PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway

 , and
, and
Abstract

1. Introduction
2. Materials and Methods
2.1. C2C12 Cell Culture, Transfection, and Differentiation
2.2. Construction of a PFN2a-Overexpressing Cell Line by CRISPR/Cas9
2.3. Identification of a PFN2a-Overexpressing Cell Line
2.4. RNA Extraction, Complementary DNA (cDNA) Synthesis, and Quantitative Real-Time PCR (qPCR)
2.5. 5-Ethynyl-2′-deoxyuridine (Edu) Assays
2.6. Flow Cytometry Analysis of the Cell Cycle
2.7. Flow Cytometry Analysis of the Cell Apoptosis during C2C12 Myogenic Development
2.8. Western Blot Analysis
2.9. Isolation of Nuclear and Cytoplasmic Extracts
2.10. Immunofluorescent Staining and Confocal Microscopy
2.11. Primers
2.12. Statistical Analysis
3. Results
3.1. PFN2a Knock-in at ROSA26 Locus of C2C12 Cells was Made with CRISPR/Cas9
3.2. PFN2a Overexpression Suppressed C2C12 Proliferation
3.3. PFN2a Overexpression Inhibited C2C12 Myogenic Differentiation and Disturbed Sarcomere Structural Assembly
3.4. PFN2a Overexpression Promoted Apoptosis during C2C12 Myogenic Differentiation
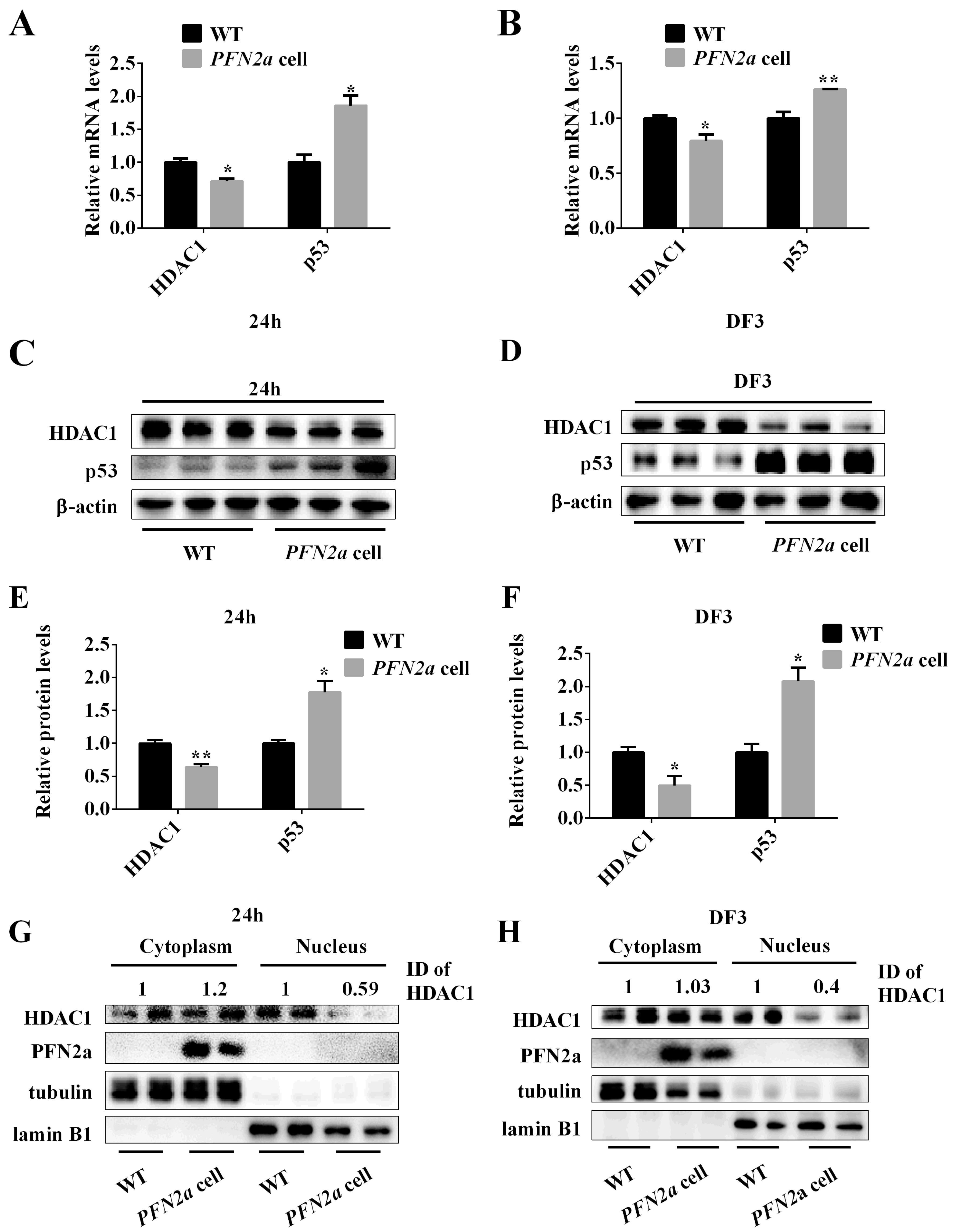
3.5. PFN2a Overexpression Suppressed the Protein Content of HDAC1 in the Nucleus and Promoted the Content of p53 during C2C12 Myogenic Development
3.6. PFN2a Overexpression Downregulated Myogenic Development through p53
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hou, L.; Xu, J.; Jiao, Y.; Li, H.; Pan, Z.; Duan, J.; Gu, T.; Hu, C.; Wang, C. Mir-27b promotes muscle development by inhibiting mdfi expression. Cell. Physiol. Biochem. 2018, 46, 2271–2283. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Campos, N.; Lopez-Pedrosa, J.M.; Rueda, R.; Rodriguez-Manas, L. Skeletal muscle regulates metabolism via interorgan crosstalk: Roles in health and disease. J. Am. Med. Dir. Assoc. 2016, 17, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Beaudart, C.; Rizzoli, R.; Bruyere, O.; Reginster, J.Y.; Biver, E. Sarcopenia: Burden and challenges for public health. Arch. Public Health 2014, 72, 45. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite cells and skeletal muscle regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [PubMed]
- Shea, K.L.; Xiang, W.; LaPorta, V.S.; Licht, J.D.; Keller, C.; Basson, M.A.; Brack, A.S. Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 2010, 6, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Lee, J.D.; Mula, J.; Kirby, T.J.; Jackson, J.R.; Liu, F.; Yang, L.; Mendias, C.L.; Dupont-Versteegden, E.E.; McCarthy, J.J.; et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2015, 21, 76–80. [Google Scholar] [CrossRef]
