Cell Host Microbe 2008, 4, 425–433. [Google Scholar] [CrossRef]
- Song, P.; Wang, Z.; Zhang, X.; Fan, J.; Li, Y.; Chen, Q.; Wang, S.; Liu, P.; Luan, J.; Ye, L.; et al. The role of autophagy in asparaginase-induced immune suppression of macrophages. Cell Death Dis. 2017, 8, e2721. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Stunault, M.I.; Bories, G.; Guinamard, R.R.; Ivanov, S. Metabolism Plays a Key Role during Macrophage Activation. Mediat. Inflamm. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Philpott, D.J.; Girardin, S.E. The bacterial and cellular determinants controlling the recruitment of mTOR to the Salmonella-containing vacuole. Biol. Open 2012, 1, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Hos, N.J.; Gutierrez, S.; Fischer, J.; Stepek, J.M.; Daglidu, E.; Krönke, M.; Robinson, N. Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS Pathog. 2017, 13, e1006227. [Google Scholar] [CrossRef]
- Kudchodkar, S.B.; Del Prete, G.Q.; Maguire, T.G.; Alwine, J.C. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J. Virol. 2007, 81, 3649–3651. [Google Scholar] [CrossRef]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 2004, 78, 11030–11039. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef]
- Guerrini, V.; Prideaux, B.; Blanc, L.; Bruiners, N.; Arrigucci, R.; Singh, S.; Ho-Liang, H.P.; Salamon, H.; Chen, P.Y.; Lakehal, K.; et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. 2018, 14, e1007223. [Google Scholar] [CrossRef] [PubMed]
- den Brok, M.H.; Raaijmakers, T.K.; Collado-Camps, E.; Adema, G.J. Lipid Droplets as Immune Modulators in Myeloid Cells. Trends Immunol. 2018, 39, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Saka, H.A.; Valdivia, R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 2012, 28, 411–437. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.K.; Knoll, L.J. Patatin-like phospholipases in microbial infections with emerging roles in fatty acid metabolism and immune regulation by Apicomplexa. Mol. Microbiol. 2018, 107, 34–46. [Google Scholar] [CrossRef] [PubMed]
- D’Avila, H.; Freire-de-Lima, C.G.; Roque, N.R.; Teixeira, L.; Barja-Fidalgo, C.; Silva, A.R.; Melo, R.C.; DosReis, G.A.; Castro-Faria-Neto, H.C.; Bozza, P.T. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E₂ generation and increased parasite growth. J. Infect. Dis. 2011, 204, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Steer, S.A.; Corbett, J.A. The Role and Regulation of COX-2 during Viral Infection. Viral Immunol. 2003, 16, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Sander, W.J.; O’Neill, H.G.; Pohl, C.H. Prostaglandin E2 as a Modulator of Viral Infections. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Vallochi, A.L.; Teixeira, L.; Oliveira K da, S.; Maya-Monteiro, C.M.; Bozza, P.T. Lipid Droplet, a Key Player in Host-Parasite Interactions. Front. Immunol. 2018, 9, 1022. [Google Scholar] [CrossRef]
- Libbing, C.L.; McDevitt, A.R.; Azcueta, R.-M.P.; Ahila, A.; Mulye, M. Lipid Droplets: A Significant but Understudied Contributor of Host–Bacterial Interactions. Cells 2019, 8, 354. [Google Scholar] [CrossRef]
- Agard, M.; Asakrah, S.; Morici, L.A. PGE (2) suppression of innate immunity during mucosal bacterial infection. Front. Cell Infect. Microbiol. 2013, 3, 45. [Google Scholar] [CrossRef]
- Bozza, P.T.; Payne, J.L.; Morham, S.G.; Langenbach, R.; Smithies, O.; Weller, P.F. Leukocyte lipid body formation and eicosanoid generation: Cyclooxygenase-independent inhibition by aspirin. Proc. Natl. Acad. Sci. USA 1996, 93, 11091–11096. [Google Scholar] [CrossRef]
