17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse Model

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Drug Treatment
2.3. Behavioral Analysis
2.4. Y-Maze Test
2.5. Cell Culturing and Drug Treatment
2.6. Cell Viability Assay
2.7. Oxidative Stress (ROS) Detection in Vitro
2.8. Protein Extraction from Mouse Brain
2.9. Western Blot Analysis
2.10. Oxidative Stress (ROS) Detection in Vivo
2.11. Determination of Lipid Peroxidation
2.12. GSH Assays
2.13. Immunofluorescence
2.14. Antibodies
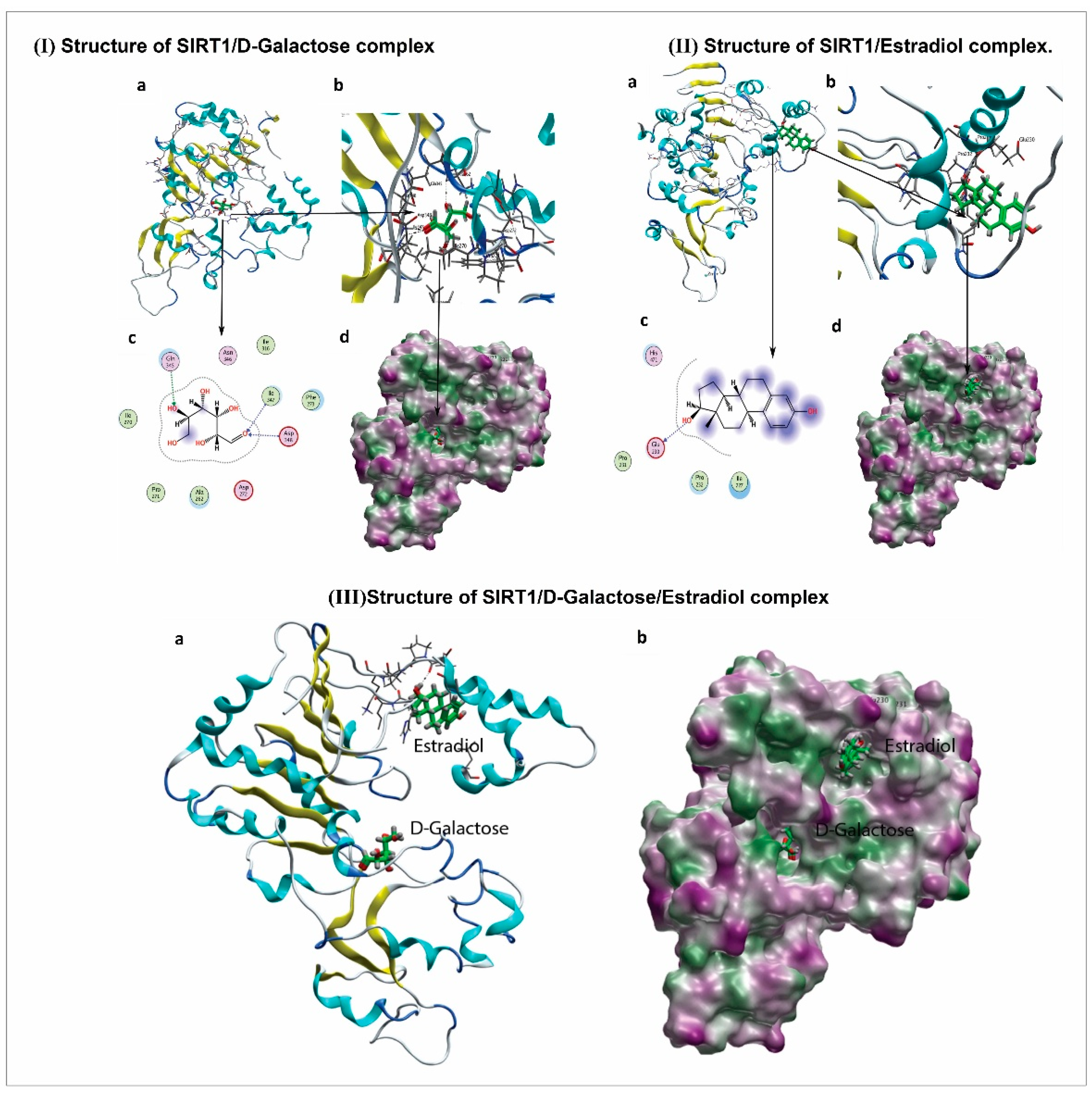
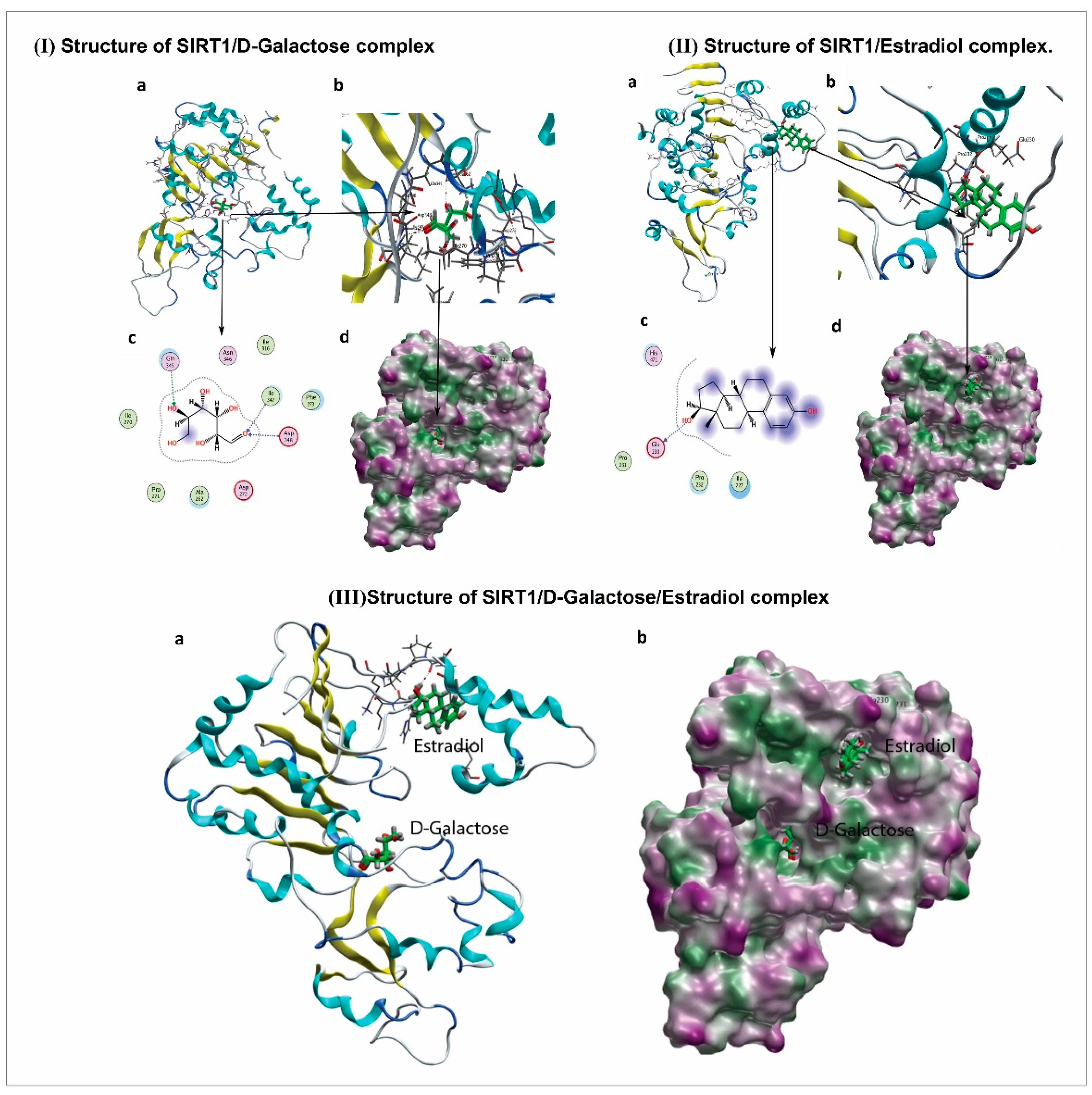
2.15. Molecular Docking Methodology
2.16. Statistical Analysis
3. Results
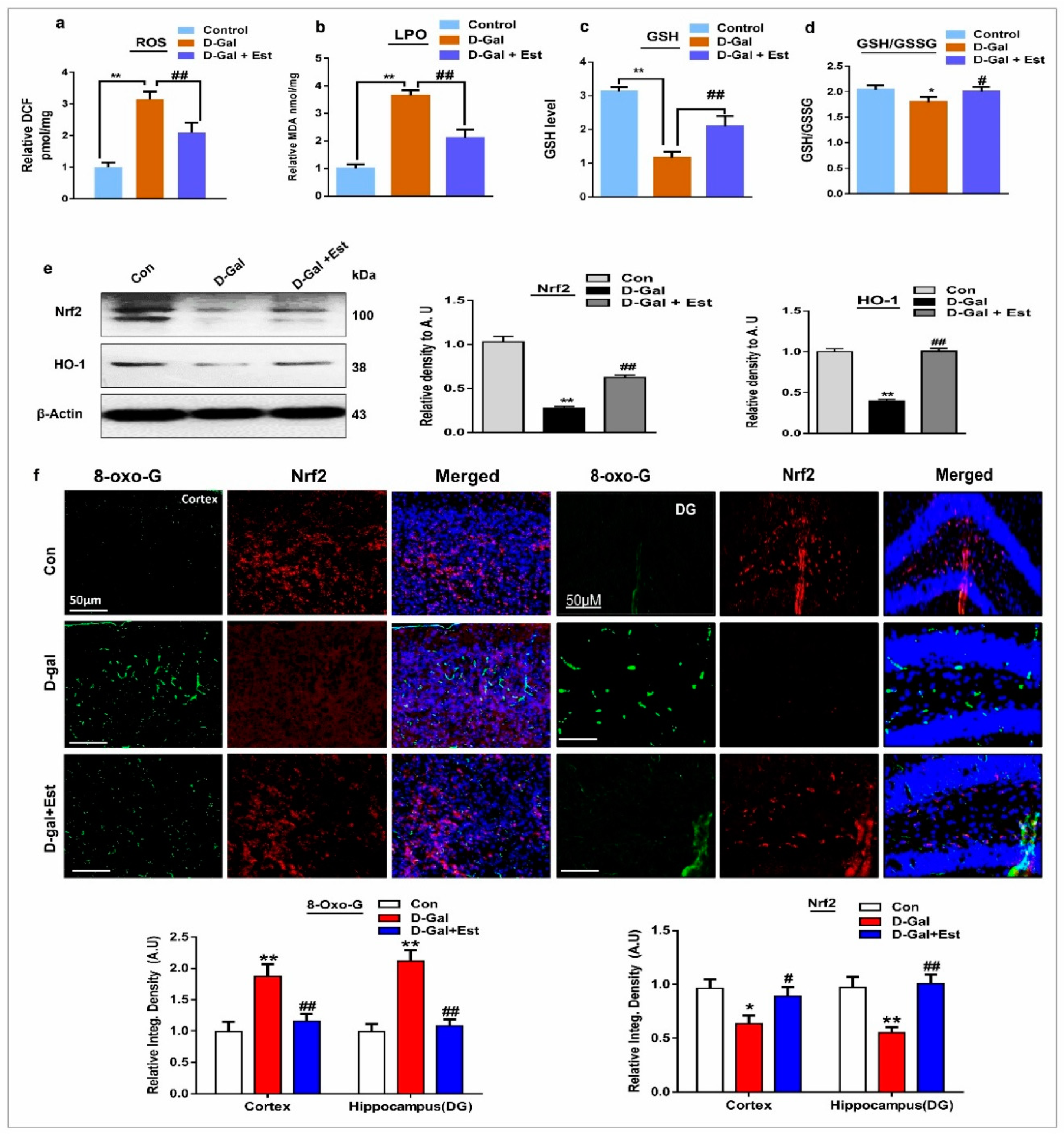
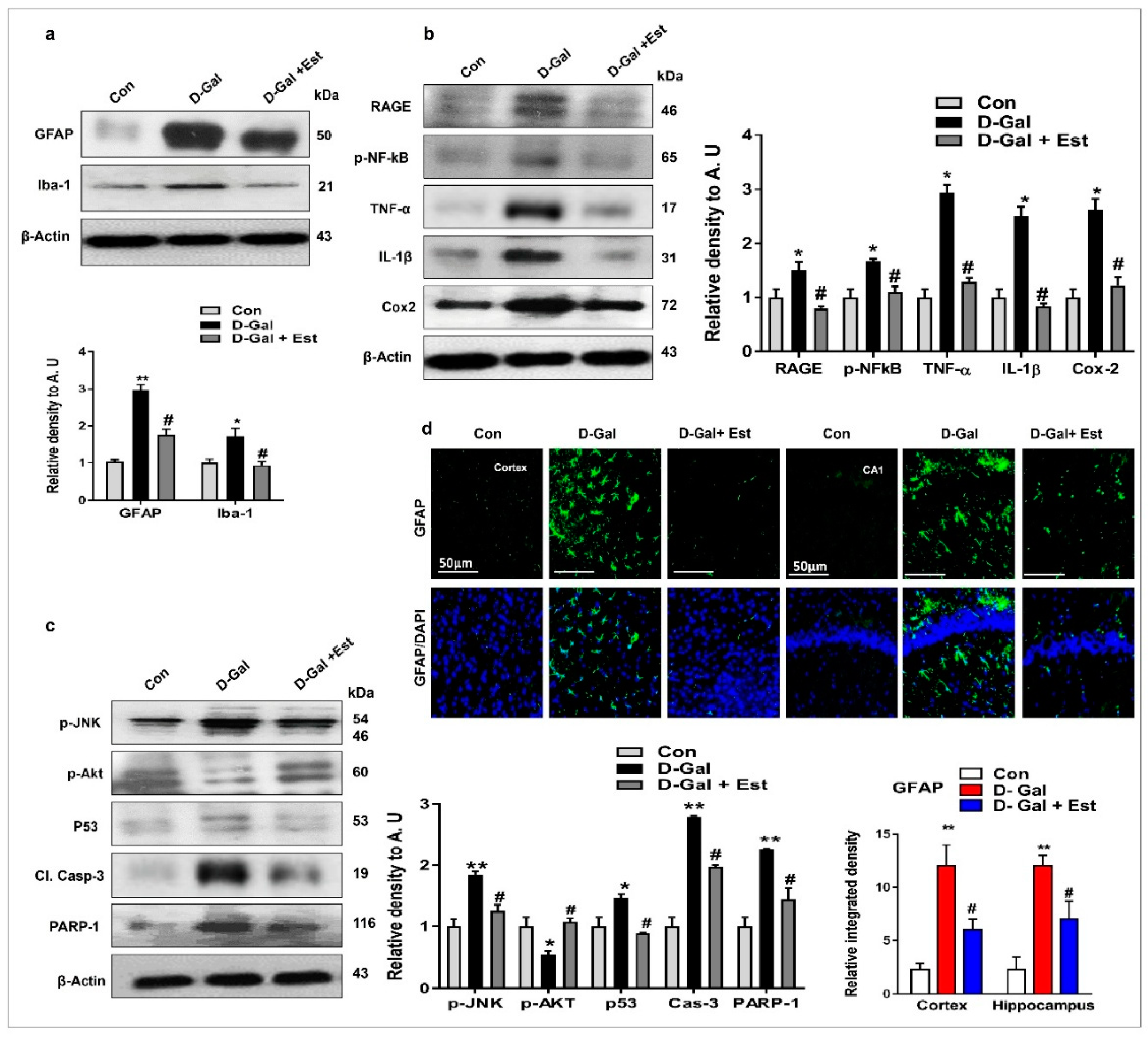
3.1. 17β-Estradiol Limits d-gal-Induced Oxidative Stress in Adult Mouse Brain
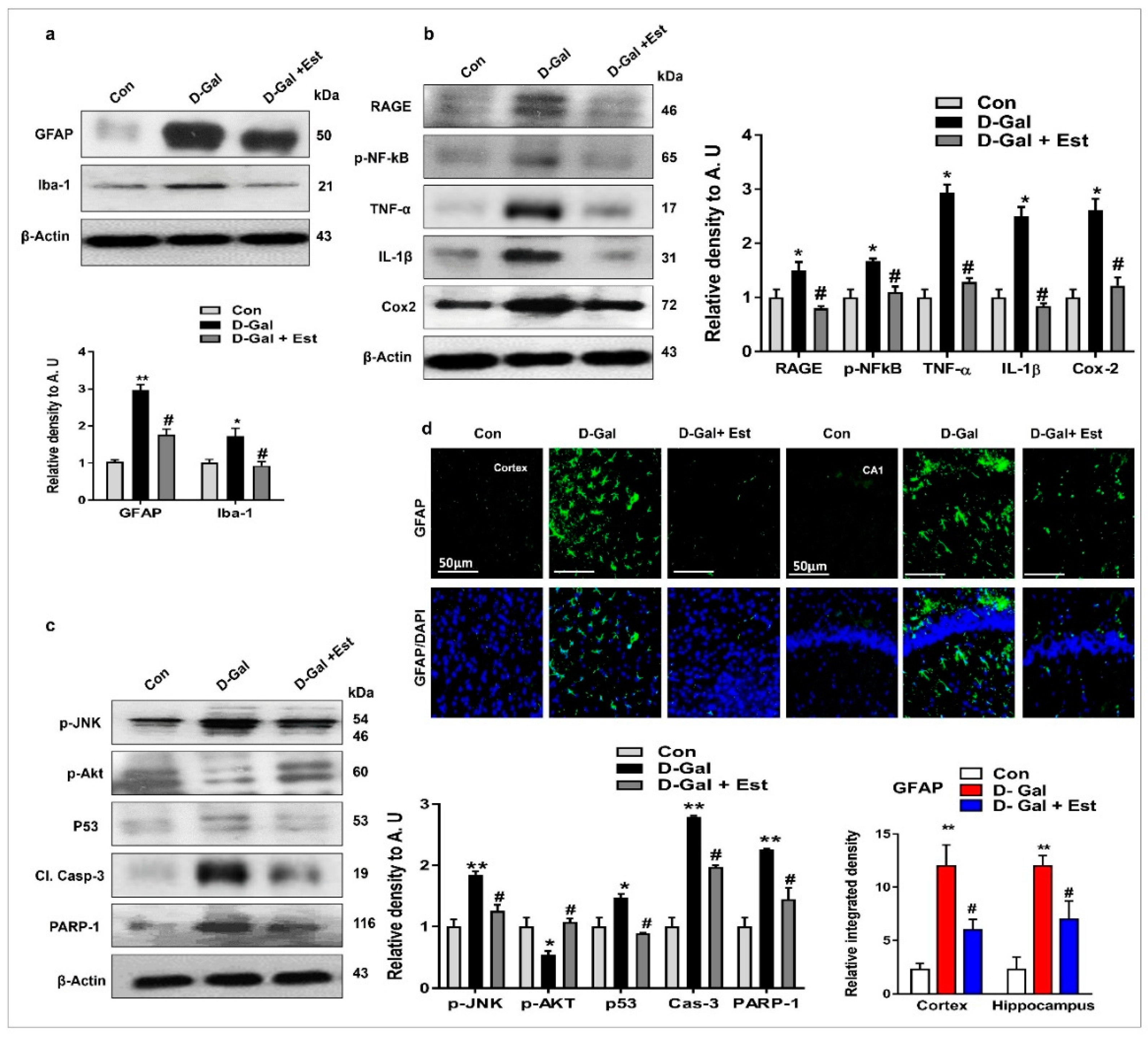
3.2. 17β-Estradiol Reduced d-gal -Induced Neuroinflammation-Mediated Neurodegeneration via JNK/Akt/NF-κB/p53 Signaling in Vivo
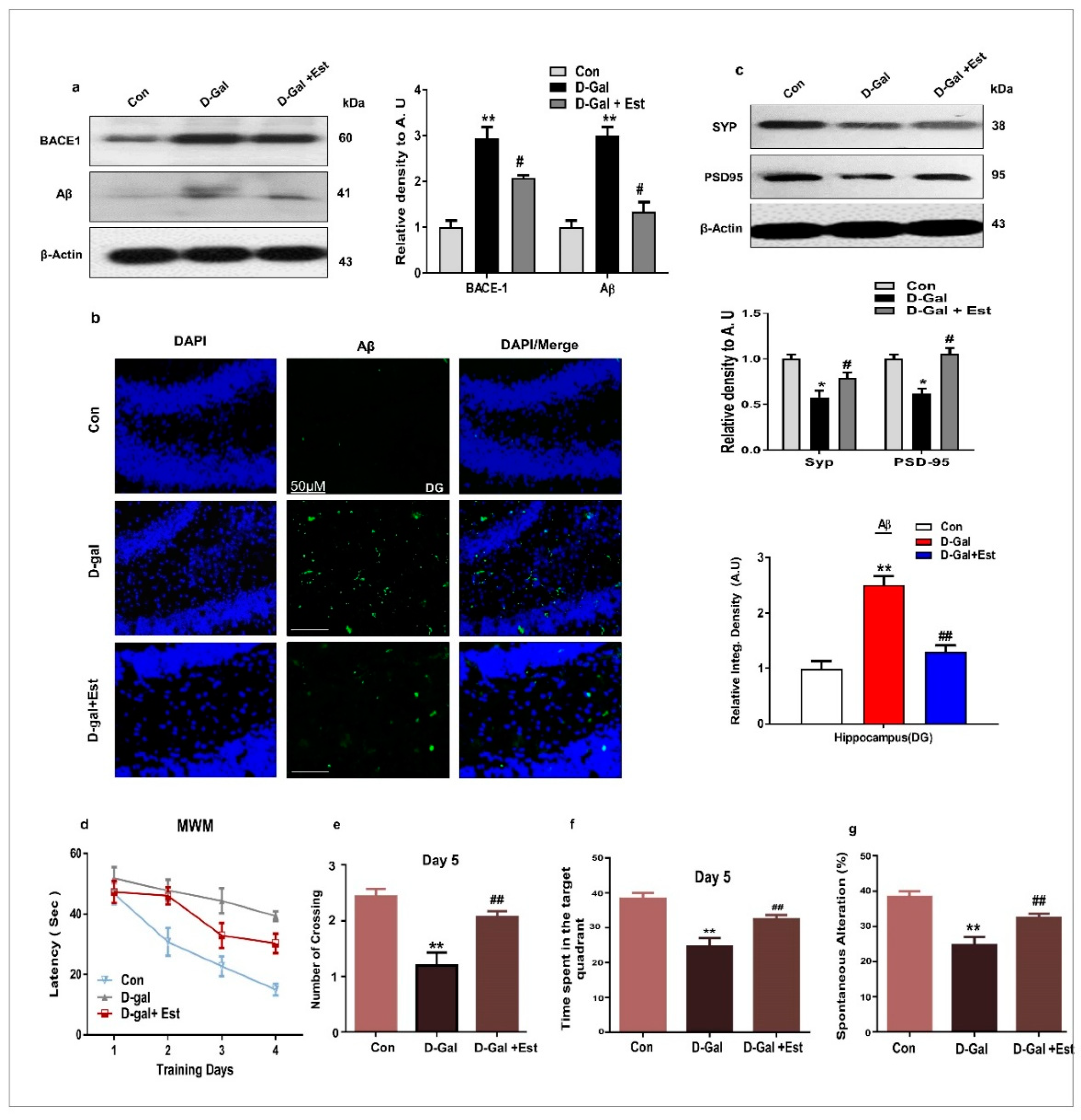
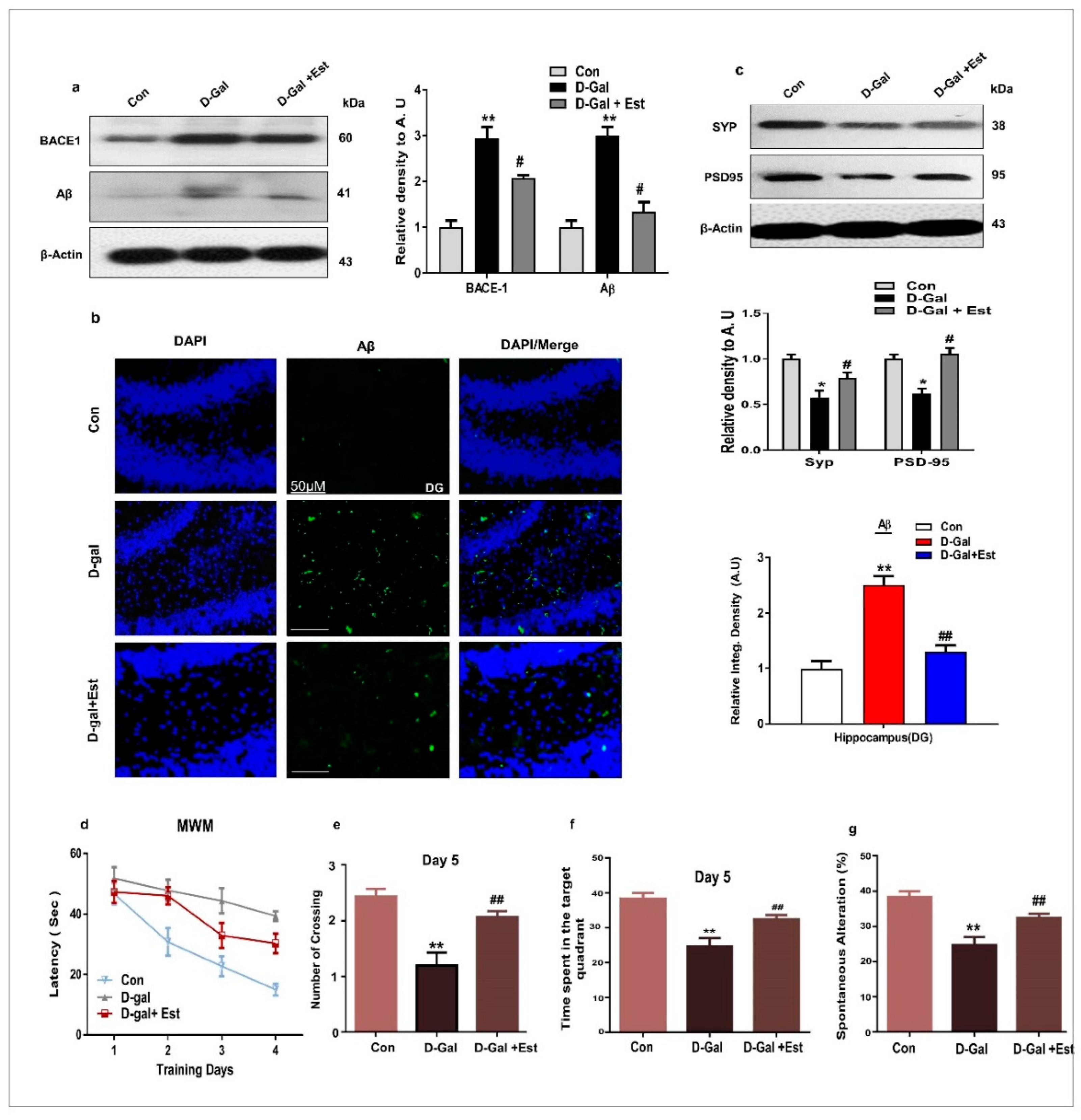
3.3. 17β-Estradiol Reversed Alzheimer Disease-Like Pathology in d-gal-Treated Adult Mice
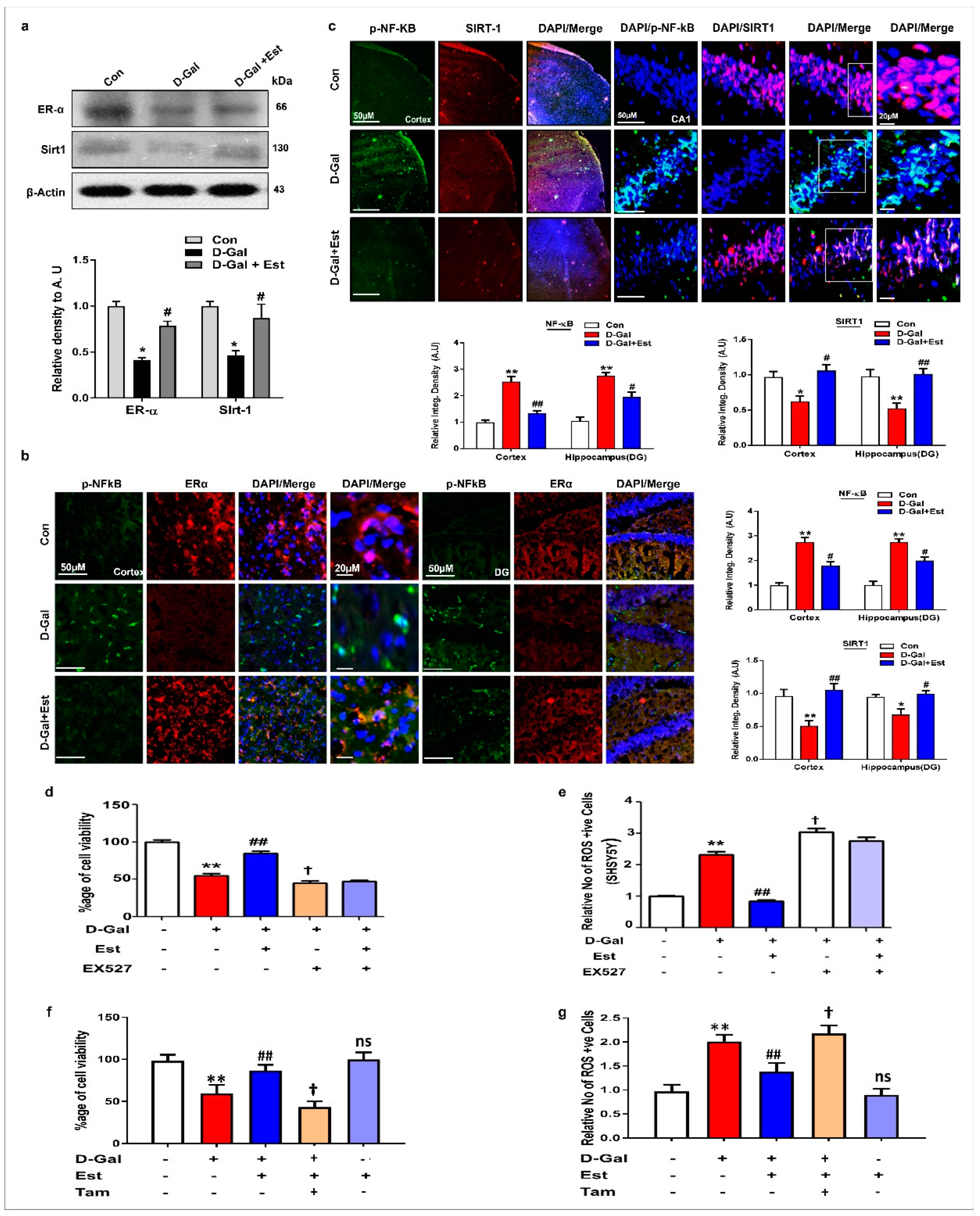
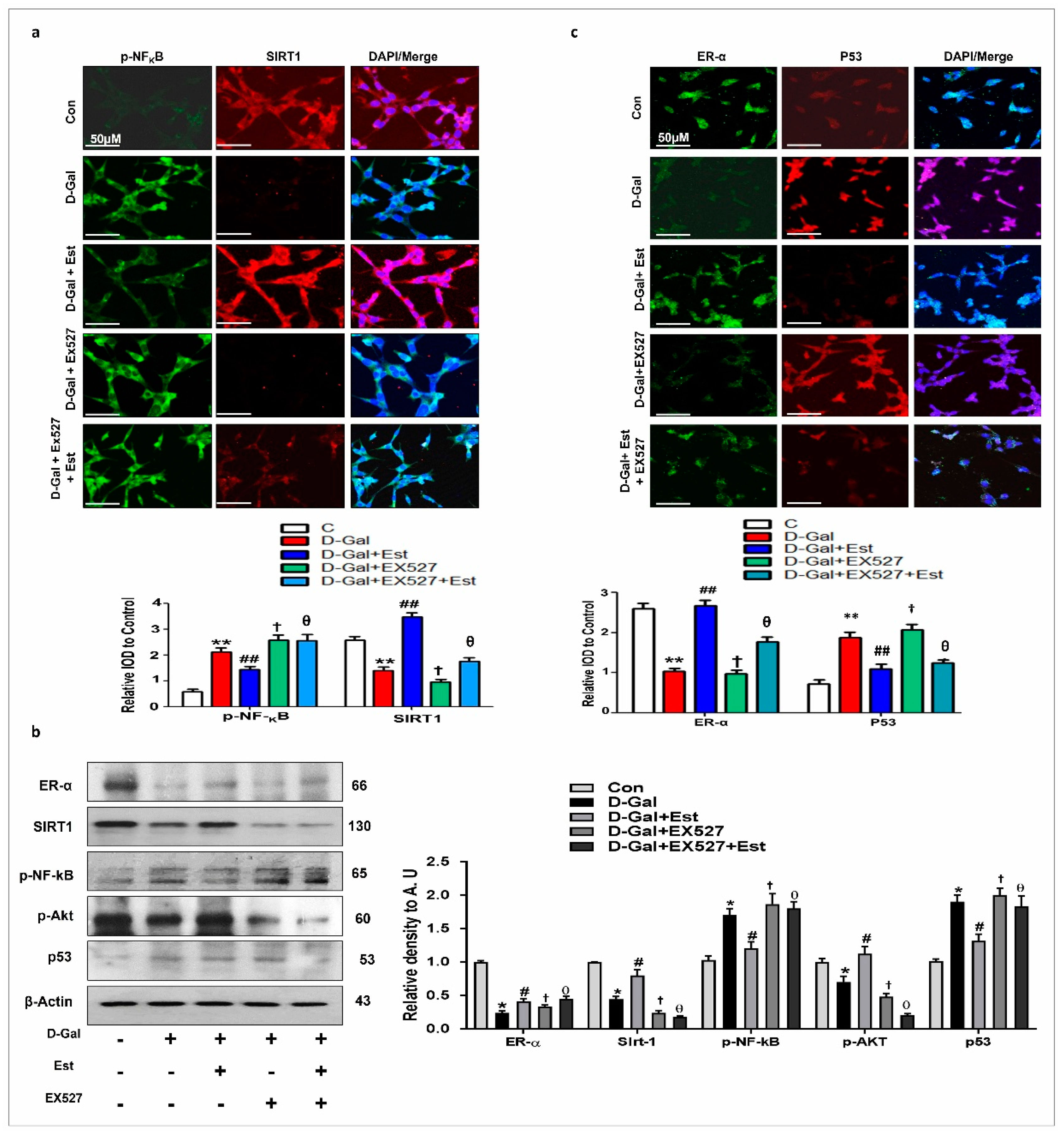
3.4. 17β-Estradiol Enhances the SIRT1 and Its Downstream Signaling Through ER-α against d-gal-Induced Neuroinflammation and Neurodegeneration
3.5. 17β-Estradiol Reactivates SIRT1 after Deactivation by d-gal
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Khan, M.; Shah, S.A.; Saeed, K.; Kim, M.O. Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration. Nutrients 2019, 11, 1195. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondria and aging. Adv. Exp. Med. Biol. 2012, 942, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Bereiter-Hahn, J. Do we age because we have mitochondria? Protoplasma 2014, 251, 3–23. [Google Scholar] [CrossRef] [PubMed]
- De Iuliis, A.; Grigoletto, J.; Recchia, A.; Giusti, P.; Arslan, P. A proteomic approach in the study of an animal model of Parkinson’s disease. Clin. Chim. Acta 2005, 357, 202–209. [Google Scholar] [CrossRef]
