H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Quantitative Scores of Genes and Pathways Regulatory Evolution
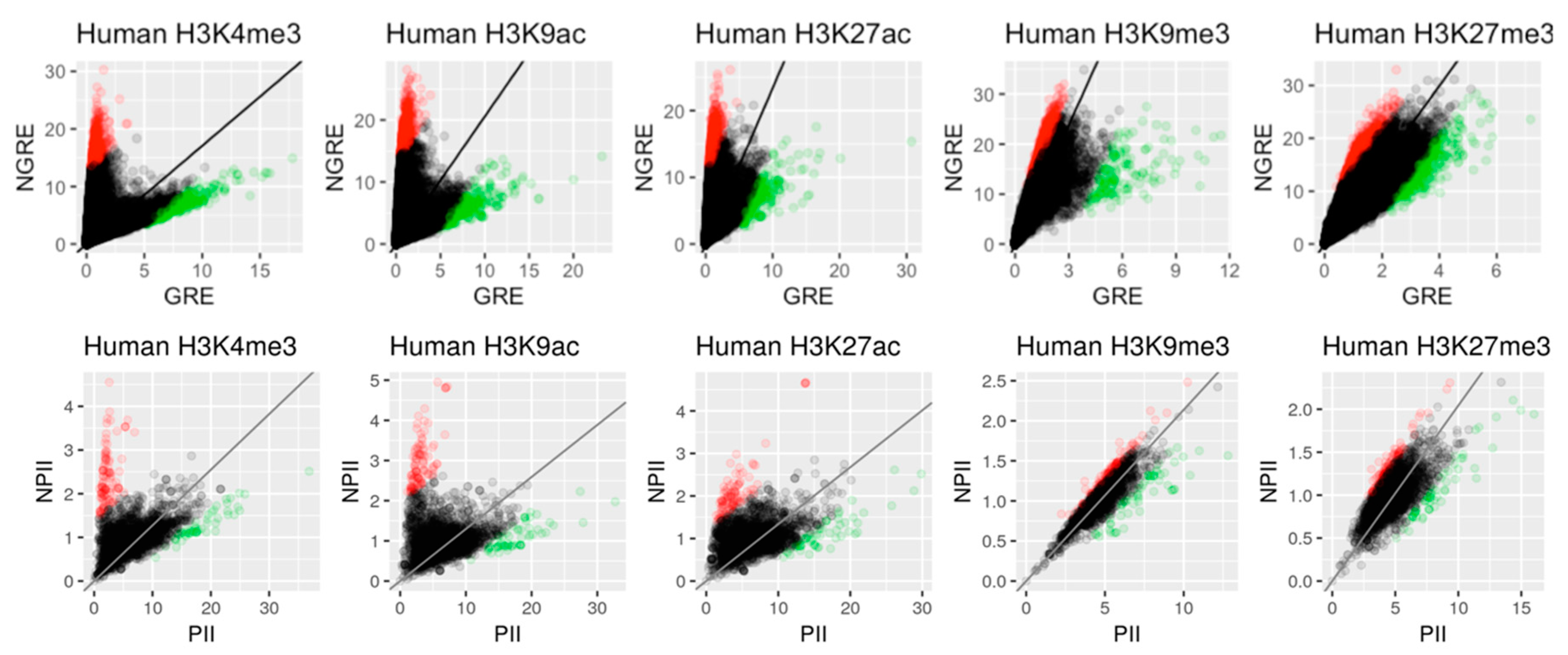
2.1.1. GRE and NGRE Scores
2.1.2. PII and NPII Scores
2.2. Primary Data
2.3. Gene Expression Data
2.4. Enrichment Analysis for Groups of Differential Genes
2.5. Measuring Pathway Enrichment by RE-Linked Hits
2.6. Combination of Gene- and Pathway-Based Enrichment Scores
3. Results
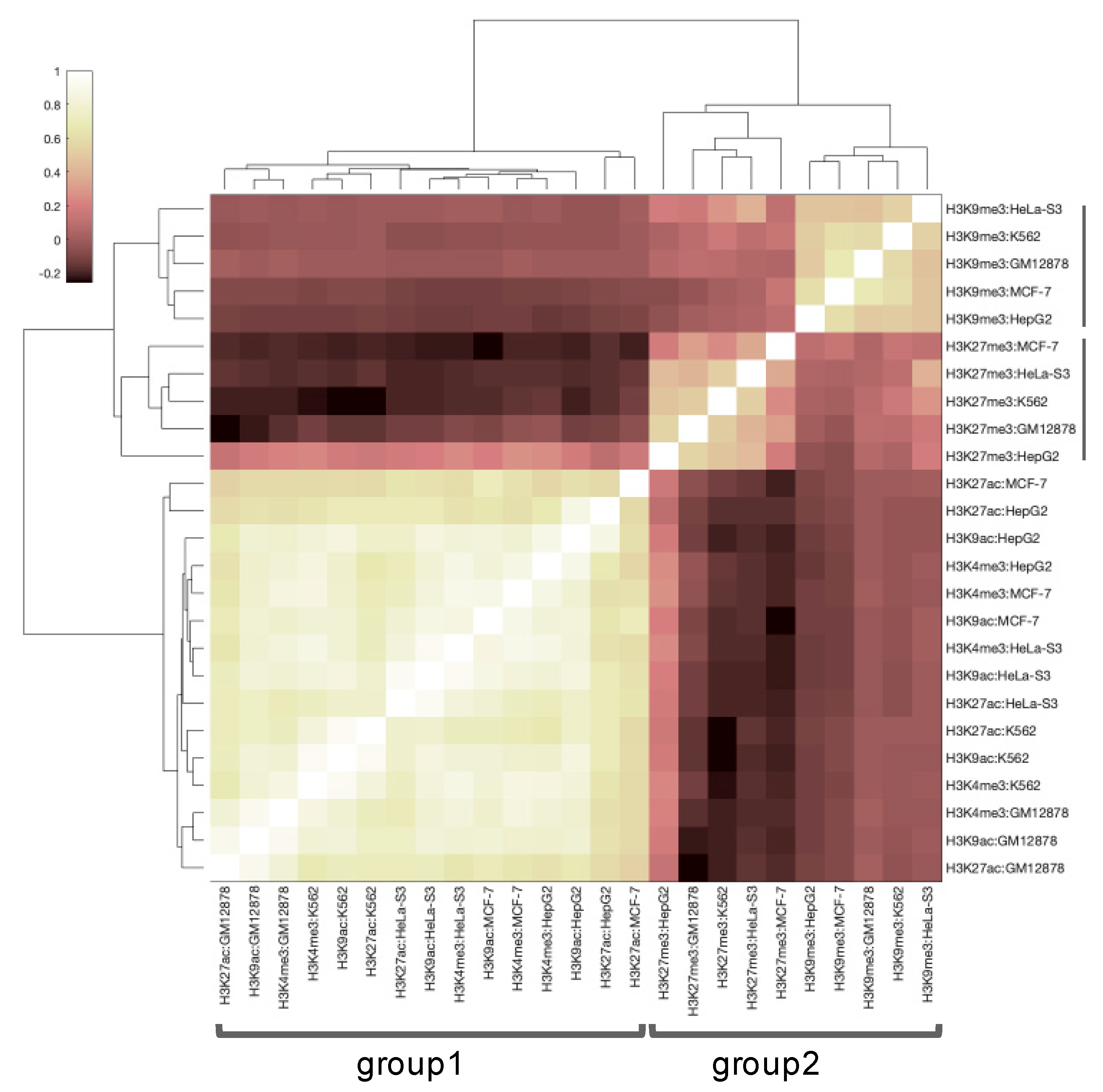
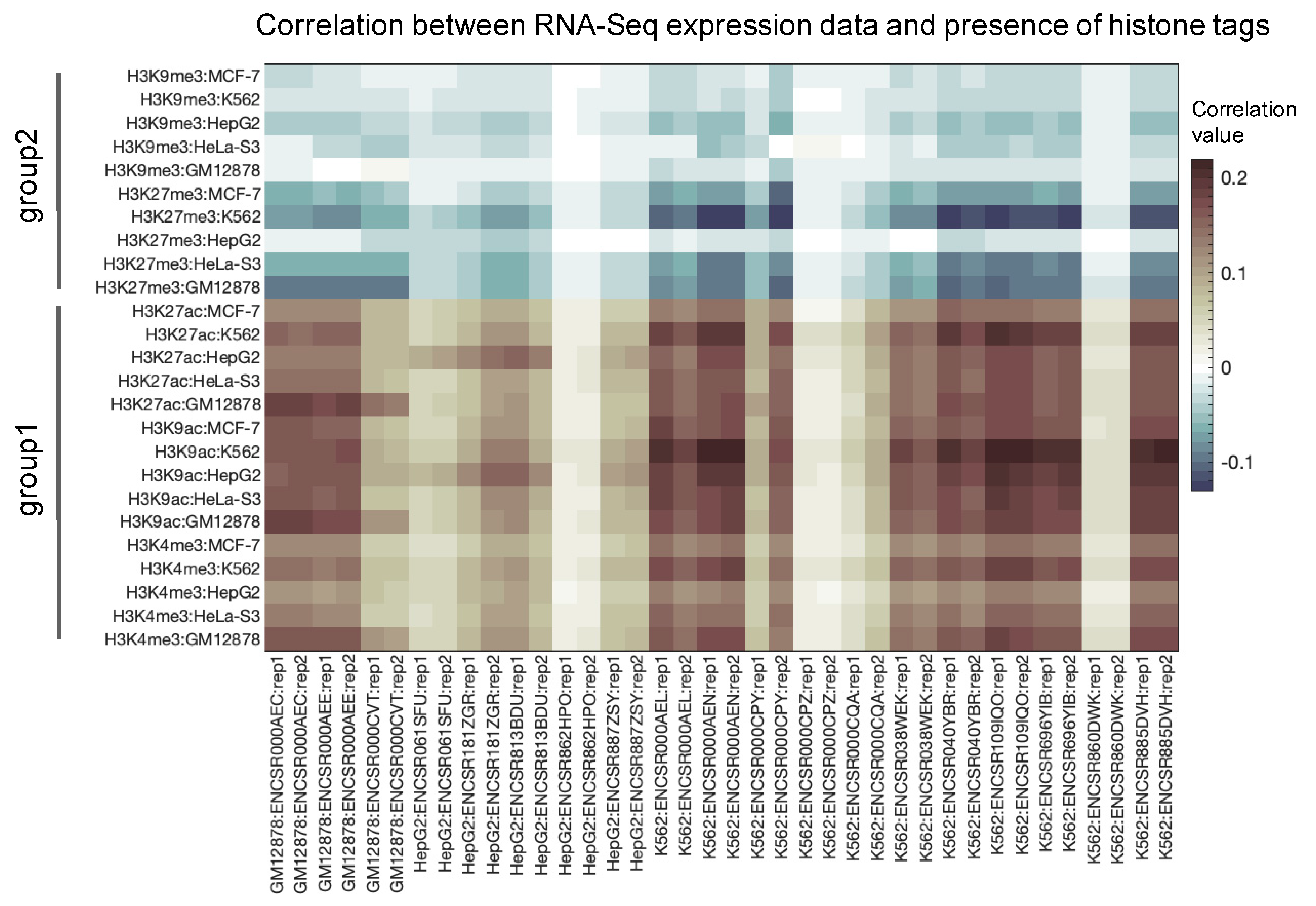
3.1. Primary Data Analysis
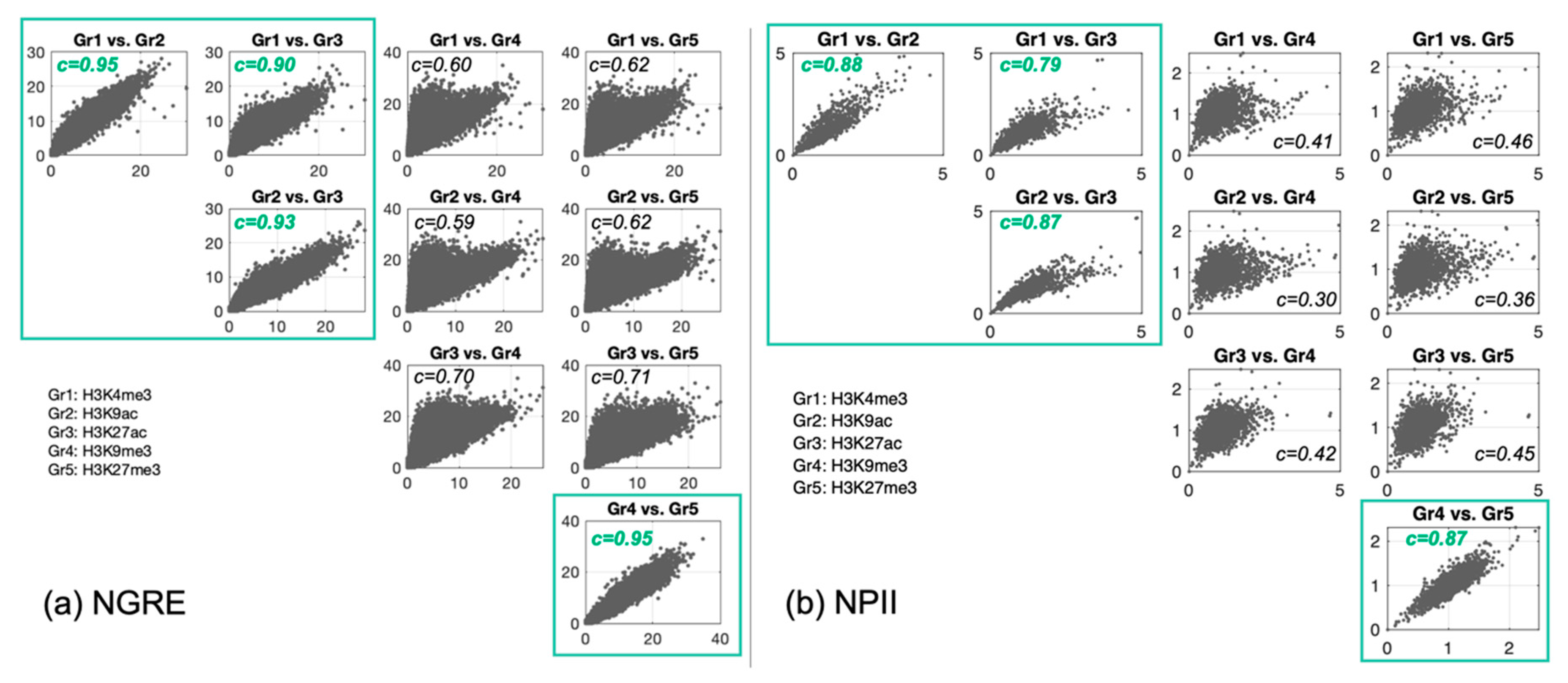
3.2. Correlation between Histone Tags Based on GRE/NGRE and PII/NPII Scores
3.3. Genes and Molecular Pathways Enriched or Deficient in RE-Linked Regulation
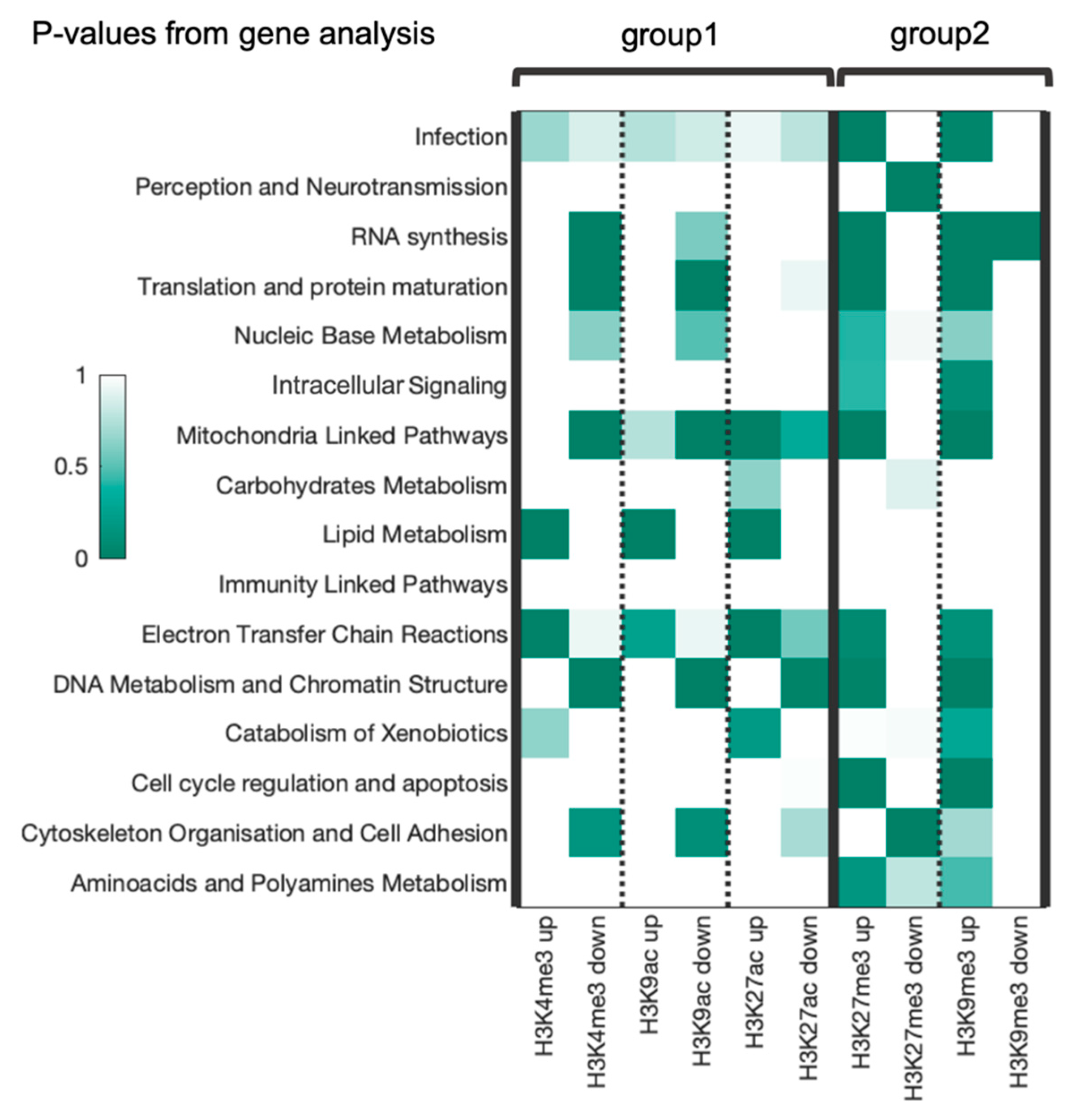
3.4. Gene Ontology (GO) Annotation of Top RRE-Enriched and Deficient Genes
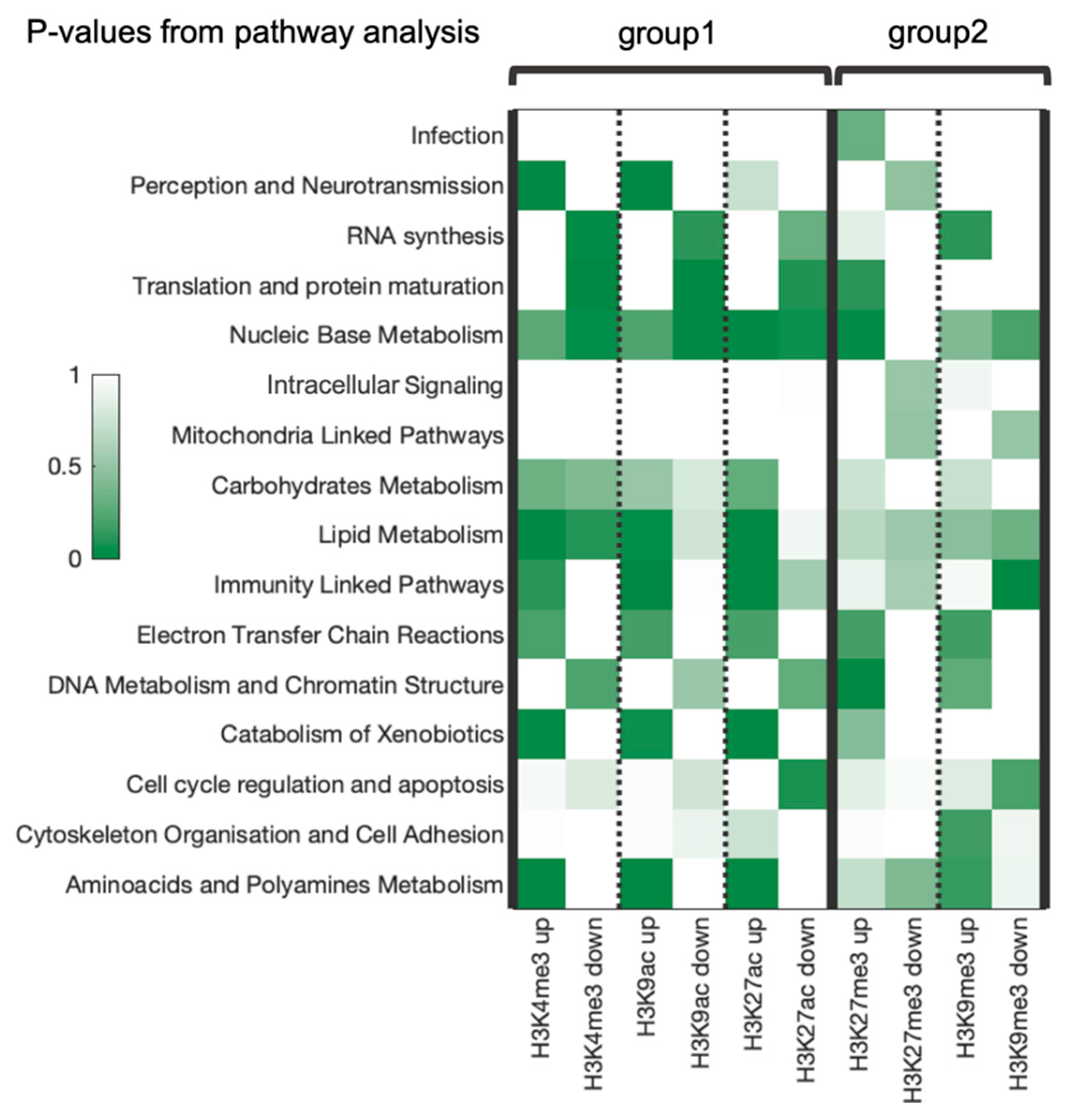
3.5. Top RRE-Enriched and Deficient Molecular Pathways
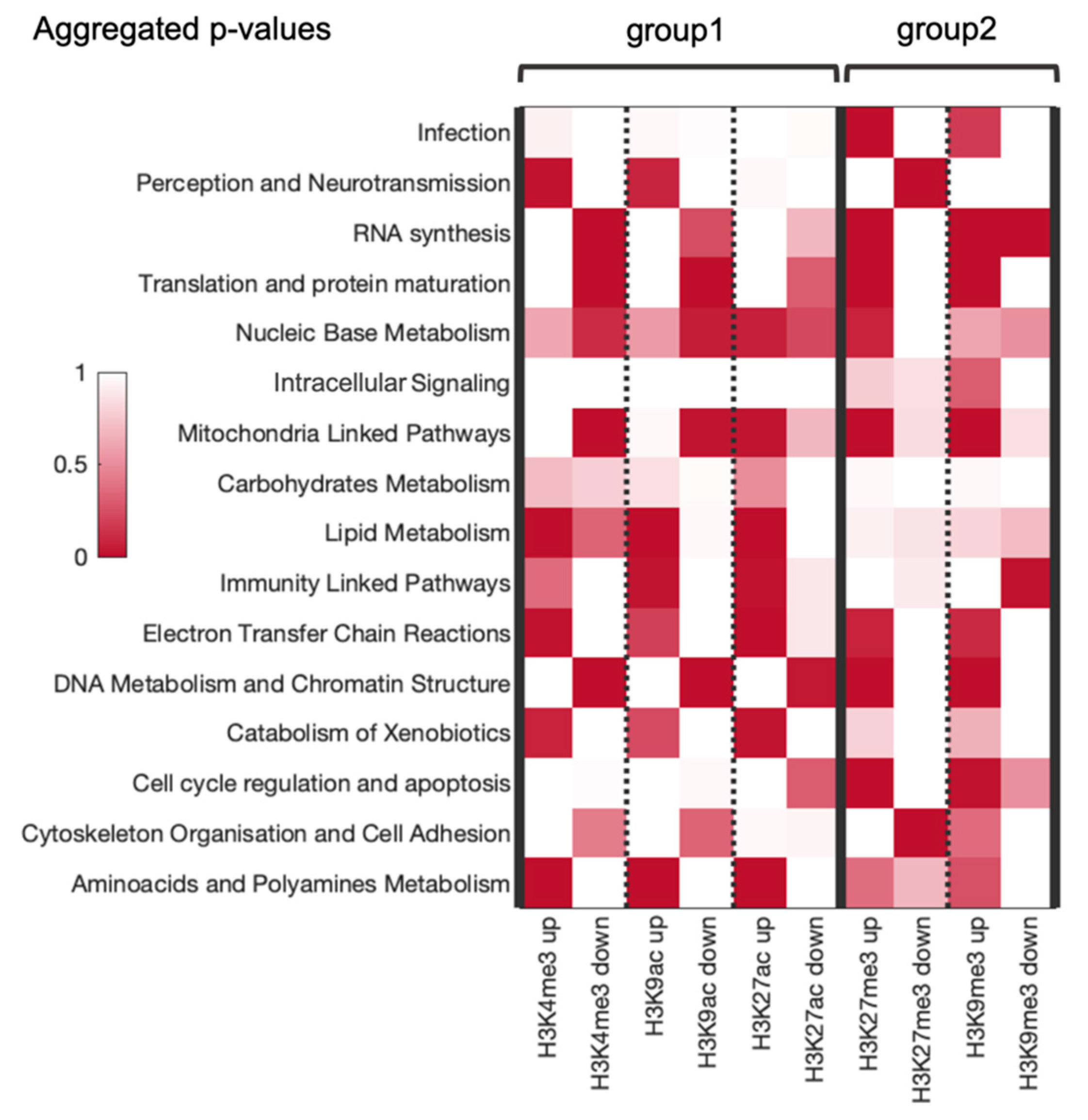
3.6. Combined Analysis of Gene and Pathway Level Trends
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deininger, P.L. Mammalian retroelements. Genome Res. 2002, 12, 1455–1465. [Google Scholar] [CrossRef]
