Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects?

 , , , ,
, , , , 

Abstract
1. Introduction
1.1. Mechanisms Contributing to Aβ Deposition in HIV-Infected subjects
1.1.1. Neuroinflammation
1.1.2. HIV Proteins Gag, GP-120, and TAT
Tat and Aβ
Gp-120 and Aβ
Gag and Aβ
1.1.3. Excitotoxicity and Oxidative Stress
1.1.4. BBB Damage
1.1.5. Antiretroviral Therapy (ART)
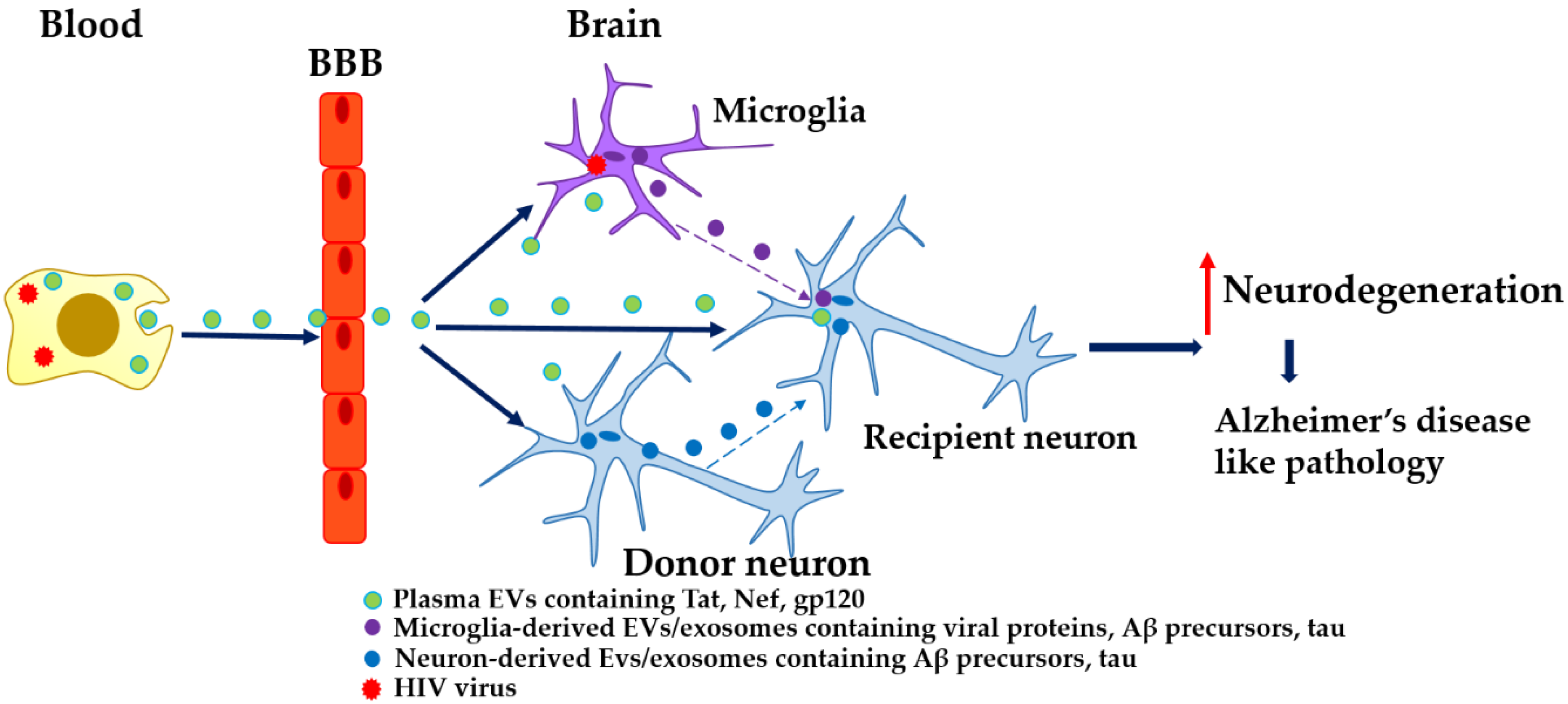
2. EVs, HIV, and AD
3. Interventions to Target Aβ Mediated via EVs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global HIV & AIDS Statistics—2018 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 18 June 2019).
- Teeraananchai, S.; Kerr, S.J.; Amin, J.; Ruxrungtham, K.; Law, M.G. Life expectancy of HIV-positive people after starting combination antiretroviral therapy: A meta-analysis. HIV Med. 2017, 18, 256–266. [Google Scholar] [CrossRef] [PubMed]
- HIV Surveillance Report. 2016. Available online: https://www.cdc.gov/hiv/pdf/library/reports/surveillance/cdc-hiv-surveillance-report-2016-vol-28.pdf (accessed on 18 June 2019).
