IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors


Abstract
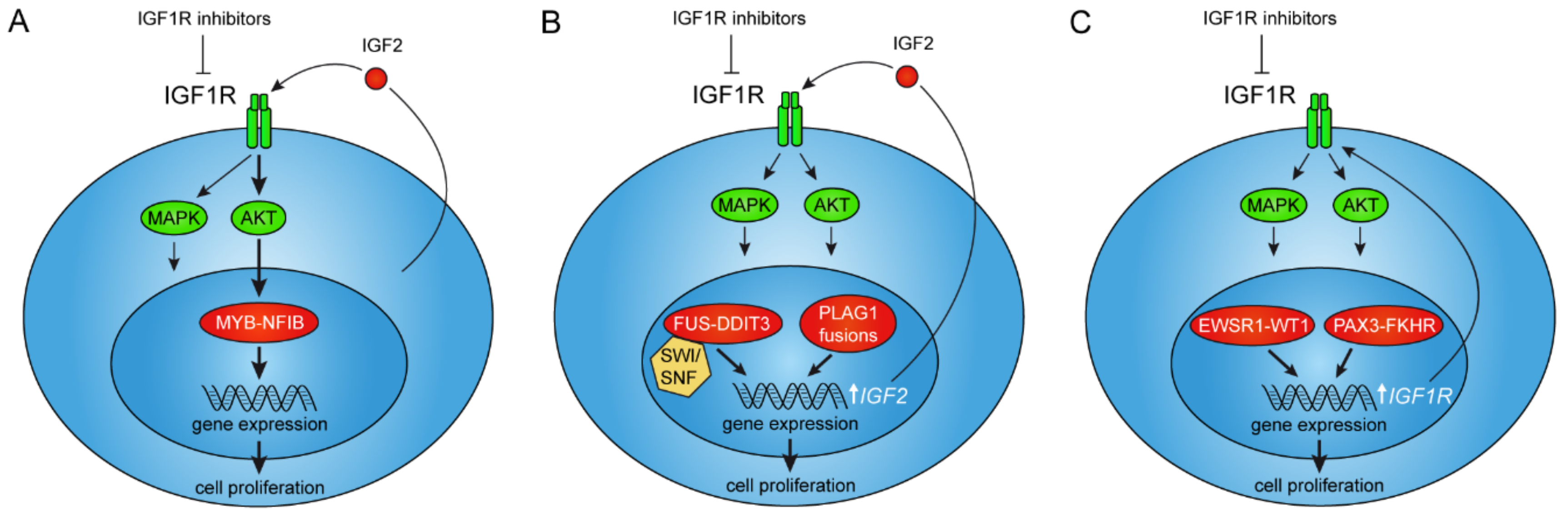
1. Introduction
2. The MYB–NFIB Gene Fusion in Adenoid Cystic Carcinoma
2.1. The MYB–NFIB Fusion Is Regulated by IGF1R in an AKT-Dependent Manner
2.2. Treatment of ACC Patients with IGF1R Inhibitors
3. IGF Signaling as a Therapeutic Target in Tumors with Activation of the PLAG1 Oncogene
4. IGF1R Signaling Is Required for ETV6–NTRK3-Mediated Tumorigenesis
5. The FET Fusion Oncogenes in Sarcomas and Leukemias
5.1. IGF/IGF1R Signaling in FET Fusion Oncogene Sarcomas
5.2. Anti-IGF Treatment of FET Fusion Oncogene Sarcomas
6. IGF1R Is a Downstream Target of the PAX3–FKHR Fusion in Rhabdomyosarcoma
7. Gene Fusions Involving IGF1R
8. Concluding Remarks
Funding
Conflicts of Interest
References
- Prensner, J.R.; Chinnaiyan, A.M. Oncogenic Gene Fusions in Epithelial Carcinomas. Curr. Opin. Genet. Dev. 2009, 19, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available online: http://cgap.nci.nih.gov/Chromosomes/Mitelman (accessed on 3 July 2019).