- Niranjan, R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: Focus on astrocytes. Mol. Neurobiol. 2014, 49, 28–38. [Google Scholar] [CrossRef]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef]
- An, X.; Fu, Z.; Mai, C.; Wang, W.; Wei, L.; Li, D.; Li, C.; Jiang, L.-H. Increasing the TRPM2 Channel Expression in Human Neuroblastoma SH-SY5Y Cells Augments the Susceptibility to ROS-Induced Cell Death. Cells 2019, 8, 28. [Google Scholar] [CrossRef]
- Chen, C.; Lang, S.; Zuo, P.; Yang, N.; Wang, X.; Xia, C. Effects of d-galactose on the expression of hippocampal peripheral-type benzodiazepine receptor and spatial memory performances in rats. Psychoneuroendocrinology 2006, 31, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, Y.L.; Luo, L.; Wu, D.M.; Sun, D.X.; Feng, Y.J. Quercetin reverses d-galactose induced neurotoxicity in mouse brain. Behav. Brain Res. 2006, 171, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.J.; Fu, Y.R.; Li, M.; Gao, K.S.; Zhang, X.G. Effect of LTA isolated from bifidobacteria on d-galactose-induced aging. Exp. Gerontol. 2009, 44, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zuo, P.; Zhang, Q.; Li, X.; Hu, Y.; Long, J.; Packer, L.; Liu, J. Chronic systemicD-galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: Protective effects of R-α-lipoic acid. J. Neurosci. Res. 2006, 84, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Amadeo, J.; Lawrence, A. Clinical Chemistry: Theory, Analysis, and Correlation, 3rd ed.; Mosby: St. Louis, MO, USA, 1944–1996. [Google Scholar]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef]
- Morris, B.J. Seven sirtuins for seven deadly diseases ofaging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Clifton, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.I. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Yao, Y.; Li, H.; Gu, Y.; Davidson, N.E.; Zhou, Q. Inhibition of SIRT1 deacetylase suppresses estrogen receptor signaling. Carcinogenesis 2010, 31, 382–387. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, S.; Gan, L.; Vosler, P.S.; Gao, Y.; Chen, J. Protective effects and mechanisms of sirtuins in the nervous system. Prog. Neurobiol. 2011, 95, 373–395. [Google Scholar] [CrossRef] [Green Version]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; Croix, C.M.S.; Usas, A.; Vo, N.; et al. NF-κB inhibition delays DNA damage–induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef]