- Canestri, A.; Lescure, F.-X.; Jaureguiberry, S.; Moulignier, A.; Amiel, C.; Marcelin, A.G.; Peytavin, G.; Tubiana, R.; Pialoux, G.; Katlama, C. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 50, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Ferretti, F.; Peterson, J.; Lee, E.; Fuchs, D.; Boschini, A.; Gisslén, M.; Angoff, N.; Price, R.W.; Cinque, P.; et al. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well controlled plasma viral load. AIDS Lond. Engl. 2012, 26, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.H.; Underwood, J.; Caan, M.W.A.; De Francesco, D.; van Zoest, R.A.; Leech, R.; Wit, F.W.N.M.; Portegies, P.; Geurtsen, G.J.; Schmand, B.A.; et al. Increased brain-predicted aging in treated HIV disease. Neurology 2017, 88, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.A.; Seider, T.R.; Navia, B. HIV effects on age-associated neurocognitive dysfunction: Premature cognitive aging or neurodegenerative disease? Alzheimers Res. Ther. 2015, 7, 37. [Google Scholar] [CrossRef]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Facts and Figures. Available online: https://alz.org/alzheimers-dementia/facts-figures (accessed on 19 June 2019).
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology 2017, 61, 143–187. [Google Scholar] [CrossRef]
- Kocahan, S.; Doğan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Green, D.A.; Masliah, E.; Vinters, H.V.; Beizai, P.; Moore, D.J.; Achim, C.L. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS Lond. Engl. 2005, 19, 407–411. [Google Scholar] [CrossRef]
- Esiri, M.M.; Biddolph, S.C.; Morris, C.S. Prevalence of Alzheimer plaques in AIDS. J. Neurol. Neurosurg. Psychiatry 1998, 65, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, J.; Milanini, B.; Masliah, E.; Tartaglia, M.C.; Dunlop, M.B.; Moore, D.J.; Javandel, S.; DeVaughn, S.; Valcour, V. A neuropathologic diagnosis of Alzheimer’s disease in an older adult with HIV-associated neurocognitive disorder. Neurocase 2018, 24, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Sami Saribas, A.; Cicalese, S.; Ahooyi, T.M.; Khalili, K.; Amini, S.; Sariyer, I.K. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: Evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2017, 8, e2542. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Yang, L.; Cai, Y.; Niu, F.; Mezzacappa, F.; Callen, S.; Fox, H.S.; Buch, S. Emerging roles of extracellular vesicles in neurodegenerative disorders: Focus on HIV-associated neurological complications. Cell Death Dis. 2016, 7, e2481. [Google Scholar] [CrossRef] [PubMed]
- Patters, B.J.; Kumar, S. The role of exosomal transport of viral agents in persistent HIV pathogenesis. Retrovirology 2018, 15, 79. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, P.; He, J.J. Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein. J. Neurovirol. 2016, 22, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.N.; Okeoma, C.M. Exosomes: Implications in HIV-1 Pathogenesis. Viruses 2015, 7, 4093–4118. [Google Scholar] [CrossRef]
- Crenshaw, B.J.; Gu, L.; Sims, B.; Matthews, Q.L. Exosome Biogenesis and Biological Function in Response to Viral Infections. Open Virol. J. 2018, 12, 134–148. [Google Scholar] [CrossRef]
- Zheng, T.; Pu, J.; Chen, Y.; Mao, Y.; Guo, Z.; Pan, H.; Zhang, L.; Zhang, H.; Sun, B.; Zhang, B. Plasma Exosomes Spread and Cluster Around β-Amyloid Plaques in an Animal Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 12. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. (Berl.) 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Pulliam, L.; Sun, B.; Mustapic, M.; Chawla, S.; Kapogiannis, D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J. Neurovirol. 2019. [Google Scholar] [CrossRef]
- Knopman, D.S.; Petersen, R.C.; Jack, C.R. A brief history of “Alzheimer disease”: Multiple meanings separated by a common name. Neurology 2019, 92, 1053–1059. [Google Scholar] [CrossRef]
- Hategan, A.; Masliah, E.; Nath, A. HIV and Alzheimer’s disease: Complex interactions of HIV-Tat with amyloid β peptide and Tau protein. J. Neurovirol. 2019. [Google Scholar] [CrossRef]