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of gene fusions in epithelial cancers: seq and ye shall find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.K.; Stenman, G. The landscape of gene fusions and somatic mutations in salivary gland neoplasms—Implications for diagnosis and therapy. Oral Oncol. 2016, 57, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J. Imatinib as a Paradigm of Targeted Therapies. Adv. Cancer Res.h 2004, 91, 1–30. [Google Scholar]
- Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef]
- De Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef]
- Heldin, C.H.; Lennartsson, J.; Westermark, B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef]
- Noujaim, J.; Thway, K.; Fisher, C.; Jones, R.L. Dermatofibrosarcoma protuberans: from translocation to targeted therapy. Cancer Boil. Med. 2015, 12, 375–384. [Google Scholar]
- Liu, D.; Tadokoro, H.; De Mello, R.; Aguiar, P., Jr. EGFR and EML4-ALK updated therapies in non-small cell lung cancer. Recent Patents Anti-Cancer Drug Discov. 2016, 11, 393–400. [Google Scholar]
- Gerber, D.E.; Minna, J.D. ALK Inhibition for Non-Small Cell Lung Cancer: From Discovery to Therapy in Record Time. Cancer Cell 2010, 18, 548–551. [Google Scholar] [CrossRef]
- Werner, H.; Meisel-Sharon, S.; Bruchim, I. Oncogenic fusion proteins adopt the insulin-like growth factor signaling pathway. Mol. Cancer 2018, 17, 28. [Google Scholar] [CrossRef]
- Tognon, C.E.; Sorensen, P.H. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin. Ther. Targets 2012, 16, 33–48. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Stenman, G.; Licitra, L.; Said-Al-Naief, N.; van Zante, A.; Yarbrough, W.G. Tumours of salivary glands: Adenoid cystic carcinoma. In WHO Classification of Head and Neck Tumours, 4th ed.; El-Naggar, A.K., Chan, J.K.C., Grandis, J.R., Takata, T., Slootweg, P.J., Eds.; IARC: Lyon, France, 2017; Volume 9, pp. 164–165. [Google Scholar]
- Carlson, J.; Licitra, L.; Locati, L.D.; Raben, D.; Persson, F.; Stenman, G. Salivary Gland Cancer: An Update on Present and Emerging Therapies. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, 257–263. [Google Scholar] [CrossRef]
- Andry, G.; Hamoir, M.; Locati, L.D.; Licitra, L.; Langendijk, J.A. Management of salivary gland tumors. Expert Rev. Anticancer Ther. 2012, 12, 1161–1168. [Google Scholar] [CrossRef]
- Laurie, S.A.; Ho, A.L.; Fury, M.G.; Sherman, E.; Pfister, D.G. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: A systematic review. Lancet Oncol. 2011, 12, 815–824. [Google Scholar] [CrossRef]
- Stenman, G.; Sandros, J.; Dahlenfors, R.; Juberg-Ode, M.; Mark, J. 6q- and loss of the Y chromosome—Two common deviations in malignant human salivary gland tumors. Cancer Genet. Cytogenet. 1986, 22, 283–293. [Google Scholar] [CrossRef]
- Persson, M.; Andrén, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef]
- Bahr, C.; Von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018, 553, 515–520. [Google Scholar] [CrossRef]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef]
- Gronostajski, R.M. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef]
- Becker-Santos, D.D.; Lonergan, K.M.; Gronostajski, R.M.; Lam, W.L. Nuclear Factor I/B: A Master Regulator of Cell Differentiation with Paradoxical Roles in Cancer. EBioMedicine 2017, 22, 2–9. [Google Scholar] [CrossRef]
- Adam, R.C.; Yang, H.; Rockowitz, S.; Larsen, S.B.; Nikolova, M.; Oristian, D.S.; Polak, L.; Kadaja, M.; Asare, A.; Zheng, D.; et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 2015, 521, 366–370. [Google Scholar] [CrossRef]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef]