- Rajendran, R.; Garva, R.; Krstic-Demonacos, M.; Demonacos, C. Sirtuins: Molecular Traffic Lights in the Crossroad of Oxidative Stress, Chromatin Remodeling, and Transcription. J. Biomed. Biotechnol. 2011, 2011, 1–17. [Google Scholar] [CrossRef]
- Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2018, 8, 11. [Google Scholar] [CrossRef]
- Huang, G.Z.; Woolley, C.S. Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR dependent mechanism. Neuron 2012, 74, 801–808. [Google Scholar] [CrossRef]
- Hyeon, J.Y.; Hur, S.P.; Kim, B.H.; Byun, J.H.; Kim, E.S.; Lim, B.S.; Lee, B.I.; Kim, S.K.; Takemura, A.; Kim, S.J. Involvement of Estrogen and Its Receptors in Morphological Changes in the Eyes of the Japanese Eel, Anguilla japonica, in the Process of Artificially-Induced Maturation. Cells 2019, 8, 310. [Google Scholar] [CrossRef]
- Arevalo, M.A.; Azcoitia, I.; Garcia-Segura, L.M. The neuroprotective actions of oestradiol and oestrogen receptors. Nat. Rev. Neurosci. 2015, 16, 17–29. [Google Scholar] [CrossRef]
- Baulieu, E. Neurosteroids: A novel function of the brain. Psychoneuroendocrinology 1998, 23, 963–987. [Google Scholar] [CrossRef]
- Brown, C.M.; Mulcahey, T.A.; Filipek, N.C.; Wise, P.M. Production of Proinflammatory Cytokines and Chemokines During Neuroinflammation: Novel Roles for Estrogen Receptors α and β. Endocrinology 2010, 151, 4916–4925. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-kappaB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-kappaB Signaling in an Abeta Mouse Model. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Khan, M.; Shah, S.A.; Kim, M.O. 17beta-Estradiol via SIRT1/Acetyl-p53/NF-kB Signaling Pathway Rescued Postnatal Rat Brain Against Acute Ethanol Intoxication. Mol. Neurobiol. 2018, 55, 3067–3078. [Google Scholar] [CrossRef]
- Khan, M.; Rutten, B.P.F.; Kim, M.O. MST1 Regulates Neuronal Cell Death via JNK/Casp3 Signaling Pathway in HFD Mouse Brain and HT22 Cells. Int. J. Mol. Sci. 2019, 20, 2504. [Google Scholar] [CrossRef]