- Raulin, J. Lipids and retroviruses. Lipids 2000, 35, 123–130. [Google Scholar] [CrossRef]
- Mattos, K.A.; Oliveira, V.G.; D’Avila, H.; Rodrigues, L.S.; Pinheiro, R.O.; Sarno, E.N.; Pessolani, M.C.; Bozza, P.T. TLR6-driven lipid droplets in Mycobacterium leprae-infected Schwann cells: Immunoinflammatory platforms associated with bacterial persistence. J. Immunol. 2011, 187, 2548–2558. [Google Scholar] [CrossRef]
- Rodríguez-Sánchez, I.; Munger, J. Meal for Two: Human Cytomegalovirus-Induced Activation of Cellular Metabolism. Viruses 2019, 11, 273. [Google Scholar] [CrossRef]
- Foley, P.; Kazazi, F.; Biti, R.; Sorrell, T.C.; Cunningham, A.L. HIV infection of monocytes inhibits the T-lymphocyte proliferative response to recall antigens, via production of eicosanoids. Immunology 1992, 75, 391–397. [Google Scholar]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 3941. [Google Scholar] [CrossRef]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection induces adipocyte-like lipogenesis through activation of sterol regulatory element binding protein 1. J. Virol. 2012, 86, 2942–2949. [Google Scholar] [CrossRef]
- Bowman, C.C.; Bost, K.L. Cyclooxygenase-2-mediated prostaglandin E2 production in mesenteric lymph nodes and in cultured macrophages and dendritic cells after infection with Salmonella. J. Immunol. 2004, 172, 2469–2475. [Google Scholar] [CrossRef]
- Chakraborty, T.; Thuer, E.; Heijink, M.; Tóth, R.; Bodai, L.; Vágvölgyi, C.; Giera, M.; Gabaldón, T.; Gácser, A. Eicosanoid biosynthesis influences the virulence of Candida parapsilosis. Virulence 2018, 9, 1019–1035. [Google Scholar] [CrossRef]
- Murphy, D.J. The dynamic roles of intracellular lipid droplets: From archaea to mammals. Protoplasma 2012, 249, 541–585. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Del Poeta, M. Lipid signalling in pathogenic fungi. Cell. Microbiol. 2011, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Desselberger, U. Lipid droplets form complexes with viroplasms and are crucial for rotavirus replication. Curr. Opin. Virol. 2016, 19, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Leier, H.C.; Messer, W.B.; Tafesse, F.G. Lipids and pathogenic flaviviruses: An intimate union. PLoS Pathog. 2018, 14, e1006952. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.X.; Randall, G. Dengue Virus Activates the AMP Kinase-mTOR Axis to Stimulate a Proviral Lipophagy. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Binns, D.; Reese, M.L. The coccidian parasites and dysregulate mammalian lipid droplet biogenesis. J. Biol. Chem. 2017, 292, 11009–11020. [Google Scholar] [CrossRef]
- Fernández, N.; González, A.; Valera, I.; Alonso, S.; Crespo, M.S. Mannan and peptidoglycan induce COX-2 protein in human PMN via the mammalian target of rapamycin. Eur. J. Immunol. 2007, 37, 2572–2582. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Pinheiro, R.O.; Nunes, M.P.; Pinheiro, C.S.; D’Avila, H.; Bozza, P.T.; Takiya, C.M.; Côrte-Real, S.; Freire-de-Lima, C.G.; DosReis, G.A. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect. 2009, 11, 181–190. [Google Scholar] [CrossRef]
- Madeira, J.B.; Masuda, C.A.; Maya-Monteiro, C.M.; Matos, G.S.; Montero-Lomelí, M.; Bozaquel-Morais, B.L. TORC1 inhibition induces lipid droplet replenishment in yeast. Mol. Cell Biol. 2015, 35, 737–746. [Google Scholar] [CrossRef]