- Brill, L.B.; Kanner, W.A.; Fehr, A.; Andren, Y.; Moskaluk, C.A.; Loning, T.; Stenman, G.; Frierson, H.F., Jr. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod. Pathol. 2011, 24, 1169–1176. [Google Scholar] [CrossRef]
- Andersson, M.K.; Afshari, M.K.; Andrén, Y.; Wick, M.J.; Stenman, G. Targeting the Oncogenic Transcriptional Regulator MYB in Adenoid Cystic Carcinoma by Inhibition of IGF1R/AKT Signaling. J. Natl. Cancer Inst. 2017, 109, djx017. [Google Scholar] [CrossRef]
- Yan, C.; Higgins, P.J. Drugging the undruggable: Transcription therapy for cancer. Biochim. Biophys. Acta 2013, 1835, 76–85. [Google Scholar] [CrossRef]
- Yeh, J.E.; Toniolo, P.A.; Frank, D.A. Targeting transcription factors: Promising new strategies for cancer therapy. Curr. Opin. Oncol. 2013, 25, 652–658. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Mitani, Y.; Diao, L.; Guijarro, I.; Wang, J.; Zweidler-McKay, P.; Bell, D.; William, W.N., Jr.; Glisson, B.S.; Wick, M.J.; et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J. Clin. Oncol. 2017, 35, 352–360. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Morelli, M.P.; Calvo, E.; Ordonez, E.; Wick, M.J.; Viqueira, B.R.; Lopez-Casas, P.P.; Bruckheimer, E.; Calles-Blanco, A.; Sidransky, D.; Hidalgo, M. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J. Clin. Oncol. 2012, 30, e45–e48. [Google Scholar] [CrossRef]
- Calvo, E.; Soria, J.C.; Ma, W.W.; Wang, T.; Bahleda, R.; Tolcher, A.W.; Gernhardt, D.; O’Connell, J.; Millham, R.; Giri, N.; et al. A Phase I Clinical Trial and Independent Patient-Derived Xenograft Study of Combined Targeted Treatment with Dacomitinib and Figitumumab in Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 1177–1185. [Google Scholar] [CrossRef]
- Mahadevan, D.; Sutton, G.R.; Arteta-Bulos, R.; Bowden, C.J.; Miller, P.J.E.; Swart, R.E.; Walker, M.S.; Haluska, P.; Munster, P.N.; Marshall, J.; et al. Phase 1b study of safety, tolerability and efficacy of R1507, a monoclonal antibody to IGF-1R in combination with multiple standard oncology regimens in patients with advanced solid malignancies. Cancer Chemother. Pharmacol. 2014, 73, 467–473. [Google Scholar] [CrossRef]
- Bell, D.; Bullerdiek, J.; Gnepp, D.R.; Schwartz, M.; Stenman, G.; Trintafyllou, A. Tumours of salivary glands: Pleomorphic adenoma. In WHO Classification of Head and Neck Tumours, 4th ed.; El-Naggar, A.K., Chan, J.K.C., Grandis, J.R., Takata, T., Slootweg, P.J., Eds.; IARC: Lyon, France, 2017; Volume 9, pp. 185–186. [Google Scholar]
- Kas, K.; Voz, M.L.; Röijer, E.; Åström, A.-K.; Meyen, E.; Stenman, G.; Van De Ven, W.J. Promoter swapping between the genes for a novel zinc finger protein and β-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet. 1997, 15, 170–174. [Google Scholar] [CrossRef]
- Geurts, J.M.; Schoenmakers, E.F.; Röijer, E.; Åström, A.-K.; Stenman, G.; Van De Ven, W.J. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene 1998, 16, 865–872. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. Chromosomal patterns in a benign human neoplasm, the mixed salivary gland tumour. Hereditas 1982, 96, 141–148. [Google Scholar] [CrossRef]
- Aström, A.K.; Voz, M.L.; Kas, K.; Röijer, E.; Wedell, B.; Mandahl, N.; Van De Ven, W.; Mark, J.; Stenman, G. Conserved mechanism of PLAG1 activation in salivary gland tumors with and without chromosome 8q12 abnormalities: Identification of SII as a new fusion partner gene. Cancer Res. 1999, 59, 918–923. [Google Scholar]
- Kas, K.; Voz, M.L.; Hensen, K.; Meyen, E.; Van De Ven, W.J.M. Transcriptional Activation Capacity of the Novel PLAG Family of Zinc Finger Proteins. J. Boil. Chem. 1998, 273, 23026–23032. [Google Scholar] [CrossRef]