- Achim, C.L.; Adame, A.; Dumaop, W.; Everall, I.P.; Masliah, E. Neurobehavioral Research Center Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2009, 4, 190–199. [Google Scholar] [CrossRef]
- Soontornniyomkij, V.; Moore, D.J.; Gouaux, B.; Soontornniyomkij, B.; Sinsheimer, J.S.; Levine, A.J. Associations of regional amyloid-β plaque and phospho-tau pathology with biological factors and neuropsychological functioning among HIV-infected adults. J. Neurovirol. 2019. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- De Almeida, S.M.; Tang, B.; Ribeiro, C.E.; Rotta, I.; Vaida, F.; Piovesan, M.; Batistela Fernandes, M.S.; Letendre, S.; Potter, M.; Ellis, R.J.; et al. Neprilysin in the Cerebrospinal Fluid and Serum of Patients Infected with HIV1-Subtypes C and B. J. Acquir. Immune Defic. Syndr. 1999 2018, 78, 248–256. [Google Scholar] [CrossRef]
- Milanini, B.; Valcour, V. Differentiating HIV-Associated Neurocognitive Disorders from Alzheimer’s Disease: An Emerging Issue in Geriatric NeuroHIV. Curr. HIV/AIDS Rep. 2017, 14, 123–132. [Google Scholar] [CrossRef]
- Thompson, P.M.; Dutton, R.A.; Hayashi, K.M.; Toga, A.W.; Lopez, O.L.; Aizenstein, H.J.; Becker, J.T. Thinning of the cerebral cortex visualized in HIV/AIDS reflects CD4+ T lymphocyte decline. Proc. Natl. Acad. Sci. USA 2005, 102, 15647–15652. [Google Scholar] [CrossRef]
- Bakkour, A.; Morris, J.C.; Wolk, D.A.; Dickerson, B.C. The effects of aging and Alzheimer’s disease on cerebral cortical anatomy: Specificity and differential relationships with cognition. NeuroImage 2013, 76, 332–344. [Google Scholar] [CrossRef]
- Ciccarelli, N.; Fabbiani, M.; Baldonero, E.; Fanti, I.; Cauda, R.; Di Giambenedetto, S.; Silveri, M.C. Effect of aging and human immunodeficiency virus infection on cognitive abilities. J. Am. Geriatr. Soc. 2012, 60, 2048–2055. [Google Scholar] [CrossRef]
- Murji, S.; Rourke, S.B.; Donders, J.; Carter, S.L.; Shore, D.; Rourke, B.P. Theoretically derived CVLT subtypes in HIV-1 infection: Internal and external validation. J. Int. Neuropsychol. Soc. JINS 2003, 9, 1–16. [Google Scholar] [CrossRef]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006171. [Google Scholar] [CrossRef]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef]
- Mäkitalo, S.; Mellgren, Å.; Borgh, E.; Kilander, L.; Skillbäck, T.; Zetterberg, H.; Gisslén, M. The cerebrospinal fluid biomarker profile in an HIV-infected subject with Alzheimer’s disease. AIDS Res. Ther. 2015, 12, 23. [Google Scholar] [CrossRef]
- Turner, R.S.; Chadwick, M.; Horton, W.A.; Simon, G.L.; Jiang, X.; Esposito, G. An individual with human immunodeficiency virus, dementia, and central nervous system amyloid deposition. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 4, 1–5. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Zhou, L.; Saksena, N.K. HIV Associated Neurocognitive Disorders. Infect. Dis. Rep. 2013, 5, e8. [Google Scholar] [CrossRef]
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van de Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell. Neurosci. 2018, 12, 307. [Google Scholar] [CrossRef]
- Flammang, B.; Pardossi-Piquard, R.; Sevalle, J.; Debayle, D.; Dabert-Gay, A.-S.; Thévenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimers Dis. JAD 2012, 30, 145–153. [Google Scholar] [CrossRef]
- Cummings, J.L. Treatment of Alzheimer’s disease: Current and future therapeutic approaches. Rev. Neurol. Dis. 2004, 1, 60–69. [Google Scholar]
- Tanahashi, H.; Tabira, T. X11L2, a new member of the X11 protein family, interacts with Alzheimer’s beta-amyloid precursor protein. Biochem. Biophys. Res. Commun. 1999, 255, 663–667. [Google Scholar] [CrossRef]
- Tanahashi, H.; Tabira, T. Genome structure and chromosomal mapping of the gene for Fe65L2 interacting with Alzheimer’s beta-amyloid precursor protein. Biochem. Biophys. Res. Commun. 1999, 258, 385–389. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Ferrell, D.; Giunta, B. The impact of HIV-1 on neurogenesis: Implications for HAND. Cell. Mol. Life Sci. CMLS 2014, 71, 4387–4392. [Google Scholar] [CrossRef]
- András, I.E.; Pu, H.; Tian, J.; Deli, M.A.; Nath, A.; Hennig, B.; Toborek, M. Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2005, 25, 1159–1170. [Google Scholar] [CrossRef]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef]