- Abou-Khalil, R.; Le Grand, F.; Pallafacchina, G.; Valable, S.; Authier, F.J.; Rudnicki, M.A.; Gherardi, R.K.; Germain, S.; Chretien, F.; Sotiropoulos, A.; et al. Autocrine and paracrine angiopoietin 1/tie-2 signaling promotes muscle satellite cell self-renewal. Cell Stem Cell 2009, 5, 298–309. [Google Scholar] [CrossRef]
- Thakur, S.S.; James, J.L.; Cranna, N.J.; Chhen, V.L.; Swiderski, K.; Ryall, J.G.; Lynch, G.S. Expression and localization of heat-shock proteins during skeletal muscle cell proliferation and differentiation and the impact of heat stress. Cell Stress Chaperones 2019, 24, 749–761. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- McLoughlin, T.J.; Smith, S.M.; DeLong, A.D.; Wang, H.; Unterman, T.G.; Esser, K.A. Foxo1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am. J. Physiol. Cell Physiol. 2009, 297, C548–C555. [Google Scholar] [CrossRef] [PubMed]
- Schoneich, C.; Dremina, E.; Galeva, N.; Sharov, V. Apoptosis in differentiating c2c12 muscle cells selectively targets bcl-2-deficient myotubes. Apoptosis 2014, 19, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; McFarland, D.C.; Strasburg, G.M.; Velleman, S.G. Function of death-associated protein 1 in proliferation, differentiation, and apoptosis of chicken satellite cells. Muscle Nerve 2013, 48, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.V.; McMahon, J.M.; Howley, B.A.; Stanley, A.; Ritter, T.; Mohr, A.; Zwacka, R.; Fearnhead, H.O. A non-apoptotic role for caspase-9 in muscle differentiation. J. Cell Sci. 2008, 121, 3786–3793. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.Y.; Zhang, C.P.; Zhong, L.P.; Pan, H.Y.; Chen, W.T.; Wang, L.Z.; Andrew, O.W.; Ji, T.; Han, W. Decreased expression of profilin 2 in oral squamous cell carcinoma and its clinicopathological implications. Oncol. Rep. 2011, 26, 813–823. [Google Scholar]
- Jeong, D.H.; Choi, Y.N.; Seo, T.W.; Lee, J.S.; Yoo, S.J. Ubiquitin-proteasome dependent regulation of profilin2 (pfn2) by a cellular inhibitor of apoptotic protein 1 (ciap1). Biochem. Biophys. Res. Commun. 2018, 506, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Gareus, R.; Kwiatkowski, D.; Witke, W. Alternative splicing of the mouse profilin ii gene generates functionally different profilin isoforms. J. Cell Sci. 2000, 113 Pt 21, 3795–3803. [Google Scholar]