- Dalin, M.G.; Katabi, N.; Persson, M.; Lee, K.-W.; Makarov, V.; Desrichard, A.; Walsh, L.A.; West, L.; Nadeem, Z.; Ramaswami, D.; et al. Multi-dimensional genomic analysis of myoepithelial carcinoma identifies prevalent oncogenic gene fusions. Nat. Commun. 2017, 8, 1197. [Google Scholar] [CrossRef]
- Åström, A.; D’Amore, E.S.; Sainati, L.; Panarello, C.; Morerio, C.; Mark, J.; Stenman, G. Evidence of involvement of the PLAG1 gene in lipoblastomas. Int. J. Oncol. 2000, 16, 1107–1110. [Google Scholar]
- Yoshida, H.; Miyachi, M.; Ouchi, K.; Kuwahara, Y.; Tsuchiya, K.; Iehara, T.; Konishi, E.; Yanagisawa, A.; Hosoi, H. Identification ofCOL3A1andRAB2Aas novel translocation partner genes ofPLAG1in lipoblastoma. Genes Chromosom. Cancer 2014, 53, 606–611. [Google Scholar] [CrossRef]
- Nitta, Y.; Miyachi, M.; Tomida, A.; Sugimoto, Y.; Nakagawa, N.; Yoshida, H.; Ouchi, K.; Tsuchiya, K.; Iehara, T.; Konishi, E.; et al. Identification of a novel BOC-PLAG1 fusion gene in a case of lipoblastoma. Biochem. Biophys. Res. Commun. 2019, 512, 49–52. [Google Scholar] [CrossRef]
- Zatkova, A.; Rouillard, J.M.; Hartmann, W.; Lamb, B.J.; Kuick, R.; Eckart, M.; von Schweinitz, D.; Koch, A.; Fonatsch, C.; Pietsch, T.; et al. Amplification and overexpression of the IGF2 regulator PLAG1 in hepatoblastoma. Genes Chromosomes Cancer 2004, 39, 126–137. [Google Scholar] [CrossRef]
- Regel, I.; Eichenmüller, M.; Joppien, S.; Liebl, J.; Häberle, B.; Müller-Höcker, J.; Vollmar, A.; Von Schweinitz, D.; Kappler, R. IGFBP3 impedes aggressive growth of pediatric liver cancer and is epigenetically silenced in vascular invasive and metastatic tumors. Mol. Cancer 2012, 11, 9. [Google Scholar] [CrossRef]
- Van Dyck, F.; Declercq, J.; Braem, C.V.; Van De Ven, W.J. PLAG1, the prototype of the PLAG gene family: versatility in tumour development (review). Int. J. Oncol. 2007, 30, 765–774. [Google Scholar] [CrossRef]
- Juma, A.R.; Damdimopoulou, P.E.; Grommen, S.V.; Van de Ven, W.J.; De Groef, B. Emerging role of PLAG1 as a regulator of growth and reproduction. J. Endocrinol. 2016, 228, R45–R56. [Google Scholar] [CrossRef]
- Hensen, K.; Braem, C.; Declercq, J.; Van Dyck, F.; Dewerchin, M.; Fiette, L.; Denef, C.; Van De Ven, W.J.M. Targeted disruption of the murine Plag1 proto-oncogene causes growth retardation and reduced fertility. Dev. Growth Differ. 2004, 46, 459–470. [Google Scholar] [CrossRef]
- Voz, M.L.; Agten, N.S.; Van De Ven, W.J.; Kas, K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000, 60, 106–113. [Google Scholar]
- DeChiara, T.M.; Robertson, E.J.; Efstratiadis, A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991, 64, 849–859. [Google Scholar] [CrossRef]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; Krailo, M.; Villaluna, D.; Adamson, P.C.; Blaney, S.M. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef]
- Knezevich, S.R.; McFadden, D.E.; Tao, W.; Lim, J.F.; Sorensen, P.H. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet. 1998, 18, 184–187. [Google Scholar] [CrossRef]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Skálová, A.; Stenman, G.; Slouka, D.; Svoboda, T.; Rinaldo, A.; Steiner, P.; Bishop, J.A.; Hunt, J.L.; Nibu, K.-I.; Poorten, V.V.; et al. The Role of Molecular Testing in the Differential Diagnosis of Salivary Gland Carcinomas. Am. J. Surg. Pathol. 2018, 42, e11–e27. [Google Scholar] [CrossRef]
- Vokuhl, C.; Nourkami-Tutdibi, N.; Furtwangler, R.; Gessler, M.; Graf, N.; Leuschner, I. ETV6-NTRK3 in congenital mesoblastic nephroma: A report of the SIOP/GPOH nephroblastoma study. Pediatr. Blood Cancer 2018, 65, e26925. [Google Scholar] [CrossRef]
- Eguchi, M.; Eguchi-Ishimae, M.; Tojo, A.; Morishita, K.; Suzuki, K.; Sato, Y.; Kudoh, S.; Tanaka, K.; Setoyama, M.; Nagamura, F.; et al. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood 1999, 93, 1355–1363. [Google Scholar]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar]