- Hategan, A.; Bianchet, M.A.; Steiner, J.; Karnaukhova, E.; Masliah, E.; Fields, A.; Lee, M.-H.; Dickens, A.M.; Haughey, N.; Dimitriadis, E.K.; et al. HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat. Struct. Mol. Biol. 2017, 24, 379–386. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef]
- Rempel, H.C.; Pulliam, L. HIV-1 Tat inhibits neprilysin and elevates amyloid beta. AIDS Lond. Engl. 2005, 19, 127–135. [Google Scholar] [CrossRef]
- Giunta, B.; Zhou, Y.; Hou, H.; Rrapo, E.; Fernandez, F.; Tan, J. HIV-1 TAT inhibits microglial phagocytosis of Abeta peptide. Int. J. Clin. Exp. Pathol. 2008, 1, 260–275. [Google Scholar]
- Aksenov, M.Y.; Aksenova, M.V.; Mactutus, C.F.; Booze, R.M. HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci. Lett. 2010, 475, 174–178. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, J.-H.; Kim, Y.-S. HIV-1 Tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS ONE 2013, 8, e77972. [Google Scholar] [CrossRef]
- Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, N.; Tanzi, R.E.; Moir, R.D.; Nath, A.; He, J.J. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 2000, 6, 1380–1387. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, W.; Jiang, W.; Wu, X.; Ye, B.; Zhou, X. HIV-1 Tat Regulates Occludin and Aβ Transfer Receptor Expression in Brain Endothelial Cells via Rho/ROCK Signaling Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 4196572. [Google Scholar] [CrossRef]
- Jiang, W.; Huang, W.; Chen, Y.; Zou, M.; Peng, D.; Chen, D. HIV-1 Transactivator Protein Induces ZO-1 and Neprilysin Dysfunction in Brain Endothelial Cells via the Ras Signaling Pathway. Oxid. Med. Cell. Longev. 2017, 2017, 3160360. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Larbi, A.; Khalil, A.; Herbein, G.; Frost, E.H. Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J. Neurovirol. 2019. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Katafiasz, B.; Fox, H.; Xiong, H. HIV-1 gp120-induced axonal injury detected by accumulation of β-amyloid precursor protein in adult rat corpus callosum. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2011, 6, 650–657. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 gag proteins: Diverse functions in the virus life cycle. Virology 1998, 251, 1–15. [Google Scholar] [CrossRef]
- Chai, Q.; Jovasevic, V.; Malikov, V.; Sabo, Y.; Morham, S.; Walsh, D.; Naghavi, M.H. HIV-1 counteracts an innate restriction by amyloid precursor protein resulting in neurodegeneration. Nat. Commun. 2017, 8, 1522. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. Vienna Austria 1996 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef]
- Potter, M.C.; Figuera-Losada, M.; Rojas, C.; Slusher, B.S. Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2013, 8, 594–607. [Google Scholar] [CrossRef]
- Nath, A.; Haughey, N.J.; Jones, M.; Anderson, C.; Bell, J.E.; Geiger, J.D. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: Protection by memantine. Ann. Neurol. 2000, 47, 186–194. [Google Scholar] [CrossRef]
- Connolly, N.M.C.; Prehn, J.H.M. The metabolic response to excitotoxicity-lessons from single-cell imaging. J. Bioenerg. Biomembr. 2015, 47, 75–88. [Google Scholar] [CrossRef]
- Sanchez, A.B.; Kaul, M. Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sci. 2017, 7, 25. [Google Scholar] [CrossRef]
- Mastrantonio, R.; D’Ezio, V.; Colasanti, M.; Persichini, T. Nrf2-Mediated System xc- Activation in Astroglial Cells Is Involved in HIV-1 Tat-Induced Neurotoxicity. Mol. Neurobiol. 2019, 56, 3796–3806. [Google Scholar] [CrossRef]
- Thomas, S.; Mayer, L.; Sperber, K. Mitochondria influence Fas expression in gp120-induced apoptosis of neuronal cells. Int. J. Neurosci. 2009, 119, 157–165. [Google Scholar] [CrossRef]
- Dreyer, E.B.; Kaiser, P.K.; Offermann, J.T.; Lipton, S.A. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science 1990, 248, 364–367. [Google Scholar] [CrossRef]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef]
- Capone, C.; Cervelli, M.; Angelucci, E.; Colasanti, M.; Macone, A.; Mariottini, P.; Persichini, T. A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic. Biol. Med. 2013, 63, 99–107. [Google Scholar] [CrossRef]