- Menon, D.; Singh, K.; Pinto, S.M.; Nandy, A.; Jaisinghani, N.; Kutum, R.; Dash, D.; Prasad, T.K.; Gandotra, S. Quantitative Lipid Droplet Proteomics Reveals Mycobacterium tuberculosis Induced Alterations in Macrophage Response to Infection. ACS Infect. Dis. 2019, 5, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Mattos, K.A.; Lara, F.A.; Oliveira, V.G.; Rodrigues, L.S.; D’Avila, H.; Melo, R.C.; Manso, P.P.; Sarno, E.N.; Bozza, P.T.; Pessolani, M.C. Modulation of lipid droplets by Mycobacterium leprae in Schwann cells: A putative mechanism for host lipid acquisition and bacterial survival in phagosomes. Cell Microbiol. 2011, 13, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Schumann, J. The Impact of Macrophage Membrane Lipid Composition on Innate Immune Response Mechanisms; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef]
- Olsson, S.; Sundler, R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol. Immunol. 2006, 43, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Khromykh, A.A.; Parton, R.G. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2007, 2, 229–239. [Google Scholar] [CrossRef]
- Rub, A.; Dey, R.; Jadhav, M.; Kamat, R.; Chakkaramakkil, S.; Majumdar, S.; Mukhopadhyaya, R.; Saha, B. Cholesterol depletion associated with Leishmania major infection alters macrophage CD40 signalosome composition and effector function. Nat. Immunol. 2009, 10, 273–280. [Google Scholar] [CrossRef]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef]
- Yin, H.; Zhou, H.; Kang, Y.; Zhang, X.; Duan, X.; Alnabhan, R.; Liang, S.; Scott, D.A.; Lamont, R.J.; Shang, J.; et al. Syk negatively regulates TLR4-mediated IFNβ and IL-10 production and promotes inflammatory responses in dendritic cells. Biochim. Biophys. Acta 2016, 1860, 588–598. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; Van Der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; Van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, E.V.; Diaz, K.; Griffin, A.J.; Rasmussen, J.A.; Crane, D.D.; Jones, B.D.; Bosio, C.M. Metabolic Reprogramming of Host Cells by Virulent Francisella tularensis for Optimal Replication and Modulation of Inflammation. J. Immunol. 2016, 196, 4227–4236. [Google Scholar] [CrossRef] [PubMed]
- Sanman, L.E.; Qian, Y.; Eisele, N.A.; Ng, T.M.; van der Linden, W.A.; Monack, D.M.; Weerapana, E.; Bogyo, M. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 2016, 5, e13663. [Google Scholar] [CrossRef] [PubMed]
- Wynosky-Dolfi, M.A.; Snyder, A.G.; Philip, N.H.; Doonan, P.J.; Poffenberger, M.C.; Avizonis, D.; Zwack, E.E.; Riblett, A.M.; Hu, B.; Strowig, T.; et al. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J. Exp. Med. 2014, 211, 653–668. [Google Scholar] [CrossRef]
- Tucey, T.M.; Verma, J.; Harrison, P.F.; Snelgrove, S.L.; Lo, T.L.; Scherer, A.K.; Barugahare, A.A.; Powell, D.R.; Wheeler, R.T.; Hickey, M.J.; et al. Glucose Homeostasis Is Important for Immune Cell Viability during Candida Challenge and Host Survival of Systemic Fungal Infection. Cell Metab. 2018, 27, 988–1006. [Google Scholar] [CrossRef]
- Moreira, D.; Rodrigues, V.; Abengozar, M.; Rivas, L.; Rial, E.; Laforge, M.; Li, X.; Foretz, M.; Viollet, B.; Estaquier, J.; et al. Leishmania infantum modulates host macrophage mitochondrial metabolism by hijacking the SIRT1-AMPK axis. PLoS Pathog. 2015, 11, e1004684. [Google Scholar] [CrossRef]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. Adenosine 5′-Monophosphate-Activated Protein Kinase Promotes Macrophage Polarization to an Anti-Inflammatory Functional Phenotype. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef]
- Escoll, P.; Song, O.R.; Viana, F.; Steiner, B.; Lagache, T.; Olivo-Marin, J.C.; Impens, F.; Brodin, P.; Hilbi, H.; Buchrieser, C. Legionella pneumophila Modulates Mitochondrial Dynamics to Trigger Metabolic Repurposing of Infected Macrophages. Cell Host Microbe 2017, 22, 302–316. [Google Scholar] [CrossRef]