- Lambrechts, A.; Braun, A.; Jonckheere, V.; Aszodi, A.; Lanier, L.M.; Robbens, J.; Van Colen, I.; Vandekerckhove, J.; Fassler, R.; Ampe, C. Profilin ii is alternatively spliced, resulting in profilin isoforms that are differentially expressed and have distinct biochemical properties. Mol. Cell. Biol. 2000, 20, 8209–8219. [Google Scholar] [CrossRef] [PubMed]
- Murk, K.; Buchmeier, S.; Jockusch, B.M.; Rothkegel, M. In birds, profilin-2a is ubiquitously expressed and contributes to actin-based motility. J. Cell Sci. 2009, 122, 957–964. [Google Scholar] [CrossRef]
- Tariq, N.; Basharat, Z.; Butt, S.; Baig, D.N. Distribution analysis of profilin isoforms at transcript resolution with mrna-seq and secondary structure in various organs of rattus norvegicus. Gene 2016, 589, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, Y.S.; Han, G.Y.; Lee, H.N.; Ahn, C.; Kim, C.W. Profilin 2 promotes migration, invasion, and stemness of ht29 human colorectal cancer stem cells. Biosci. Biotechnol. Biochem. 2015, 79, 1438–1446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pilo Boyl, P.; Di Nardo, A.; Mulle, C.; Sassoe-Pognetto, M.; Panzanelli, P.; Mele, A.; Kneussel, M.; Costantini, V.; Perlas, E.; Massimi, M.; et al. Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J. 2007, 26, 2991–3002. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Viswanathan, M.C.; Lee, D.I.; Rainer, P.P.; Schmidt, W.; Kronert, W.A.; Harding, S.E.; Kass, D.A.; Bernstein, S.I.; Van Eyk, J.E.; et al. Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 110, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.; Rubenstein, P.A. Control of profilin and actin expression in muscle and nonmuscle cells. Cell Motil. Cytoskelet. 1993, 24, 179–188. [Google Scholar] [CrossRef]
- Tang, Y.N.; Ding, W.Q.; Guo, X.J.; Yuan, X.W.; Wang, D.M.; Song, J.G. Epigenetic regulation of smad2 and smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, Y.; Specht, S.; Lunz, J.G., 3rd; Isse, K.; Corbitt, N.; Takizawa, T.; Demetris, A.J. Sprr2a enhances p53 deacetylation through hdac1 and down regulates p21 promoter activity. BMC Mol. Biol. 2012, 13, 20. [Google Scholar] [CrossRef]
- Hou, L.; Shi, J.; Cao, L.; Xu, G.; Hu, C.; Wang, C. Pig has no uncoupling protein 1. Biochem. Biophys. Res. Commun. 2017, 487, 795–800. [Google Scholar] [CrossRef]
- Jockusch, B.M.; Murk, K.; Rothkegel, M. The profile of profilins. Rev. Physiol. Biochem. Pharmacol. 2007, 159, 131–149. [Google Scholar]
- Zhang, L.; Bu, L.; Hu, J.; Xu, Z.; Ruan, L.; Fang, Y.; Wang, P. Hdac1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018, 399, 603–610. [Google Scholar] [CrossRef]