- Ferrucci, A.; Nonnemacher, M.R.; Cohen, E.A.; Wigdahl, B. Extracellular human immunodeficiency virus type 1 viral protein R causes reductions in astrocytic ATP and glutathione levels compromising the antioxidant reservoir. Virus Res. 2012, 167, 358–369. [Google Scholar] [CrossRef]
- Toborek, M.; Lee, Y.W.; Pu, H.; Malecki, A.; Flora, G.; Garrido, R.; Hennig, B.; Bauer, H.-C.; Nath, A. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J. Neurochem. 2003, 84, 169–179. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurodegeneration and the neurovascular unit. Nat. Med. 2010, 16, 1370–1371. [Google Scholar] [CrossRef]
- Singh, V.B.; Singh, M.V.; Gorantla, S.; Poluektova, L.Y.; Maggirwar, S.B. Smoothened Agonist Reduces Human Immunodeficiency Virus Type-1-Induced Blood-Brain Barrier Breakdown in Humanized Mice. Sci. Rep. 2016, 6, 26876. [Google Scholar] [CrossRef]
- Calabria, A.R.; Shusta, E.V. Blood-brain barrier genomics and proteomics: Elucidating phenotype, identifying disease targets and enabling brain drug delivery. Drug Discov. Today 2006, 11, 792–799. [Google Scholar] [CrossRef]
- An, S.F.; Groves, M.; Gray, F.; Scaravilli, F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J. Neuropathol. Exp. Neurol. 1999, 58, 1156–1162. [Google Scholar] [CrossRef]
- Ivey, N.S.; MacLean, A.G.; Lackner, A.A. AIDS and the blood-brain barrier. J. Neurovirol. 2009, 15, 111–122. [Google Scholar] [CrossRef]
- Nottet, H.S.; Persidsky, Y.; Sasseville, V.G.; Nukuna, A.N.; Bock, P.; Zhai, Q.H.; Sharer, L.R.; McComb, R.D.; Swindells, S.; Soderland, C.; et al. Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. J. Immunol. Baltim. Md 1950 1996, 156, 1284–1295. [Google Scholar]
- András, I.E.; Pu, H.; Deli, M.A.; Nath, A.; Hennig, B.; Toborek, M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J. Neurosci. Res. 2003, 74, 255–265. [Google Scholar] [CrossRef]
- Kim, T.-A.; Avraham, H.K.; Koh, Y.-H.; Jiang, S.; Park, I.-W.; Avraham, S. HIV-1 Tat-mediated apoptosis in human brain microvascular endothelial cells. J. Immunol. Baltim. Md 1950 2003, 170, 2629–2637. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: A potential mechanism of HIV-CNS invasion and NeuroAIDS. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Duan, F.; Morsey, B.; Persidsky, Y.; Kanmogne, G.D. HIV-1 activates proinflammatory and interferon-inducible genes in human brain microvascular endothelial cells: Putative mechanisms of blood-brain barrier dysfunction. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2008, 28, 697–711. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Schall, K.; Leibhart, J.; Knipe, B.; Gendelman, H.E.; Persidsky, Y. HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: Implication for viral neuropathogenesis. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2007, 27, 123–134. [Google Scholar] [CrossRef]
- Rao, V.R.; Ruiz, A.P.; Prasad, V.R. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res. Ther. 2014, 11, 13. [Google Scholar] [CrossRef]
- Roberts, T.K.; Buckner, C.M.; Berman, J.W. Leukocyte transmigration across the blood-brain barrier: Perspectives on neuroAIDS. Front. Biosci. Landmark Ed. 2010, 15, 478–536. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease—Systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef]
- Sharma, H.S.; Castellani, R.J.; Smith, M.A.; Sharma, A. The blood-brain barrier in Alzheimer’s disease: Novel therapeutic targets and nanodrug delivery. Int. Rev. Neurobiol. 2012, 102, 47–90. [Google Scholar]
- Xu, J.; Ikezu, T. The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: A foreseeable medical challenge in post-HAART era. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2009, 4, 200–212. [Google Scholar] [CrossRef]
- Hamel, F.G.; Fawcett, J.; Tsui, B.T.; Bennett, R.G.; Duckworth, W.C. Effect of nelfinavir on insulin metabolism, proteasome activity and protein degradation in HepG2 cells. Diabetes Obes. Metab. 2006, 8, 661–668. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- González-Scarano, F.; Martín-García, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Rodrigue, K.M.; Kennedy, K.M.; Park, D.C. Beta-amyloid deposition and the aging brain. Neuropsychol. Rev. 2009, 19, 436–450. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release Off. J. Control. Release Soc. 2015, 219, 396–405. [Google Scholar] [CrossRef]