- Schumann, G.G.; Gogvadze, E.V.; Osanai-Futahashi, M.; Kuroki, A.; Münk, C.; Fujiwara, H.; Ivics, Z.; Buzdin, A.A. Unique functions of repetitive transcriptomes. Int. Rev. Cell Mol. Biol. 2010, 285, 115–188. [Google Scholar]
- Gogvadze, E.; Buzdin, A. Retroelements and their impact on genome evolution and functioning. Cell. Mol. Life Sci. 2009, 66, 3727–3742. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Goncalves, I. Nature and structure of human genes that generate retropseudogenes. Genome Res. 2000, 10, 672–678. [Google Scholar] [CrossRef][Green Version]
- Buzdin, A.; Kovalskaya-Alexandrova, E.; Gogvadze, E.; Sverdlov, E. At Least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J. Virol. 2006, 80, 10752–10762. [Google Scholar] [CrossRef]
- Chuong, E.B.; Rumi, M.A.K.; Soares, M.J.; Baker, J.C. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 2013, 45, 325–329. [Google Scholar] [CrossRef]
- Suntsova, M.; Gogvadze, E.V.; Salozhin, S.; Gaifullin, N.; Eroshkin, F.; Dmitriev, S.E.; Martynova, N.; Kulikov, K.; Malakhova, G.; Tukhbatova, G.; et al. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc. Natl. Acad. Sci. USA 2013, 110, 19472–19477. [Google Scholar] [CrossRef]