- Shah, S.A.; Amin, F.U.; Khan, M.; Abid, M.N.; Rehman, S.U.; Kim, T.H.; Kim, M.W.; Kim, M.O. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflammation 2016, 13, 19. [Google Scholar] [CrossRef]
- Shah, F.A.; Zeb, A.; Ali, T.; Muhammad, T.; Faheem, M.; Alam, S.I.; Saeed, K.; Koh, P.O.; Lee, K.W.; Kim, M.O. Identification of Proteins Differentially Expressed in the Striatum by Melatonin in a Middle Cerebral Artery Occlusion Rat Model-a Proteomic and in silico Approach. Front. Neurosci. 2018, 12, 888. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef]
- Ikram, M.; Saeed, K.; Khan, A.; Muhammad, T.; Khan, M.S.; Jo, M.G.; Rehman, S.U.; Kim, M.O. Natural Dietary Supplementation of Curcumin Protects Mice Brains against Ethanol-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment via Nrf2/TLR4/RAGE Signaling. Nutrients 2019, 11, 1082. [Google Scholar] [CrossRef]
- Shah, A.S.; Lee, H.Y.; Bressan, A.R.; Yun, D.J.; Kim, O.M. Novel osmotin attenuates glutamate-induced synaptic dysfunction and neurodegeneration via the JNK/PI3K/Akt pathway in postnatal rat brain. Cell Death Dis. 2014, 5, e1026. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cereb. Cortex 2018, 28, 2854–2872. [Google Scholar] [CrossRef]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins Reversed D-Galactose-Induced Oxidative Stress and Neuroinflammation Mediated Cognitive Impairment in Adult Rats. Mol. Neurobiol. 2017, 54, 255–271. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative Stress Increases Expression and Activity of BACE in NT2 Neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Micevych, P.; Dominguez, R. Membrane estradiol signaling in the brain. Front. Neuroendocr. 2009, 30, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Alhazzani, A.; Rajagopalan, P.; Albarqi, Z.; Devaraj, A.; Mohamed, M.H.; Al-Hakami, A.; Chandramoorthy, H.C. Mesenchymal Stem Cells (MSCs) Coculture Protects [Ca2+]i Orchestrated Oxidant Mediated Damage in Differentiated Neurons In Vitro. Cells 2018, 7, 250. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Amrani, A.; Gris, D. NLRX1 Enhances Glutamate Uptake and Inhibits Glutamate Release by Astrocytes. Cells 2019, 8, 400. [Google Scholar] [CrossRef]