- Kodidela, S.; Wang, Y.; Patters, B.J.; Gong, Y.; Sinha, N.; Ranjit, S.; Gerth, K.; Haque, S.; Cory, T.; McArthur, C.; et al. Proteomic Profiling of Exosomes Derived from Plasma of HIV-Infected Alcohol Drinkers and Cigarette Smokers. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2019. [Google Scholar] [CrossRef]
- Rahman, M.A.; Kodidela, S.; Sinha, N.; Haque, S.; Shukla, P.K.; Rao, R.; Kumar, S. Plasma exosomes exacerbate alcohol- and acetaminophen-induced toxicity via CYP2E1 pathway. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Haque, S.; Sinha, N.; Ranjit, S.; Midde, N.M.; Kashanchi, F.; Kumar, S. Monocyte-derived exosomes upon exposure to cigarette smoke condensate alter their characteristics and show protective effect against cytotoxicity and HIV-1 replication. Sci. Rep. 2017, 7, 16120. [Google Scholar] [CrossRef]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of Uptake. J. Circ. Biomark. 2015, 4. [Google Scholar] [CrossRef]
- Horibe, S.; Tanahashi, T.; Kawauchi, S.; Murakami, Y.; Rikitake, Y. Mechanism of recipient cell-dependent differences in exosome uptake. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Rahman, M.A.; Patters, B.J.; Kodidela, S.; Kumar, S. Extracellular Vesicles: Intercellular Mediators in Alcohol-Induced Pathologies. J. Neuroimmune Pharmacol. 2019. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. CMLS 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Dias, M.V.S.; Costa, C.S.; daSilva, L.L.P. The Ambiguous Roles of Extracellular Vesicles in HIV Replication and Pathogenesis. Front. Microbiol. 2018, 9, 2411. [Google Scholar] [CrossRef]
- Arakelyan, A.; Fitzgerald, W.; Zicari, S.; Vanpouille, C.; Margolis, L. Extracellular Vesicles Carry HIV Env and Facilitate Hiv Infection of Human Lymphoid Tissue. Sci. Rep. 2017, 7, 1695. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic Cph. Den. 2010, 11, 110–122. [Google Scholar] [CrossRef]
- Kodidela, S.; Ranjit, S.; Sinha, N.; McArthur, C.; Kumar, A.; Kumar, S. Cytokine profiling of exosomes derived from the plasma of HIV-infected alcohol drinkers and cigarette smokers. PLoS ONE 2018, 13, e0201144. [Google Scholar] [CrossRef]
- Li, M.; Aliotta, J.M.; Asara, J.M.; Tucker, L.; Quesenberry, P.; Lally, M.; Ramratnam, B. Quantitative proteomic analysis of exosomes from HIV-1-infected lymphocytic cells. Proteomics 2012, 12, 2203–2211. [Google Scholar] [CrossRef]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Sun, B.; Dalvi, P.; Abadjian, L.; Tang, N.; Pulliam, L. Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS Lond. Engl. 2017, 31, F9–F17. [Google Scholar] [CrossRef]
- Kadiu, I.; Narayanasamy, P.; Dash, P.K.; Zhang, W.; Gendelman, H.E. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J. Immunol. Baltim. Md 1950 2012, 189, 744–754. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release Off. J. Control. Release Soc. 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Qin, J.; Xu, Q. Functions and application of exosomes. Acta Pol. Pharm. 2014, 71, 537–543. [Google Scholar]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Fröhlich, D.; Kuo, W.P.; Frühbeis, C.; Sun, J.-J.; Zehendner, C.M.; Luhmann, H.J.; Pinto, S.; Toedling, J.; Trotter, J.; Krämer-Albers, E.-M. Multifaceted effects of oligodendroglial exosomes on neurons: Impact on neuronal firing rate, signal transduction and gene regulation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Xiao, T.; Zhang, W.; Jiao, B.; Pan, C.-Z.; Liu, X.; Shen, L. The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl. Neurodegener. 2017, 6. [Google Scholar] [CrossRef]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell. Mol. Life Sci. CMLS 2018, 75, 757–773. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Exosomes as possible spread factor and potential biomarkers in Alzheimer’s disease: Current concepts. Biomark. Med. 2018, 12, 1025–1033. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.-I.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Sergeant, N.; Buée, L. Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer’s disease. Front. Physiol. 2012, 3, 229. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. J. Alzheimers Assoc. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflammation 2014, 11, 68. [Google Scholar] [CrossRef]