- Nikitin, D.; Garazha, A.; Sorokin, M.; Penzar, D.; Tkachev, V.; Markov, A.; Gaifullin, N.; Borger, P.; Poltorak, A.; Buzdin, A. Retroelement-linked transcription factor binding patterns point to quickly developing molecular pathways in human evolution. Cells 2019, 8, 130. [Google Scholar] [CrossRef]
- Garazha, A.; Ivanova, A.; Suntsova, M.; Malakhova, G.; Roumiantsev, S.; Zhavoronkov, A.; Buzdin, A. New bioinformatic tool for quick identification of functionally relevant endogenous retroviral inserts in human genome. Cell Cycle 2015, 14, 1476–1484. [Google Scholar] [CrossRef]
- Kaminker, J.S.; Bergman, C.M.; Kronmiller, B.; Carlson, J.; Svirskas, R.; Patel, S.; Frise, E.; Wheeler, D.A.; Lewis, S.E.; Rubin, G.M.; et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002, 3, research0084. [Google Scholar] [CrossRef]
- Lippman, Z.; Gendrel, A.-V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Nikitin, D.; Penzar, D.; Garazha, A.; Sorokin, M.; Tkachev, V.; Borisov, N.; Poltorak, A.; Prassolov, V.; Buzdin, A.A. Profiling of human molecular pathways affected by retrotransposons at the level of regulation by transcription factor proteins. Front. Immunol. 2018, 9, 30. [Google Scholar] [CrossRef]