- Wang, X.Q.; Bai, H.M.; Li, S.T.; Sun, H.; Min, L.Z.; Tao, B.B.; Zhong, J.; Li, B. Knockdown of hdac1 expression suppresses invasion and induces apoptosis in glioma cells. Oncotarget 2017, 8, 48027–48040. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. Mdm2-hdac1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis-the p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Wawryk-Gawda, E.; Chylinska-Wrzos, P.; Lis-Sochocka, M.; Chlapek, K.; Bulak, K.; Jedrych, M.; Jodlowska-Jedrych, B. P53 protein in proliferation, repair and apoptosis of cells. Protoplasma 2014, 251, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Lagger, G.; Doetzlhofer, A.; Schuettengruber, B.; Haidweger, E.; Simboeck, E.; Tischler, J.; Chiocca, S.; Suske, G.; Rotheneder, H.; Wintersberger, E.; et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/waf1/cip1 gene. Mol. Cell. Biol. 2003, 23, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Petrachkova, T.; Wortinger, L.A.; Bard, A.J.; Singh, J.; Warga, R.M.; Kane, D.A. Lack of cyclin B1 in zebrafish causes lengthening of G2 and M phases. Dev. Biol. 2019, 451, 167–179. [Google Scholar] [CrossRef]
- Belso, N.; Guban, B.; Manczinger, M.; Kormos, B.; Bebes, A.; Nemeth, I.; Vereb, Z.; Szell, M.; Kemeny, L.; Bata-Csorgo, Z. Differential role of d cyclins in the regulation of cell cycle by influencing ki67 expression in hacat cells. Exp. Cell Res. 2019, 374, 290–303. [Google Scholar] [CrossRef]
- Jurikova, M.; Danihel, L.; Polak, S.; Varga, I. Ki67, pcna, and mcm proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef]
- Chuang, L.S.; Ian, H.I.; Koh, T.W.; Ng, H.H.; Xu, G.; Li, B.F. Human DNA-(cytosine-5) methyltransferase-pcna complex as a target for p21waf1. Science 1997, 277, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with pcna. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. Cdk inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Porrello, A.; Cerone, M.A.; Coen, S.; Gurtner, A.; Fontemaggi, G.; Cimino, L.; Piaggio, G.; Sacchi, A.; Soddu, S. P53 regulates myogenesis by triggering the differentiation activity of prb. J. Cell Biol. 2000, 151, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Connolly, T.; Futcher, B.; Beach, D. Human d-type cyclin. Cell 1991, 65, 691–699. [Google Scholar] [CrossRef]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and activation of a cyclin e-cdk2 complex during the g1 phase of the human cell cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin a is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef]