- Xu, B.; Lang, L.M.; Li, S.Z.; Guo, J.R.; Wang, J.F.; Wang, D.; Zhang, L.P.; Yang, H.M.; Lian, S. Cortisol Excess-Mediated Mitochondrial Damage Induced Hippocampal Neuronal Apoptosis in Mice Following Cold Exposure. Cells 2019, 8, 612. [Google Scholar] [CrossRef]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Chen, B.H.; Hernandez, D.G.; Singleton, A.B.; Ferrucci, L.; Bandinelli, S.; Salfati, E.; Manson, J.E.; Quach, A.; et al. Menopause accelerates biological aging. Proc. Natl. Acad. Sci. USA 2016, 113, 9327–9332. [Google Scholar] [CrossRef] [Green Version]
- Greendale, G.A.; Derby, C.A.; Maki, P.M. Perimenopause and Cognition. Obstet. Gynecol. Clin. N. Am. 2011, 38, 519–535. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F.; Ye, Q.; Liu, C.M.; Shan, Q.; Wang, Y.J. Ursolic Acid Attenuates D-Galactose-Induced Inflammatory Response in Mouse Prefrontal Cortex through Inhibiting AGEs/RAGE/NF-B Pathway Activation. Cereb. Cortex 2010, 20, 2540–2548. [Google Scholar] [CrossRef]
- Mehta, N.J.; Marwah, P.K.; Njus, D. Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin? Cells 2019, 8, 59. [Google Scholar] [CrossRef]
- Bourne, K.Z.; Ferrari, D.C.; Roßner, S.; Wood, T.G.; Lange-Dohna, C.; Perez-Polo, J.R.; Lange-Dohna, C.; Perez-Polo, J.R. Differential regulation of BACE1 promoter activity by nuclear factor-κB in neurons and glia upon exposure to β-amyloid peptides. J. Neurosci. Res. 2007, 85, 1194–1204. [Google Scholar] [CrossRef]
- Tian, J.; Ishibashi, K.; Ishibashi, K.; Reiser, K.; Grebe, R.; Biswal, S.; Gehlbach, P.; Handa, J.T. Advanced glycation endproduct-induced aging of the retinal pigment epithelium and choroid: A comprehensive transcriptional response. Proc. Natl. Acad. Sci. USA 2005, 102, 11846–11851. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Hua, X.; Xiao, M.; Ding, J.; Han, Q.; Hu, G. Impairments of astrocytes are involved in the d-galactose-induced brain aging. Biochem. Biophys. Res. Commun. 2008, 369, 1082–1087. [Google Scholar] [CrossRef]