- Borisov, N.; Suntsova, M.; Sorokin, M.; Garazha, A.; Kovalchuk, O.; Aliper, A.; Ilnitskaya, E.; Lezhnina, K.; Korzinkin, M.; Tkachev, V.; et al. Data aggregation at the level of molecular pathways improves stability of experimental transcriptomic and proteomic data. Cell Cycle 2017, 16, 1810–1823. [Google Scholar] [CrossRef]
- Borisov, N.M.; Terekhanova, N.V.; Aliper, A.M.; Venkova, L.S.; Smirnov, P.Y.; Roumiantsev, S.; Korzinkin, M.B.; Zhavoronkov, A.A.; Buzdin, A.A. Signaling pathways activation profiles make better markers of cancer than expression of individual genes. Oncotarget 2014, 5, 10198. [Google Scholar] [CrossRef]
- Harris, B.H.L.; Barberis, A.; West, C.M.L.; Buffa, F.M. Gene expression signatures as biomarkers of tumour hypoxia. Clin. Oncol. 2015, 27, 547–560. [Google Scholar] [CrossRef]
- Yuryev, A. Gene expression profiling for targeted cancer treatment. Exp. Opin. Drug Discov. 2015, 10, 91–99. [Google Scholar] [CrossRef]
- Aliper, A.M.; Korzinkin, M.B.; Kuzmina, N.B.; Zenin, A.A.; Venkova, L.S.; Smirnov, P.Y.; Zhavoronkov, A.A.; Buzdin, A.A.; Borisov, N.M. Mathematical justification of expression-based pathway activation scoring (PAS). In Biological Networks and Pathway Analysis; Humana Press: New York, NY, USA, 2017; pp. 31–51. [Google Scholar]