- Seethala, R.R.; Chiosea, S.I.; Liu, C.Z.; Nikiforova, M.; Nikiforov, Y.E. Clinical and Morphologic Features of ETV6-NTRK3 Translocated Papillary Thyroid Carcinoma in an Adult Population without Radiation Exposure. Am. J. Surg. Pathol. 2017, 41, 446–457. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Zehir, A.; Yaeger, R.; Wang, L.; Middha, S.; Zheng, T.; Hyman, D.M.; Solit, D.; Arcila, M.E.; Borsu, L.; et al. Identification of Targetable Kinase Alterations in Patients with Colorectal Carcinoma That are Preferentially Associated with Wild-Type RAS/RAF. Mol. Cancer Res. 2016, 14, 296–301. [Google Scholar] [CrossRef]
- Alassiri, A.H.; Ali, R.H.; Shen, Y.; Lum, A.; Strahlendorf, C.; Deyell, R.; Rassekh, R.; Sorensen, P.H.; Laskin, J.; Marra, M.; et al. ETV6-NTRK3 Is Expressed in a Subset of ALK-Negative Inflammatory Myofibroblastic Tumors. Am. J. Surg. Pathol. 2016, 40, 1–1061. [Google Scholar] [CrossRef]
- Tognon, C.E.; Martin, M.J.; Moradian, A.; Trigo, G.; Rotblat, B.; Cheng, S.W.; Pollard, M.; Uy, E.; Chow, C.; Carboni, J.M.; et al. A tripartite complex composed of ETV6-NTRK3, IRS1 and IGF1R is required for ETV6-NTRK3-mediated membrane localization and transformation. Oncogene 2012, 31, 1334–1340. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Albert, C.M.; Davis, J.L.; Federman, N.; Casanova, M.; Laetsch, T.W. TRK Fusion Cancers in Children: A Clinical Review and Recommendations for Screening. J. Clin. Oncol. 2019, 37, 513–524. [Google Scholar] [CrossRef]
- Martin, M.J.; Melnyk, N.; Pollard, M.; Bowden, M.; Leong, H.; Podor, T.J.; Gleave, M.; Sorensen, P.H.B. The Insulin-Like Growth Factor I Receptor Is Required for Akt Activation and Suppression of Anoikis in Cells Transformed by the ETV6-NTRK3 Chimeric Tyrosine Kinase. Mol. Cell. Boil. 2006, 26, 1754–1769. [Google Scholar] [CrossRef][Green Version]
- Tognon, C.E.; Somasiri, A.M.; Evdokimova, V.E.; Trigo, G.; Uy, E.E.; Melnyk, N.; Carboni, J.M.; Gottardis, M.M.; Roskelley, C.D.; Pollak, M.; et al. ETV6-NTRK3-mediated breast epithelial cell transformation is blocked by targeting the IGF1R signaling pathway. Cancer Res. 2011, 71, 1060–1070. [Google Scholar] [CrossRef]
- Tognon, C.E.; Rafn, B.; Cetinbas, N.M.; Kamura, T.; Trigo, G.; Rotblat, B.; Okumura, F.; Matsumoto, M.; Chow, C.; Davare, M.; et al. Insulin-like growth factor 1 receptor stabilizes the ETV6–NTRK3 chimeric oncoprotein by blocking its KPC1/Rnf123-mediated proteasomal degradation. J. Boil. Chem. 2018, 293, 12502–12515. [Google Scholar] [CrossRef]
- Åman, P. Fusion Oncogenes of Sarcomas. In Chromosomal Translocations and Genome Rearrangements in Cancer, 1st ed.; Rowley, J.D., Le Beau, M.M., Rabbitts, T.H., Eds.; Springer International Publishing: Basel, Switzerland, 2015; pp. 321–331. [Google Scholar]
- Riggi, N.; Cironi, L.; Suvà, M.-L.; Stamenkovic, I. Sarcomas: genetics, signalling, and cellular origins. Part 1: The fellowship of TET. J. Pathol. 2007, 213, 4–20. [Google Scholar] [CrossRef]
- Aman, P. Fusion genes in solid tumors. Semin. Cancer Boil. 1999, 9, 303–318. [Google Scholar] [CrossRef]
- Andersson, M.K.; Ståhlberg, A.; Arvidsson, Y.; Olofsson, A.; Semb, H.; Stenman, G.; Nilsson, O.; Åman, P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Boil. 2008, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Blechingberg, J.; Luo, Y.; Bolund, L.; Damgaard, C.K.; Nielsen, A.L. Gene Expression Responses to FUS, EWS, and TAF15 Reduction and Stress Granule Sequestration Analyses Identifies FET-Protein Non-Redundant Functions. PLoS ONE 2012, 7, e46251. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Cironi, L.; Provero, P.; Suvà, M.