- Xiao, H.; Deng, M.; Yang, B.; Hu, Z.; Tang, J. Pretreatment with 17beta-Estradiol Attenuates Cerebral Ischemia-Induced Blood-Brain Barrier Disruption in Aged Rats: Involvement of Antioxidant Signaling. Neuroendocrinology 2018, 106, 20–29. [Google Scholar] [CrossRef]
- Vivacqua, A.; Sebastiani, A.; Miglietta, A.M.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Santolla, M.F.; Lappano, R.; Giordano, F.; et al. miR-338-3p Is Regulated by Estrogens through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts (CAFs). Cells 2018, 7, 203. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Yu, L.J.; Hui, X.C.; Wu, Z.Z.; Yin, K.L.; Yang, H.; Xu, Y. Hydroxy-safflor yellow A attenuates Aβ1-42-induced inflammation by modulating the JAK2/STAT3/NF-κB pathway. Brain Res. 2014, 1563, 72–80. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, Y.; Tang, F.L.; Xiong, L.; Su, C.; Mei, L.; Zhu, X.J.; Xiong, W.C. pHluorin-BACE1-mCherry Acts as a Reporter for the Intracellular Distribution of Active BACE1 In Vitro and In Vivo. Cells 2019, 8, 474. [Google Scholar] [CrossRef]
- Arendash, G.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.; Zacharia, L.; Cracchiolo, J.; Shippy, D.; Tan, J.; Zacharia, L. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef]
- Yun, J.; Yeo, I.J.; Hwang, C.J.; Choi, D.Y.; Im, H.S.; Kim, J.Y.; Choi, W.R.; Jung, M.H.; Han, S.B.; Hong, J.T. Estrogen deficiency exacerbates Aβ-induced memory impairment through enhancement of neuroinflammation, amyloidogenesis and NF-ĸB activation in ovariectomized mice. Brain Behav. Immun. 2018, 73, 282–293. [Google Scholar] [CrossRef]
- Vegeto, E.; Belcredito, S.; Ghisletti, S.; Meda, C.; Etteri, S.; Maggi, A. The Endogenous Estrogen Status Regulates Microglia Reactivity in Animal Models of Neuroinflammation. Endocrinology 2006, 147, 2263–2272. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; Yen, E.S.; Lamming, D.W.; et al. Evidence for a Common Mechanism of SIRT1 Regulation by Allosteric Activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Balestrieri, M.L.; Rienzo, M.; Felice, F.; Rossiello, R.; Grimaldi, V.; Milone, L.; Casamassimi, A.; Servillo, L.; Farzati, B.; Giovane, A.; et al. High glucose downregulates endothelial progenitor cell number via SIRT1. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 936–945. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef]