- Levine, A.J. P53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequences 1 (5′ to 3′) | Size (bp) | Application |
|---|---|---|---|
| PFN2a donor primer | F: AGGCGCGCCACCGCCTCTGCTCCTGC | 673 | Amplification of the ORF of PFN2a for constructing PFN2a donor |
| R: CGCGGATCCCCGCCTCTAACCAATGCTG | |||
| pCDNA3.1 (+)-PFN2a primer | F: GGGGTACCATGGCCGGTTGGCAGAGCTA | 439 | Amplification of the ORF of PFN2a for constructing pCDNA3.1 (+)-PFN2a. This vector was used to establish standard curve line for absolute quantitative analysis of PFN2a- overexpressing cell line. |
| R: CGGAATTCCTAGAACCCAGAGTCTCTCAAGT |
| Primer Name | Primer Sequences 1 (5′ to 3′) | Size (bp) | Application |
|---|---|---|---|
| 5′HR | F: AGGGAGCGGAAAAGTCTCCA | 1280 | Accurate detection of integration of PFN2a donor into ROSA26 locus. |
| R: CTCCCACCGTACACGCCTAC | |||
| 3′HR | F: GGTGCCTGAAATCAACCTCTGGA | 1596 | |
| R: TCAAGCCAGTCCAAGAGAAAGCA | |||
| F2R2 | F2: ACCGCCTCTGCTCCTGC | 655 | Amplification of the ORF of PFN2a. |
| R2: CCGCCTCTAACCAATGCTG | |||
| F3R3 | F3: CAGGCAGACCTCCATCGC | 354 or 4163 | Genotype analysis of PFN2a-overexpressing cell line by PCR. |
| R3: GACAACGCCCACACACCAG | |||
| F4R4 | F: CAACGGCAAAATACTTGAGAGA | 120 | Genotype analysis of PFN2a-overexpressing cell line by absolute quantitative analysis |
| R: CCGACGGATACAAAGGAGAC |
| Primer Name | Primer Sequences 1 (5′ to 3′) | Size (bp) | Accession Number |
|---|---|---|---|
| PFN2a | F: CAACGGCAAAATACTTGAGAGA | 120 | NM_019410.3 |
| R: CCGACGGATACAAAGGAGAC | |||
| PCNA | F: AGCGGAGAAGGTGCTGGAG | 157 | NM_011045.2 |
| R: ATAGCGGCGGTATGTGTCG | |||
| CCNB1 | F: TAGGGCGAGGTCAGTATGGC | 211 | NM_172301.3 |
| R: ACTTCCCGACAGGTTTTGGTAG | |||
| CCND1 | F: GCCACGCCCTCCGTATCT | 190 | NM_007631.2 |
| R: GTAACCAGCGGCTCTTCTTCA | |||
| p21 | F: CGAGAACGGTGGAACTTTGAC | 107 | NM_007669.5 |
| R: CCAGGGCTCAGGTAGACCTT | |||
| MyHC | F: CCCGCTAAGGGTCTTCGTA | 176 | NM_030679.2 |
| R: GCCCCGTTGACATTGGA | |||
| myogenin | F: CCTGGAAGAAAAGGGACTGG | 246 | NM_031189.2 |
| R: CGCTCAATGTACTGGATGGC | |||
| α-actinin | F: ATGCCTCGCTGCTGAATGA | 191 | NM_134156.2 |
| R: GCTCGCAAAAGCCTCGTG | |||
| tropomyosin 1 | F: AACCCGCACAAATACTCCGA | 138 | NM_001164248.1 |
| R: AACAGCCCACTCCTCCTCAAC | |||
| titin | F: GCCGCCTGGAATCCCTAC | 324 | NM_011652.3 |
| R: ACGCATCTGGCATCAAAGTG | |||
| nebulin | F: CAGCCAAAGCCACCCCA | 348 | NM_010889.1 |
| R: TCTCACCAACCCGCCTCAT | |||
| caspase 9 | F: CGCCATCTGGGTCTCGG | 241 | NM_001277932.1 |