- Daily, A.; Nath, A.; Hersh, L.B. Tat peptides inhibit neprilysin. J. Neurovirol. 2006, 12, 153–160. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Usuki, S.; Sakai, S.; Hanamatsu, H.; Mioka, T.; Kimura, N.; Okada, M.; Tahara, H.; Furukawa, J.; et al. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-β peptide. FEBS Lett. 2015, 589, 84–88. [Google Scholar] [CrossRef]
- Soliman, M.L.; Geiger, J.D.; Chen, X. Caffeine Blocks HIV-1 Tat-Induced Amyloid Beta Production and Tau Phosphorylation. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2017, 12, 163–170. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimers Dis. JAD 2010, 20 (Suppl. 1), S167–S174. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef]
- Druzhkova, T.A.; Yakovlev, A.A. Exosome Drug Delivery through the Blood–Brain Barrier: Experimental Approaches and Potential Applications. Neurochem. J. 2018, 12, 195–204. [Google Scholar] [CrossRef]
- Morales-Prieto, D.M.; Stojiljkovic, M.; Diezel, C.; Streicher, P.-E.; Röstel, F.; Lindner, J.; Weis, S.; Schmeer, C.; Marz, M. Peripheral blood exosomes pass blood-brain-barrier and induce glial cell activation. bioRxiv 2018, 471409. [Google Scholar] [CrossRef]
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnology 2018, 16, 81. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Fields, J.A.; Metcalf, J.; Overk, C.; Adame, A.; Spencer, B.; Wrasidlo, W.; Florio, J.; Rockenstein, E.; He, J.J.; Masliah, E. The anticancer drug sunitinib promotes autophagyand protects from neurotoxicity in an HIV-1 Tat model of neurodegeneration. J. Neurovirol. 2017, 23, 290–303. [Google Scholar] [CrossRef]
- Tang, S.C.; Lagas, J.S.; Lankheet, N.A.G.; Poller, B.; Hillebrand, M.J.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int. J. Cancer 2012, 130, 223–233. [Google Scholar] [CrossRef]
- Do Rosário André, M.; Pedro, A.; Lyden, D. Cancer Exosomes as Mediators of Drug Resistance. Methods Mol. Biol. Clifton NJ 2016, 1395, 229–239. [Google Scholar]
- Zhang, H.; Jiang, L.-H.; Hou, J.-C.; Zhong, S.-L.; Zhu, L.-P.; Wang, D.-D.; Zhou, S.-Y.; Yang, S.-J.; Wang, J.-Y.; Zhang, Q.; et al. Exosome: A novel mediator in drug resistance of cancer cells. Epigenomics 2018, 10, 1499–1509. [Google Scholar] [CrossRef]
- Ali, A.; Banerjea, A.C. Curcumin inhibits HIV-1 by promoting Tat protein degradation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Zhang, L.; Fiala, M.; Cashman, J.; Sayre, J.; Espinosa, A.; Mahanian, M.; Zaghi, J.; Badmaev, V.; Graves, M.C.; Bernard, G.; et al. Curcuminoids enhance amyloid-beta uptake by macrophages of Alzheimer’s disease patients. J. Alzheimers Dis. JAD 2006, 10, 1–7. [Google Scholar] [CrossRef]
- Tang, M.; Taghibiglou, C. The Mechanisms of Action of Curcumin in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 58, 1003–1016. [Google Scholar] [CrossRef]
- Kalani, A.; Chaturvedi, P. Curcumin-primed and curcumin-loaded exosomes: Potential neural therapy. Neural Regen. Res. 2017, 12, 205–206. [Google Scholar] [CrossRef]
- Kalani, A.; Chaturvedi, P.; Kamat, P.K.; Maldonado, C.; Bauer, P.; Joshua, I.G.; Tyagi, S.C.; Tyagi, N. Curcumin-loaded embryonic stem cell exosomes restored neurovascular unit following ischemia-reperfusion injury. Int. J. Biochem. Cell Biol. 2016, 79, 360–369. [Google Scholar] [CrossRef]
- Wang, H.; Sui, H.; Zheng, Y.; Jiang, Y.; Shi, Y.; Liang, J.; Zhao, L. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3β pathway. Nanoscale 2019, 11, 7481–7496. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Chivero, E.T.; Guo, M.-L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef]
- Cypryk, W.; Nyman, T.A.; Matikainen, S. From Inflammasome to Exosome-Does Extracellular Vesicle Secretion Constitute an Inflammasome-Dependent Immune Response? Front. Immunol. 2018, 9, 2188. [Google Scholar] [CrossRef]