- Abshire, C.F.; Dragoi, A.-M.; Roy, C.R.; Ivanov, S.S. MTOR-Driven Metabolic Reprogramming Regulates Legionella pneumophila Intracellular Niche Homeostasis. PLoS Pathog. 2016, 12, e1006088. [Google Scholar] [CrossRef]
- Ivanov, S. The tug-of-war over MTOR in Legionella infections. Microb. Cell 2017, 4, 67–68. [Google Scholar] [CrossRef]
- Prevost, M.S.; Pinotsis, N.; Dumoux, M.; Hayward, R.D.; Waksman, G. The Legionella effector WipB is a translocated Ser/Thr phosphatase that targets the host lysosomal nutrient sensing machinery. Sci. Rep. 2017, 7, 9450. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.; Muralidhar, G.; Linton, P.-J. Autophagy in the Immune System. Autophagy Health Dis. 2013, 30, 41–55. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Sorbara, M.T.; Yang, C.; Tooze, S.A.; Philpott, D.J.; Girardin, S.E. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013, 32, 3066–3078. [Google Scholar] [CrossRef]
- Abdel-Nour, M.; Tsalikis, J.; Kleinman, D.; Girardin, S.E. The emerging role of mTOR signalling in antibacterial immunity. Immunol. Cell Biol. 2014, 92, 346–353. [Google Scholar] [CrossRef]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef]
- Casanova, J.E. Bacterial Autophagy: Offense and Defense at the Host–Pathogen Interface. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 237–243. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8. [Google Scholar] [CrossRef]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K–Akt–mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef]
- Li, N.; Tang, B.; Jia, Y.P.; Zhu, P.; Zhuang, Y.; Fang, Y.; Li, Q.; Wang, K.; Zhang, W.J.; Guo, G.; et al. Helicobacter pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via c-Met-PI3K/Akt-mTOR Signaling Pathway. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Geng, P.; Liu, Y.; Wu, J.; Qiao, H.; Xie, Y.; Yin, N.; Chen, L.; Lin, X.; Liu, Y.; et al. Rotavirus-encoded virus-like small RNA triggers autophagy by targeting IGF1R via the PI3K/Akt/mTOR pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, X.; Zhu, G.; Tu, H.; Liu, Z.; Li, W.; Han, S.; Yin, J.; Peng, B.; Liu, W. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PLoS ONE 2015, 10, e0122109. [Google Scholar] [CrossRef]
- Wang, Y.; Weiss, L.M.; Orlofsky, A. Host Cell Autophagy Is Induced by Toxoplasma gondii and Contributes to Parasite Growth. J. Biol. Chem. 2009, 284, 1694. [Google Scholar] [CrossRef]
- Wang, Y.; Weiss, L.M.; Orlofsky, A. Intracellular parasitism with Toxoplasma gondii stimulates mTOR-dependent host cell growth despite impaired signaling to S6K1 and 4E-BP1. Cell Microbiol. 2009, 11, 983. [Google Scholar] [CrossRef]
- Thomas, S.A.; Nandan, D.; Kass, J.; Reiner, N.E. Countervailing, time-dependent effects on host autophagy promotes intracellular survival of Leishmania. J. Biol. Chem. 2018, 293, 2617–2630. [Google Scholar] [CrossRef]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of autophagy in HIV infection and pathogenesis. J. Intern. Med. 2017, 281, 422–432. [Google Scholar] [CrossRef]
- Alwine, J.C. Modulation of host cell stress responses by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 2008, 325, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Shives, K.D.; Massey, A.R.; May, N.A.; Morrison, T.E.; Beckham, J.D. 4EBP-Dependent Signaling Supports West Nile Virus Growth and Protein Expression. Viruses 2016, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.-E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef] [PubMed]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef]