-L.; Kaloulis, K.; Garcia-Echeverria, C.; Hoffmann, F.; Trumpp, A.; Stamenkovic, I. Development of Ewing’s Sarcoma from Primary Bone Marrow-Derived Mesenchymal Progenitor Cells. Cancer Res. 2005, 65, 11459–11468. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, A.; Gustafsson, C.K.; Engtröm, K.; Thomsen, C.; Dolatabadi, S.; Jonasson, E.; Li, C.-Y.; Ruff, D.; Chen, S.-M.; Åman, P. Normal and Functional TP53 in Genetically Stable Myxoid/Round Cell Liposarcoma. PLoS ONE 2014, 9, e113110. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.A.; Mathur, M.; Raaka, B.M.; Ron, D.; Samuels, H.H. TLS (Translocated-in-Liposarcoma) Is a High-Affinity Interactor for Steroid, Thyroid Hormone, and Retinoid Receptors. Mol. Endocrinol. 1998, 12, 4–18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohno, T.; Rao, V.N.; Reddy, E.S. EWS/Fli-1 chimeric protein is a transcriptional activator. Cancer Res. 1993, 53, 5859–5863. [Google Scholar] [PubMed]
- May, W.A.; Lessnick, S.L.; Braun, B.S.; Klemsz, M.; Lewis, B.C.; Lunsford, L.B.; Hromas, R.; Denny, C.T. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol. Cell. Boil. 1993, 13, 7393–7398. [Google Scholar] [CrossRef]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schönegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef]
- Engström, K.; Willén, H.; Kåbjörn-Gustafsson, C.; Andersson, C.; Olsson, M.; Göransson, M.; Järnum, S.; Olofsson, A.; Warnhammar, E.; Åman, P. The Myxoid/Round Cell Liposarcoma Fusion Oncogene FUS-DDIT3 and the Normal DDIT3 Induce a Liposarcoma Phenotype in Transfected Human Fibrosarcoma Cells. Am. J. Pathol. 2006, 168, 1642–1653. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Lindén, M.; Thomsen, C.; Grundevik, P.; Jonasson, E.; Andersson, D.; Runnberg, R.; Dolatabadi, S.; Vannas, C.; Santamarίa, M.L.; Fagman, H.; et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019, 20, e45766. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e19. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Hodges, C.; Calarco, J.P.; Braun, S.M.; Ku, W.L.; Kadoch, C.; Zhao, K.; Crabtree, G.R. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017, 49, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Cironi, L.; Provero, P.; Suvà, M.-L.; Stehle, J.-C.; Baumer, K.; Guillou, L.; Stamenkovic, I. Expression of the FUS-CHOP Fusion Protein in Primary Mesenchymal Progenitor Cells Gives Rise to a Model of Myxoid Liposarcoma. Cancer Res. 2006, 66, 7016–7023. [Google Scholar] [CrossRef] [PubMed]
- De Alava, E.; Lessnick, S.L.; Sorensen, P.H. Ewing sarcoma. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D.M., Bridge, J.A., Hoogendorn, P.C.W., Mertens, F., Eds.; IARC: Lyon, France, 2013; Volume 5, pp. 305–309. [Google Scholar]
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. EWS/FLI-1 Silencing and Gene Profiling of Ewing Cells Reveal Downstream Oncogenic Pathways and a Crucial Role for Repression of Insulin-Like Growth Factor Binding Protein 3†. Mol. Cell. Boil. 2004, 24, 7275–7283. [Google Scholar] [CrossRef]
- Scotlandi, K.; Benini, S.; Nanni, P.; Lollini, P.L.; Nicoletti, G.; Landuzzi, L.; Serra, M.; Manara, M.C.; Picci, P.; Baldini, N. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice. Cancer Res. 1998, 58, 4127–4131. [Google Scholar]
- Toretsky, J.A.; Kalebic, T.; Blakesley, V.; Leroith, D.; Helman, L.J. The Insulin-like Growth Factor-I Receptor Is Required for EWS/FLI-1 Transformation of Fibroblasts. J. Boil. Chem. 1997, 272, 30822–30827. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Ladanyi, M. Tumours of uncertain differentiation: Desmoplastic small round cell tumour. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D.M., Bridge, J.A., Hoogendorn, P.C.W., Mertens, F., Eds.; IARC: Lyon, France, 2013; Volume 5, pp. 225–227. [Google Scholar]