- Buzdin, A.A.; Prassolov, V.; Zhavoronkov, A.A.; Borisov, N.M. Bioinformatics meets biomedicine: Oncofinder, a quantitative approach for interrogating molecular pathways using gene expression data. In Biological Networks and Pathway Analysis; Humana Press: New York, NY, USA, 2017; pp. 53–83. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Artemov, A.; Aliper, A.; Korzinkin, M.; Lezhnina, K.; Jellen, L.; Zhukov, N.; Roumiantsev, S.; Gaifullin, N.; Zhavoronkov, A.; Borisov, N.; et al. A method for predicting target drug efficiency in cancer based on the analysis of signaling pathway activation. Oncotarget 2015, 6, 29347. [Google Scholar] [CrossRef]
- Yin, H.; Wang, S.; Zhang, Y.-H.; Cai, Y.-D.; Liu, H. Analysis of important gene ontology terms and biological pathways related to pancreatic cancer. Biomed Res. Int. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Jiang, J.-C.; Upton, K.R. Human transposons are an abundant supply of transcription factor binding sites and promoter activities in breast cancer cell lines. Mob. DNA 2019, 10, 16. [Google Scholar] [CrossRef]
- Danino, Y.M.; Even, D.; Ideses, D.; Juven-Gershon, T. The core promoter: At the heart of gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 1116–1131. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Howe, F.S.; Fischl, H.; Murray, S.C.; Mellor, J. Is H3K4me3 instructive for transcription activation? BioEssays 2017, 39, e201600095. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Saksouk, N.; Simboeck, E.; Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenet. Chromatin 2015, 8, 3. [Google Scholar] [CrossRef]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef]
- Huang, D.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Estécio, M.R.H.; Gallegos, J.; Dekmezian, M.; Lu, Y.; Liang, S.; Issa, J.-P.J. SINE retrotransposons cause epigenetic reprogramming of adjacent gene promoters. Mol. Cancer Res. 2012, 10, 1332–1342. [Google Scholar] [CrossRef]
- Nault, R.; Fader, K.A.; Zacharewski, T. RNA-Seq versus oligonucleotide array assessment of dose-dependent TCDD-elicited hepatic gene expression in mice. BMC Genom. 2015, 16, 373. [Google Scholar] [CrossRef]
- SEQC/MAQC-III Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef]
- Hurst, T.P.; Magiorkinis, G. Epigenetic control of human endogenous retrovirus expression: Focus on regulation of long-terminal repeats (LTRs). Viruses 2017, 9, 130. [Google Scholar] [CrossRef]
- Lannes, R.; Rizzon, C.; Lerat, E. Does the presence of transposable elements impact the epigenetic environment of human duplicated genes? Genes 2019, 10, 249. [Google Scholar] [CrossRef]
- Renault, S.; Genty, M.; Gabori, A.; Boisneau, C.; Esnault, C.; Dugé de Bernonville, T.; Augé-Gouillou, C. The epigenetic regulation of HsMar1, a human DNA transposon. BMC Genet. 2019, 20, 17. [Google Scholar] [CrossRef]
- Jursch, T.; Miskey, C.; Izsvák, Z.; Ivics, Z. Regulation of DNA transposition by CpG methylation and chromatin structure in human cells. Mob. DNA 2013, 4, 15. [Google Scholar] [CrossRef]
- Palazzo, A.; Lorusso, P.; Miskey, C.; Walisko, O.; Gerbino, A.; Marobbio, C.M.T.; Ivics, Z.; Marsano, R.M. Transcriptionally promiscuous “blurry” promoters in Tc1/mariner transposons allow transcription in distantly related genomes. Mob. DNA 2019, 10, 13. [Google Scholar] [CrossRef]
- Bire, S.; Casteret, S.; Piégu, B.; Beauclair, L.; Moiré, N.; Arensbuger, P.; Bigot, Y. Mariner Transposons contain a silencer: Possible role of the polycomb repressive complex 2. PLoS Genet. 2016, 12, e1005902. [Google Scholar] [CrossRef]
- Ha, M.; Ng, D.W.-K.; Li, W.-H.; Chen, Z.J. Coordinated histone modifications are associated with gene expression variation within and between species. Genome Res. 2011, 21, 590–598. [Google Scholar] [CrossRef]