- De Leon, J.A.; Qiu, J.; Nicolai, C.J.; Counihan, J.L.; Barry, K.C.; Xu, L.; Lawrence, R.E.; Castellano, B.M.; Zoncu, R.; Nomura, D.K.; et al. Positive and Negative Regulation of the Master Metabolic Regulator mTORC1 by Two Families of Legionella pneumophila Effectors. Cell Rep. 2017, 21, 2031–2038. [Google Scholar] [CrossRef]
- Leroux, L.P.; Lorent, J.; Graber, T.E.; Chaparro, V.; Masvidal, L.; Aguirre, M.; Fonseca, B.D.; van Kempen, L.C.; Alain, T.; Larsson, O.; et al. The Protozoan Parasite Toxoplasma gondii Selectively Reprograms the Host Cell Translatome. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Pernas, L.; Adomako-Ankomah, Y.; Shastri, A.J.; Ewald, S.E.; Treeck, M.; Boyle, J.P.; Boothroyd, J.C. Toxoplasma effector MAF1 mediates recruitment of host mitochondria and impacts the host response. PLoS Biol. 2014, 12, e1001845. [Google Scholar] [CrossRef]
- Suzutani, T.; Nagamine, M.; Shibaki, T.; Ogasawara, M.; Yoshida, I.; Daikoku, T.; Nishiyama, Y.; Azuma, M. The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J. Gen. Virol. 2000, 81, 1763–1771. [Google Scholar] [CrossRef]
- Shi, G.; Ozog, S.; Torbett, B.E.; Compton, A.A. mTOR inhibitors lower an intrinsic barrier to virus infection mediated by IFITM3. Proc. Natl. Acad. Sci. USA 2018, 115, E10069–E10078. [Google Scholar] [CrossRef]
- Beretta, L.; Gingras, A.C.; Svitkin, Y.V.; Hall, M.N.; Sonenberg, N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996, 15, 658–664. [Google Scholar] [CrossRef]
- Li, Y.; Fang, L.; Zhou, Y.; Tao, R.; Wang, D.; Xiao, S. Porcine Reproductive and Respiratory Syndrome Virus Infection Induces both eIF2α Phosphorylation-Dependent and -Independent Host Translation Shutoff. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Zhang, L.; Wei, F.; Fang, Q.; Bao, F.; Mi, S.; Li, N.; Wang, C.; Liu, Y.; Tu, C. mTORC1 Negatively Regulates the Replication of Classical Swine Fever Virus Through Autophagy and IRES-Dependent Translation. iScience 2018, 3, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Katholnig, K.; Linke, M.; Pham, H.; Hengstschläger, M.; Weichhart, T. Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem. Soc. Trans. 2013, 41, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Peres, A.G.; Stegen, C.; Li, J.; Xu, A.Q.; Levast, B.; Surette, M.G.; Cousineau, B.; Desrosiers, M.; Madrenas, J. Uncoupling of pro- and anti-inflammatory properties of Staphylococcus aureus. Infect. Immun. 2015, 83, 1587–1597. [Google Scholar] [CrossRef]
- Cheekatla, S.S.; Aggarwal, A.; Naik, S. mTOR signaling pathway regulates the IL-12/IL-10 axis in Leishmania donovani infection. Med. Microbiol. Immunol. 2012, 201, 37–46. [Google Scholar] [CrossRef]
- Foldenauer, M.E.B.; McClellan, S.A.; Berger, E.A.; Hazlett, L.D. Mammalian target of rapamycin regulates IL-10 and resistance to Pseudomonas aeruginosa corneal infection. J. Immunol. 2013, 190, 5649–5658. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef]
- Lai, Z.W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Langan, R.A.; Japp, A.S.; Partridge, H.L.; Pierson, S.K.; Singh, A.; Arenas, D.J.; Ruth, J.R.; Nabel, C.S.; Stone, K.; et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockade-refractory idiopathic multicentric Castleman disease. J. Clin. Investig. 2019, 130, 4451–4463. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Direct Targeting mTOR | ||
|---|---|---|
| Effect | Pathogen | References |
| Induction of autophagy by secreting a rapamycin homolog and inhibiting mTOR leading to impaired human monocyte-derived DC function | B. malayi | [22] |