- Karnieli, E.; Werner, H.; Rauscher, F.J., 3rd; Benjamin, L.E.; LeRoith, D. The IGF-I receptor gene promoter is a molecular target for the Ewing’s sarcoma-Wilms’ tumor 1 fusion protein. J. Biol. Chem. 1996, 271, 19304–19309. [Google Scholar] [CrossRef]
- Idelman, G.; Glaser, T.; Roberts, C.T., Jr.; Werner, H. WT1-p53 interactions in insulin-like growth factor-I receptor gene regulation. J. Biol. Chem. 2003, 278, 3474–3482. [Google Scholar] [CrossRef]
- Werner, H.; Idelman, G.; Rubinstein, M.; Pattee, P.; Nagalla, S.R.; Roberts, C.T. A novel EWS-WT1 gene fusion product in desmoplastic small round cell tumor is a potent transactivator of the insulin-like growth factor-I receptor (IGF-IR) gene. Cancer Lett. 2007, 247, 84–90. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Ladanyi, M. Adipocytic tumours. Myxoid liposarcoma. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D.M., Bridge, J.A., Hoogendorn, P.C.W., Mertens, F., Eds.; IARC: Lyon, France, 2013; Volume 5, pp. 39–41. [Google Scholar]
- Crozat, A.; Åman, P.; Mandahl, N.; Ron, D. Pierre Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef]
- Rabbitts, T.; Forster, A.; Larson, R.; Nathan, P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 1993, 4, 175–180. [Google Scholar] [CrossRef]
- Åman, P.; Ron, D.; Mandahl, N.; Fioretos, T.; Heim, S.; Arheden, K.; Willén, H.; Rydholm, A.; Mitelman, F. Rearrangement of the transcription factor geneCHOP in myxoid liposarcomas with t(12;16)(q13;p11). Genes Chromosom. Cancer 1992, 5, 278–285. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Höglund, M.; Mertens, F.; Mandahl, N.; Mitelman, F.; Åman, P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Cancer Genet. Cytogenet. 1996, 91, 146. [Google Scholar] [CrossRef]
- Demicco, E.G.; Torres, K.E.; Ghadimi, M.P.; Colombo, C.; Bolshakov, S.; Hoffman, A.; Peng, T.; Bovee, J.V.; Wang, W.L.; Lev, D.; et al. Involvement of the PI3K/Akt pathway in myxoid/round cell liposarcoma. Mod. Pathol. 2012, 25, 212–221. [Google Scholar] [CrossRef]
- Trautmann, M.; Menzel, J.; Bertling, C.; Cyra, M.; Isfort, I.; Steinestel, K.; Elges, S.; Grünewald, I.; Altvater, B.; Rossig, C.; et al. FUS–DDIT3 Fusion Protein-Driven IGF-IR Signaling is a Therapeutic Target in Myxoid Liposarcoma. Clin. Cancer Res. 2017, 23, 6227–6238. [Google Scholar] [CrossRef]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary Efficacy of the Anti-Insulin–Like Growth Factor Type 1 Receptor Antibody Figitumumab in Patients With Refractory Ewing Sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.-P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a Monoclonal Antibody to the Insulin-Like Growth Factor 1 Receptor, in Patients With Recurrent or Refractory Ewing Sarcoma Family of Tumors: Results of a Phase II Sarcoma Alliance for Research Through Collaboration Study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II Study of Ganitumab, a Fully Human Anti–Type-1 Insulin-Like Growth Factor Receptor Antibody, in Patients With Metastatic Ewing Family Tumors or Desmoplastic Small Round Cell Tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef]
- Anderson, P.M.; Bielack, S.S.; Gorlick, R.G.; Skubitz, K.; Daw, N.C.; Herzog, C.E.; Monge, O.R.; Lassaletta, A.; Boldrini, E.; Pápai, Z.; et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr. Blood Cancer 2016, 63, 1761–1770. [Google Scholar] [CrossRef]
- Naing, A.; Lorusso, P.; Fu, S.; Hong, D.S.; Anderson, P.; Benjamin, R.S.; Ludwig, J.; Chen, H.X.; Doyle, L.A.; Kurzrock, R. Insulin Growth Factor-Receptor (IGF-1R) Antibody Cixutumumab Combined with the mTOR Inhibitor Temsirolimus in Patients with Refractory Ewing’s Sarcoma Family Tumors. Clin. Cancer Res. 2012, 18, 2625–2631. [Google Scholar] [CrossRef]