- Sun, B.; Fernandes, N.; Pulliam, L. Profile of neuronal exosomes in HIV cognitive impairment exposes gender differences. AIDS Lond. Engl. 2019. [Google Scholar] [CrossRef]


{kind=link}
| HIV Components | Mechanism | Consequences/Conclusion | References |
|---|---|---|---|
| Neuro-Inflammation | |||
| HIV-infected microglia, macrophages, astrocytes | HIV infection of CNS cells provides an inflammatory stimulus and promotes secretion of viral proteins, e.g., Tat, gp-120 | Cycle of excessive cytokine/chemokine production, Aβ production, ROS production | [48,97] |
| HIV Proteins | |||
| Tat | Forms highly neurotoxic complexes with Aβ; inhibits neprilysin; inhibits microglial phagocytosis of Aβ; stimulates Aβ 1–42 release and promotes plaque accumulation; enhances cleavage of Aβ precursors; alters BBB permeability (see BBB damage) | More Aβ is produced in the CNS while less is cleared; alteration of Aβ degradation/metabolism; BBB damage | [27,53,54,55,57,59,60] |
| Gp-120 | Like Tat, alters Aβ trafficking/accumulation and enhances cleavage of Aβ precursors; alters BBB permeability (see BBB damage) | More Aβ is produced in the CNS while less is cleared | [62,63] |
| Gag | Aβ precursor, APP, binds and sequesters Gag in lipid rafts within macrophages to prevent viral spreading. In defense, Gag enhances APP cleavage | Increased Aβ production | [65] |
| Excitotoxicity and oxidative stress | |||
| HIV-infected microglia, macrophages, astrocytes | Infected CNS cells release pro-inflammatory chemicals that activate NMDARs | Excessive activation of NMDARs promotes excitotoxicity and free radical production | [69,70,71,76] |
| Tat, gp-120, Nef, Vpr | Viral proteins injure neuronal cells directly and disrupt calcium homeostasis, activate caspases, promote ROS accumulation; alter BBB permeability via oxidative stress pathways (see BBB damage) | Excitotoxicity/Induction of oxidative stress; BBB damage | [73,74,75,77] |
| BBB Damage | |||
| HIV virus | Affect HBMECs by releasing HIV gene products, inflammatory cytokines, and adhesion molecules on brain endothelium | Induction of oxidative stress | [84] |
| Tat, gp120, Nef | Alteration of the levels of tight junction proteins, nitric oxide, pro-inflammatory and interferon-inducible genes, leukocyte adhesion, trans-endothelial electrical resistance, and matrix metalloproteinases | Increased permeability of HBMEC | [58,85,87,88,89,90] |
| Aging | β-amyloid generation | Higher Aβ | [98] |
| ART Medication | |||
| ART | Increased oxidative stress; IRIS promotes vasculitis, hyperlipidemia, diabetes, coronary artery disease; Nelfinavir inhibits Aβ degradation enzyme | Contributes to AD risk factors and Aβ accumulation | [94,95,96] |
| HIV Proteins | Source | Reference | AD | Source | Reference |
|---|---|---|---|---|---|
| Tat | Astrocytes | [19] | Aβ | Primary cortical neurons | [131] |
| Cerebrospinal fluid | [131] | ||||
| Neuroblastoma cell lines (SH-SY5Y) | [23] | ||||
| Gag | Monocyte-derived macrophages | [114] | Brain tissues from AD patients | [23] | |
| Astrocyte derived exosomes from plasma of AD patients | [122] | ||||
| NDEs from plasma of AD patients | [122] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kodidela, S.; Gerth, K.; Haque, S.; Gong, Y.; Ismael, S.; Singh, A.; Ishrat, T.; Kumar, S. Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects? Cells 2019, 8, 968. https://doi.org/10.3390/cells8090968
Kodidela S, Gerth K, Haque S, Gong Y, Ismael S, Singh A, Ishrat T, Kumar S. Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects? Cells. 2019; 8(9):968. https://doi.org/10.3390/cells8090968
Chicago/Turabian StyleKodidela, Sunitha, Kelli Gerth, Sanjana Haque, Yuqing Gong, Saifudeen Ismael, Ajay Singh, Tauheed Ishrat, and Santosh Kumar. 2019. "Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects?" Cells 8, no. 9: 968. https://doi.org/10.3390/cells8090968
APA StyleKodidela, S., Gerth, K., Haque, S., Gong, Y., Ismael, S., Singh, A., Ishrat, T., & Kumar, S. (2019). Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects? Cells, 8(9), 968. https://doi.org/10.3390/cells8090968