| Cleavage of mTOR via the protease GP63 in B10R macrophages leading to decreased type I IFN production and expression of iNOS | L. major | [23] |
| Relocating mTOR to maintain mTORC1 activation in HFFs and U373-MG cells | HCMV | [24,25] |
| Sequestering of raptor and rictor in PMA differentiated THP-1 cells leading to mTOR relocalization and inhibition of cGAS-STING activation and induction of IRGs | Poxviruses (including VacV) | [28,29] |
| Direct interaction with mTORC2 to modulate macrophage phenotype and migration (in this case PMA differentiated THP-1 cells) | HIV-1 | [30] |
| Amino Acid Metabolism | ||
| Effect | Pathogen | References |
| Synthesize tryptophan thereby counteracting IDO depletion of tryptophan | M. tuberculosis, F. Tularensis, C. trachomatis | [9,41] |
| Expresses a IL-10 homolog (cmvIL-10) that induces IDO in human monocyte-derived DCs | HCMV | [47,48] |
| Induction of IDO in langerin negative dermal dendritic cells | HPV | [49] |
| Induction of host arginases in mouse macrophages | Trypanosoma sps, Leishmania sps, T. gondii, S. typhimurium, H. pylori, S. pneumonia, C. albicans, M. tuberculosis and S. mansoni | [65,66,67,68,69,70] |
| Expression of pathogen arginase | Leishmania sps, Plasmodium sps, C. albicans, S. mansoni and H. pylori | [65,71,72,73,74,75,76] |
| Arginase 1 induction in CD33+ PBMCs mediates L-arginine depletion leading to mTOR inhibition and decreased IFNy production in co-cultured NK cells | HCV | [79] |
| Depletion of L-arginine by expression of arginine deiminase modulates cytokine production and phenotype of human monocyte-derived DCs via the inhibition of mTORC1 | G. duodenalis | [80] |
| Asparaginase expression leading to asparagine depletion and dampening of immune responses in T cells and macrophages (ANA-1 and RAW264.7 cells) | Expression: multiple pathogens including H. pylori and S. typhimurium Dampening immune response: S. typhimurium, Erwinia asparaginase | [88,89,90,91,92] [90,93] |
| Lipid Metabolism | ||
| Effect | Pathogen | References |
| Induction of LD formation and PGE2 synthesis for successful replication and modulation of the immune (reduction of antigen-stimulated lymphocyte replication, reduction of killing ability infected cells) | T. cruzi, M. leprae, HMCV, HIV | [112,113,114,115,116,117,118] |
| Active stimulation of LD formation via SseJ | S. Typhimurium | [104] |
| Accumulation of LDs in pathogen vacuoles | C. pneumoniae, T. cruzi | [104] |
| Upregulation COX-2 and PGE2 synthesis in RAW264.7 cells and murine BMDMs and BMDCs | S. typhimurium | [60,119] |
| Production of PGE2 and accumulation of LDs increasing virulence | Human pathogenic fungi | [120,121,122] |
| Induction of LDs to enhance replication efficiency and the assembly of nascent virions | Viruses, including HCV and Rotaviruses | [104,123] |
| mTOR inhibition to induce autophagy of LDs to enhance replication efficiency | Flaviviruses, including Dengue | [124,125] |
| Promoting mTORC1-dependent TAG accumulation in human macrophages that contributes to LD formation | M. tuberculosis, T. gondii | [102,126] |
| Inducing LDs and PGE2 synthesis via autophagy in BALB/c macrophages | L. amazonensis | [129] |
| Rapamycin induces LD formation | S. cerevisiae | [130] |
| Reducing cholesterol levels at the plasma membrane of Vero cells which disrupts Jak-STAT signaling | WNV | [135] |
| Altered CD40 signalosome in BALB/c derived peritoneal macrophages by depleting cholesterol leading to IL-10 production | L. major | [136] |
| Carbohydrate Metabolism | ||
| Effect | Pathogen | References |