- Quek, R.; Wang, Q.; Morgan, J.A.; Shapiro, G.I.; Butrynski, J.E.; Ramaiya, N.; Huftalen, T.; Jederlinic, N.; Manola, J.; Wagner, A.J.; et al. Combination mTOR and IGF-1R inhibition: Phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011, 17, 871–879. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Tap, W.D.; Qin, L.-X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J., 3rd; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Sublett, J.E.; Jeon, I.S.; Shapiro, D.N. The alveolar rhabdomyosarcoma PAX3/FKHR fusion protein is a transcriptional activator. Oncogene 1995, 11, 545–552. [Google Scholar]
- Fredericks, W.J.; Galili, N.; Mukhopadhyay, S.; Rovera, G.; Bennicelli, J.; Barr, F.G.; Rauscher, F.J., 3rd. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol. Cell Biol. 1995, 15, 1522–1535. [Google Scholar] [CrossRef]
- Ayalon, D.; Glaser, T.; Werner, H. Transcriptional regulation of IGF-I receptor gene expression by the PAX3–FKHR oncoprotein. Growth Horm. IGF Res. 2001, 11, 289–297. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide Identification of PAX3-FKHR Binding Sites in Rhabdomyosarcoma Reveals Candidate Target Genes Important for Development and Cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.W.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance. Cancer 2014, 120, 2448–2456. [Google Scholar] [CrossRef]
- Kang, Z.; Yu, Y.; Zhu, Y.J.; Helman, L.; Meltzer, P.; Cao, L. Abstract 1719: Down-regulation of IGFBP2 is associated with resistance to IGF1R therapy in rhabdomyosarcoma. Exp. Mol. Ther. 2014, 74, 1719. [Google Scholar]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative Clinical Genomics of Metastatic Cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Piarulli, G.; Puls, F.; Wängberg, B.; Fagman, H.; Hansson, M.; Nilsson, J.; Arbajian, E.; Mertens, F. Gene fusion involving the insulin-like growth factor 1 receptor in an ALK -negative inflammatory myofibroblastic tumour. Histopathology 2019, 74, 1098–1102. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tumor Type | Chromosome Translocation | Gene Fusion |
|---|---|---|
| Adenoid cystic carcinoma | t(6;9)(q23;p23) t(8;9)(q13.1;p23) | MYB–NFIB MYBL1–NFIB |
| Pleomorphic adenoma | t(3;8)(p21;q12) | CTNNB1–PLAG1 |
| Congenital fibrosarcoma Secretory carcinoma (breast and salivary gland) Mesoblastic nephroma Acute leukemia Pediatric non-brainstem high-grade glioma Papillary thyroid carcinoma Colorectal carcinoma Inflammatory myofibroblastic tumor | t(12;15)(p13;q25) | ETV6–NTRK3 |
| Ewing sarcoma | t(11;22)(q24;q12) | EWSR1–FLI1 |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1–WT1 |
| Myxoid liposarcoma | t(12;16)(q13;p11) t(12;22)(q13;q12) | FUS–DDIT3 EWSR1–DDIT3 |
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) | PAX3–FKHR |
| Inflammatory myofibroblastic tumor | t(2;15)(q35;q26.3) | FN1–IGF1R |
| Gastrointestinal stromal tumor | t(1;15)(p36.13;q26.3) | NBPF1–IGF1R |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, M.K.; Åman, P.; Stenman, G. IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors. Cells 2019, 8, 913. https://doi.org/10.3390/cells8080913
Andersson MK, Åman P, Stenman G. IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors. Cells. 2019; 8(8):913. https://doi.org/10.3390/cells8080913
Chicago/Turabian StyleAndersson, Mattias K., Pierre Åman, and Göran Stenman. 2019. "IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors" Cells 8, no. 8: 913. https://doi.org/10.3390/cells8080913
APA StyleAndersson, M. K., Åman, P., & Stenman, G. (2019). IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors. Cells, 8(8), 913. https://doi.org/10.3390/cells8080913