- GitHub. Available online: https://github.com/iganna/evo_epigen/ (accessed on 4 September 2019).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differential Regulation | Number of Pathways in Category | Total Number of Pathways |
|---|---|---|
| Number of Enriched/Deficient Pathways | max (0, X-1) | Y |
| Number of not Enriched/Deficient Pathways | Z-X | K-Y |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034. https://doi.org/10.3390/cells8091034
Igolkina AA, Zinkevich A, Karandasheva KO, Popov AA, Selifanova MV, Nikolaeva D, Tkachev V, Penzar D, Nikitin DM, Buzdin A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells. 2019; 8(9):1034. https://doi.org/10.3390/cells8091034
Chicago/Turabian StyleIgolkina, Anna A., Arsenii Zinkevich, Kristina O. Karandasheva, Aleksey A. Popov, Maria V. Selifanova, Daria Nikolaeva, Victor Tkachev, Dmitry Penzar, Daniil M. Nikitin, and Anton Buzdin. 2019. "H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks" Cells 8, no. 9: 1034. https://doi.org/10.3390/cells8091034
APA StyleIgolkina, A. A., Zinkevich, A., Karandasheva, K. O., Popov, A. A., Selifanova, M. V., Nikolaeva, D., Tkachev, V., Penzar, D., Nikitin, D. M., & Buzdin, A. (2019). H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells, 8(9), 1034. https://doi.org/10.3390/cells8091034