| Prevents glycolytic shift in primary macrophages by downregulating HIF1α | F. Tularensis | [143] |
| Depleting intracellular glucose to inhibit glycolysis in murine BMDMs | S. Typhimurium | [144,145] |
| Depleting extracellular glucose leading to the death of restimulated macrophages (murine BMDMs and PMA differentiated THP-1 cells) | C. albicans | [146] |
| Promotion of glucose oxidation in murine BMDMs through activation of SIRT1, LKB1 and AMPK increasing survival and proliferation | L. infantum | [147] |
| Promotes glycolysis (Warburg like metabolism) in human monocyte-derived macrophages | L. pneumophila | [149,150,151,152] |
| Autophagy | ||
| Effect | Pathogen | References |
| Inhibition by stimulating the PI3K-Akt-mTOR pathway | Viruses in general | [160,161] |
| Targeting AMPK, SIRT1 and LKB1 for degradation in murine BMDMs | S. typhimurium | [98] |
| Activation of mTOR (via CagA) and inhibition of autophagy | H. pylori | [162] |
| Induction of autophagy to increase sites of replication, to liberate nutrients and/or to protect host cell death | Flaviviruses (including Zika), coxsackievirus | [163,164,165,166] |
| Induction of autophagy independent of mTOR which contributes to parasite growth. | T. gondii | [167,168] |
| Inhibition of autophagy by stimulating mTOR early in infection in PMA differentiated THP-1 cells. Induction of autophagy during later stages of infection, regulated independent of mTOR. | L. donovani | [169] |
| Inhibiting autophagy by stimulating mTOR to decrease cross-presentation and enhance spreading of infection. | HIV-1 | [170,171] |
| Host Translation | ||
| Effect | Pathogen | References |
| Activates mTORC1 to promote translation of proteins for mitochondrial biogenesis and function, possibly to modulate the innate immune response (in BMDMs and PECs) | T. gondii | [177,178] |
| Cleavage of mTOR via the protease GP63 in B10R macrophages to prevent IFN type I production and iNOS translation | L. major | [23] |
| Mediates shut off of host translation which leads to a decreased translation of innate cytokines in U937 cells | HSV-1 | [179] |
| Shut off of host translation to favor replication of own genome and to downregulate IFITM proteins | Viruses relying on IRES-dependent translation | [23,161,180,181,182,183] |
| Innate Cytokines | ||
| Effect | Pathogen | References |
| Cell wall moieties induce the production of IL-10 via TLR2 and mTOR signaling in human PBMCs | S. aureus | [185] |
| TLR2 and mTOR dependent IL-10 production in PMA differentiated THP-1 cells | L. donovani | [186] |
| Rapamycin decreases IL-10 production in cornea of infected Balb/c mice | P. aeruginosa | [187] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nouwen, L.V.; Everts, B. Pathogens MenTORing Macrophages and Dendritic Cells: Manipulation of mTOR and Cellular Metabolism to Promote Immune Escape. Cells 2020, 9, 161. https://doi.org/10.3390/cells9010161
Nouwen LV, Everts B. Pathogens MenTORing Macrophages and Dendritic Cells: Manipulation of mTOR and Cellular Metabolism to Promote Immune Escape. Cells. 2020; 9(1):161. https://doi.org/10.3390/cells9010161
Chicago/Turabian StyleNouwen, Lonneke V., and Bart Everts. 2020. "Pathogens MenTORing Macrophages and Dendritic Cells: Manipulation of mTOR and Cellular Metabolism to Promote Immune Escape" Cells 9, no. 1: 161. https://doi.org/10.3390/cells9010161
APA StyleNouwen, L. V., & Everts, B. (2020). Pathogens MenTORing Macrophages and Dendritic Cells: Manipulation of mTOR and Cellular Metabolism to Promote Immune Escape. Cells, 9(1), 161. https://doi.org/10.3390/cells9010161