| R: ACTCGCTGCTCCTTTGCTG | |||
| caspase 8 | F: CGCCCGTGCTTGGACTAC | 152 | NM_001080126.1 |
| R: TTCTCCCGCCGACTGATGT | |||
| caspase 3 | F: GTCTGACTGGAAAGCCGAAACT | 104 | NM_001284409.1 |
| R: GCAAGCCATCTCCTCATCAGTC | |||
| p53 | F: GAACCGCCGACCTATCCTTA | 95 | NM_001127233.1 |
| R: GGCAGGCACAAACACGAAC | |||
| HDAC1 | F: AGTCTGTTACTACTACGACGGG | 101 | NM_008228.2 |
| R: TGAGCAGCAAATTGTGAGTCAT | |||
| β-actin | F: TGGTGCGAATGGGTCAGAA | 310 | NM_007393.5 |
| R: CCGCCAGAGGCATACAGG |
| Primary Antibody | Clone | Company | Catalog No. | Dilution |
|---|---|---|---|---|
| PFN2a | Polyclonal | Enogene | E93047 | 1:1000 |
| CCNB1 | Polyclonal | CST | 4138 | 1:1000 |
| CCND1 | Polyclonal | CST | 2922 | 1:1000 |
| MyHC | Monoclonal | BOSTER | BM0096 | 1:500 |
| myogenin | Polyclonal | SANTA CRUZ | J2314 | 1:200 |
| α-Actinin | Polyclonal | GeneTex | GTX103219 | 1:1000 |
| caspase 3 (H-277) | Polyclonal | SANTA CRUZ | sc-7148 | 1:1000 |
| cleaved caspase 3 (Asp175) | Polyclonal | CST | 9661 | 1:1000 |
| p53 | Monoclonal | GeneTex | GTX70214 | 1:5000 |
| HDAC1 | Polyclonal | Bioss | bs-1414R | 1:1000 |
| tubulin | Polyclonal | Bioworld | AP0064 | 1:5000 |
| lamin B1 | Polyclonal | Bioworld | AP6001 | 1:5000 |
| β-actin | Monoclonal | Bioworld | BS6007M | 1:5000 |
| Secondary Antibody | Conjugate Used | Company | Catalog No. | Dilution |
|---|---|---|---|---|
| Goat anti-Rabbit IgG 1 (H+L) | HRP 2 | Bioworld | BS13278 | 1:50000 |
| Goat anti-Mouse IgG 1 (H+L) | HRP 2 | Bioworld | BS12478 | 1:50000 |
| Goat Anti-Mouse IgG 1 (H+L) | Cy3 3 | Bioworld | BS10006 | 1:200 |
| Goat Anti-rabbit IgG 1 | Alexa Fluor 647 3 | Bioss | bs-0295G-AF647 | 1:200 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Hou, L.; Zhang, Y.; Jiang, F.; Zhu, Y.; Li, Q.X.; Hu, C.Y.; Wang, C. PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway. Cells 2019, 8, 959. https://doi.org/10.3390/cells8090959
Li H, Hou L, Zhang Y, Jiang F, Zhu Y, Li QX, Hu CY, Wang C. PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway. Cells. 2019; 8(9):959. https://doi.org/10.3390/cells8090959
Chicago/Turabian StyleLi, Huaqin, Lianjie Hou, Yu Zhang, Fangyi Jiang, Yifan Zhu, Qing X. Li, Ching Yuan Hu, and Chong Wang. 2019. "PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway" Cells 8, no. 9: 959. https://doi.org/10.3390/cells8090959
APA StyleLi, H., Hou, L., Zhang, Y., Jiang, F., Zhu, Y., Li, Q. X., Hu, C. Y., & Wang, C. (2019). PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway. Cells, 8(9), 959. https://doi.org/10.3390/cells8090959