- Sun, Q.; Jia, N.; Wang, W.; Jin, H.; Xu, J.; Hu, H. Activation of SIRT1 by curcumin blocks the neurotoxicity of amyloid-β25–35 in rat cortical neurons. Biochem. Biophys. Res. Commun. 2014, 448, 89–94. [Google Scholar] [CrossRef]
- Albani, D.; Polito, L.; Batelli, S.; De Mauro, S.; Fracasso, C.; Martelli, G.; Colombo, L.; Manzoni, C.; Salmona, M.; Caccia, S.; et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by α-synuclein or amyloid-β (1-42) peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Han, L.; Wang, P.; Zhao, G.; Wang, H.; Wang, M.; Chen, J.; Tong, T. Upregulation of SIRT1 by 17β-estradiol depends on ubiquitin-proteasome degradation of PPAR-γ mediated by NEDD4-1. Protein Cell 2013, 4, 310–321. [Google Scholar] [CrossRef]
- Wang, T.; Di, G.; Yang, L.; Dun, Y.; Sun, Z.; Wan, J.; Peng, B.; Liu, C.; Xiong, G.; Zhang, C.; et al. Saponins from P anax japonicus attenuate D-galactose-induced cognitive impairment through its anti-oxidative and anti-apoptotic effects in rats. J. Pharm. Pharmacol. 2015, 67, 1284–1296. [Google Scholar] [CrossRef]
- Wu, H.; Wang, H.; Zhang, W.; Wei, X.; Zhao, J.; Yan, P.; Liu, C. rhEPO affects apoptosis in hippocampus of aging rats by upregulating SIRT1. Int. J. Clin. Exp. Pathol. 2015, 8, 6870–6880. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Catalog | Application (Conc.) | Host | Manufacturer |
|---|---|---|---|---|
| anti-β-actin | sc-47,778 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-Nrf2 | sc-722 | WB/IF (1:1000/1:100) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-Akt | sc-514032 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-HO1 | sc-136961 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-Iba-1 | sc-32725 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-GFAP | sc-33673 | WB/IF (1:1000/1:100) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-IL-1β | sc-32294 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-TNF-α | sc-52746 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-p-NF-κB | sc-136548 | WB/IF (1:1000/1:100) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-p-JNK | sc-6254 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-PSD-95 | sc-71,933 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-PARP-1 | sc-8007 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-Cl-Casp-3 | sc-7272 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-Cox2 | sc- 7951 | WB (1:1000) | Rabbit | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-RAGE | sc-365154 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-P53 | sc-126 | WB/IF (1:1000/1:100) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-BACE1 | sc-33711 | WB (1:1000) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-Aβ | sc-28365 | WB/IF (1:1000/1:100) | Mouse | Santa Cruz Biotech (Dallas, TX, USA) |
| anti-ERα | ab75635 | WB/IF (1:1000/1:100) | Rabbit | Abcam (Cambridge, MA, USA) |
| anti-SIRT1 | #9475 | WB/IF (1:1000/1:100) | Rabbit | Cell Signaling Tech (Danvers, MA, USA) |
| 8-OXO-G | MAB3560 | IF (1:100) | Mouse | Millipore, USA (Billerica, MA, USA) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.; Ullah, R.; Rehman, S.U.; Shah, S.A.; Saeed, K.; Muhammad, T.; Park, H.Y.; Jo, M.H.; Choe, K.; Rutten, B.P.F.; et al. 17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse Model. Cells 2019, 8, 928. https://doi.org/10.3390/cells8080928
Khan M, Ullah R, Rehman SU, Shah SA, Saeed K, Muhammad T, Park HY, Jo MH, Choe K, Rutten BPF, et al. 17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse Model. Cells. 2019; 8(8):928. https://doi.org/10.3390/cells8080928
Chicago/Turabian StyleKhan, Mehtab, Rahat Ullah, Shafiq Ur Rehman, Shahid Ali Shah, Kamran Saeed, Tahir Muhammad, Hyun Young Park, Myeung Hoon Jo, Kyonghwan Choe, Bart P.F. Rutten, and et al. 2019. "17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse Model" Cells 8, no. 8: 928. https://doi.org/10.3390/cells8080928
APA StyleKhan, M., Ullah, R., Rehman, S. U., Shah, S. A., Saeed, K., Muhammad, T., Park, H. Y., Jo, M. H., Choe, K., Rutten, B. P. F., & Ok Kim, M. (2019). 17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse Model. Cells, 8(8), 928. https://doi.org/10.3390/cells8080928