Clinical and Genetic Analysis of Children with Kartagener Syndrome

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Issues
2.2. Patients
2.2.1. Patient-1
2.2.2. Patient-2
2.3. Sample Collection
2.4. Sample Processing for Transmission Electron Microscopy
2.5. Cilia Morphological Evaluation and Orientation
2.6. Whole-Exome Sequencing
2.7. Gene Expression Analysis
2.8. Immunofluorescence Analysis
3. Results
3.1. Echocardiogram and Thoracoabdominal CT Scan
3.2. Transmission Electron Microscopy of Cilia
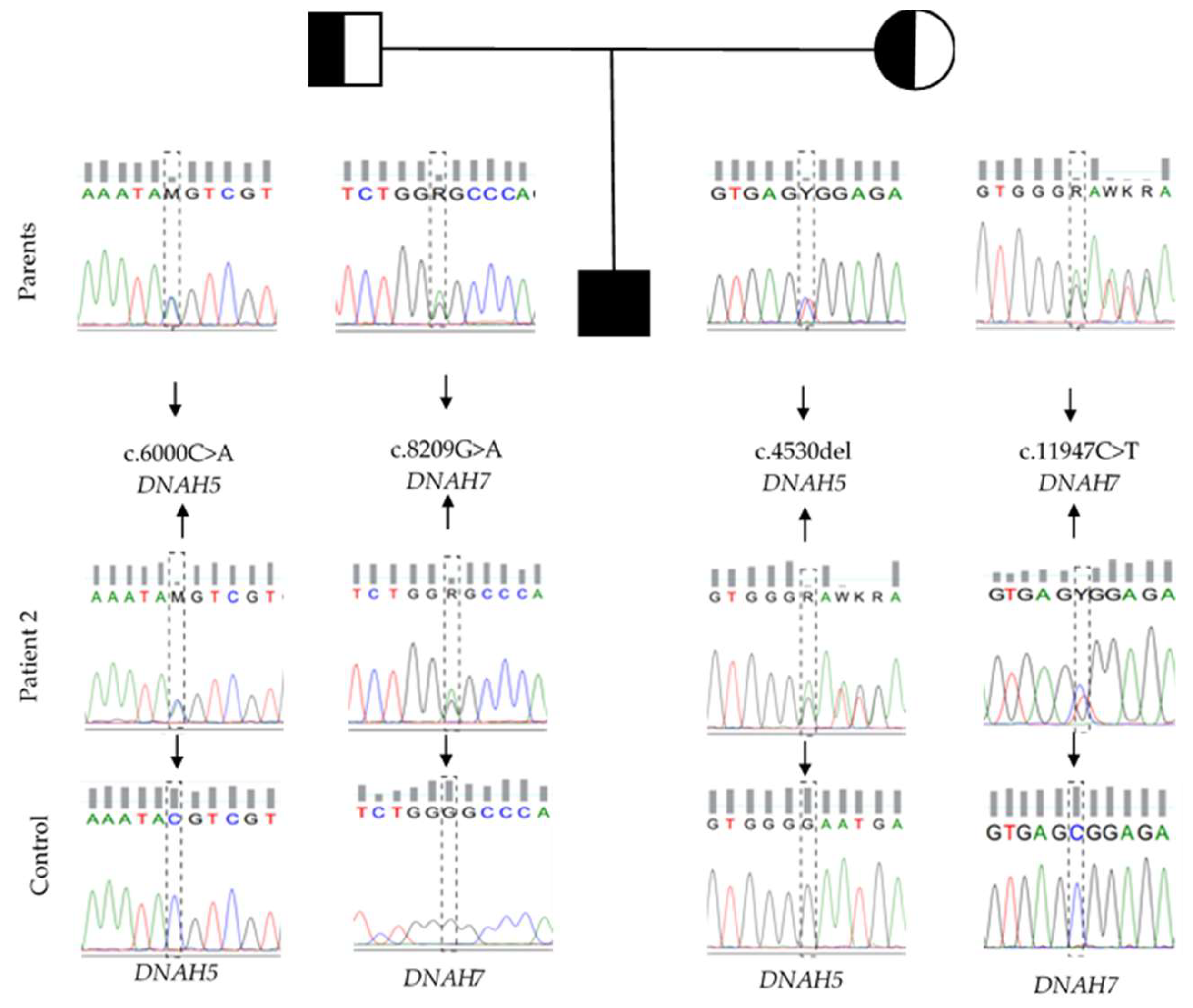
3.3. Whole-Exome Sequencing
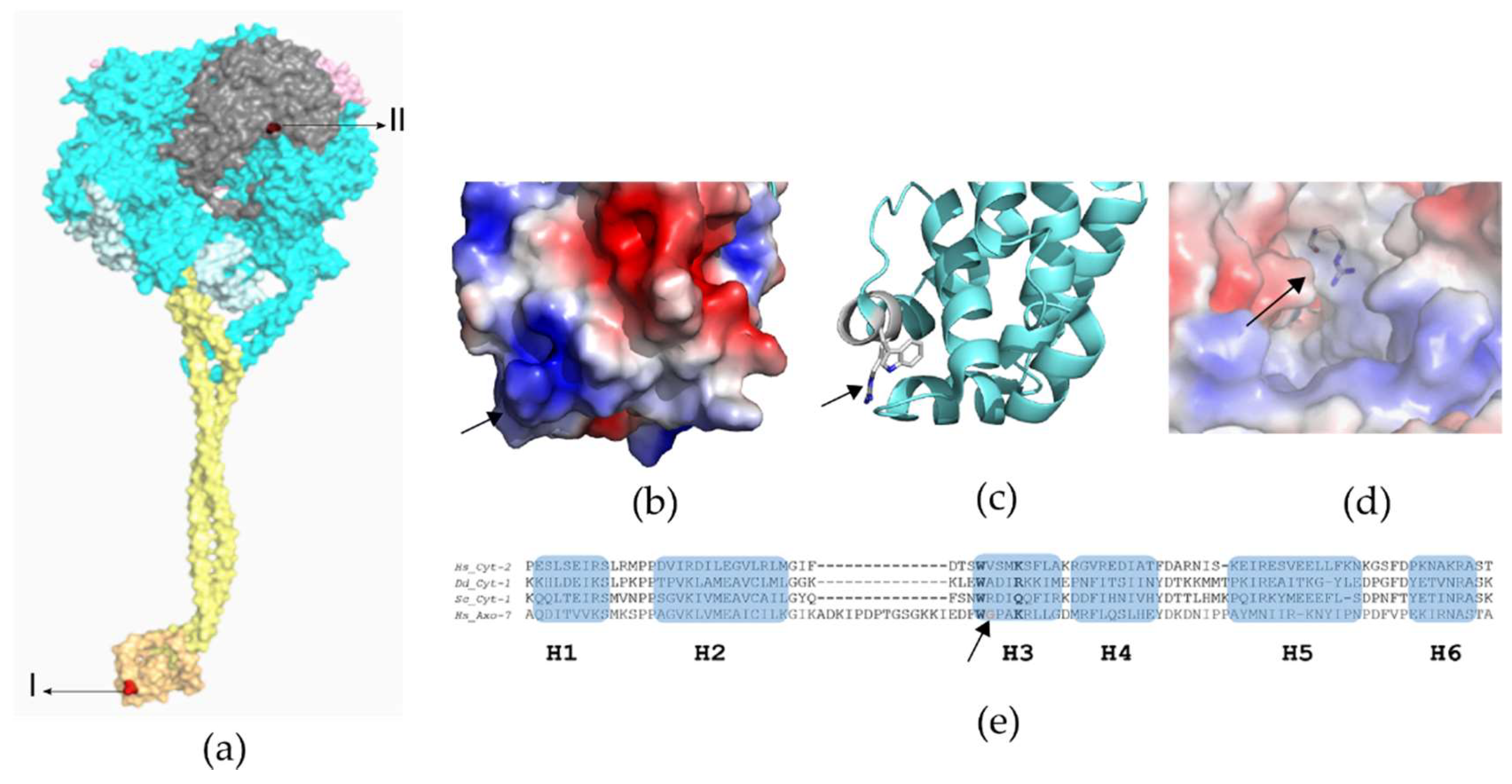
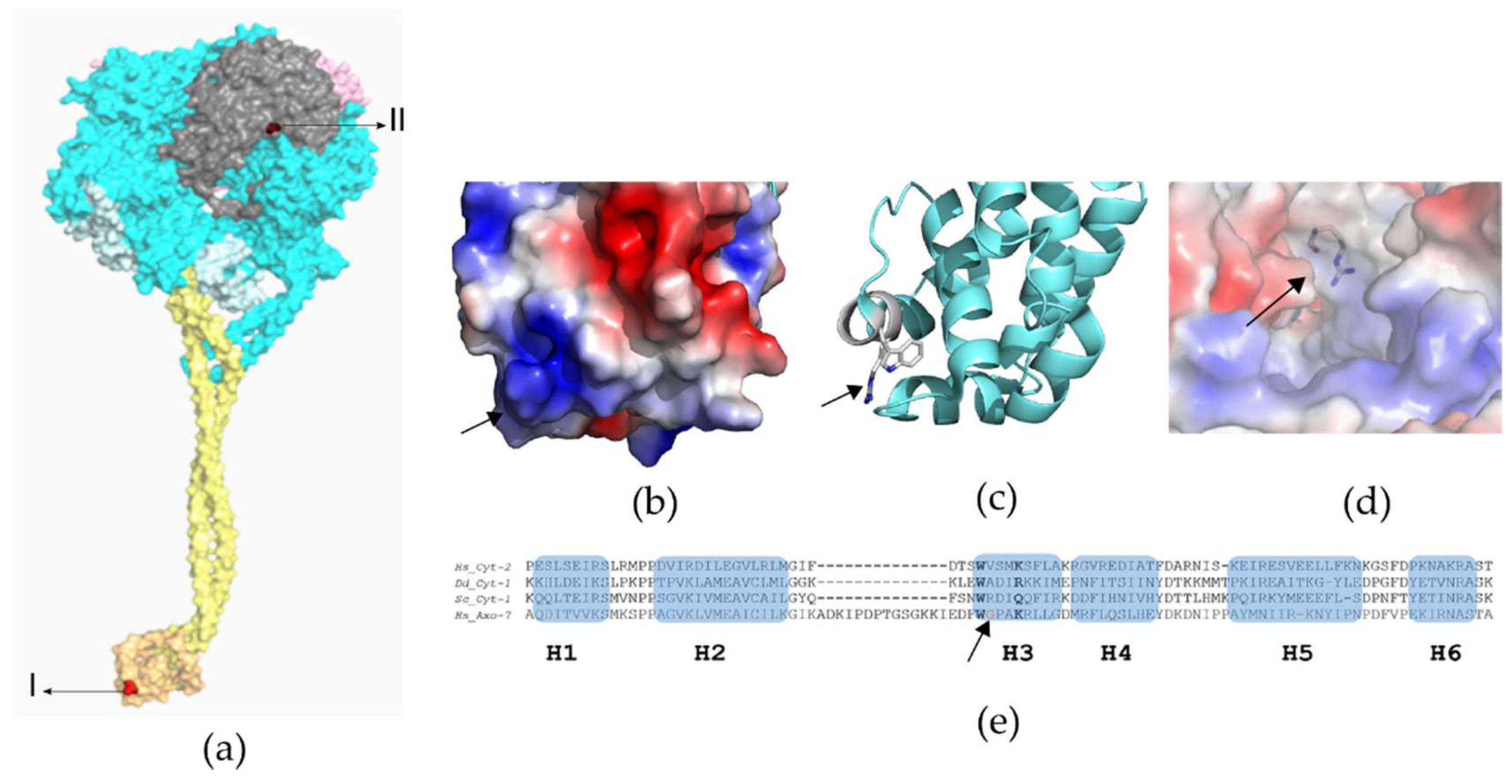
Whole-Exome Sequencing Variant Interpretation and Bioinformatic Analysis
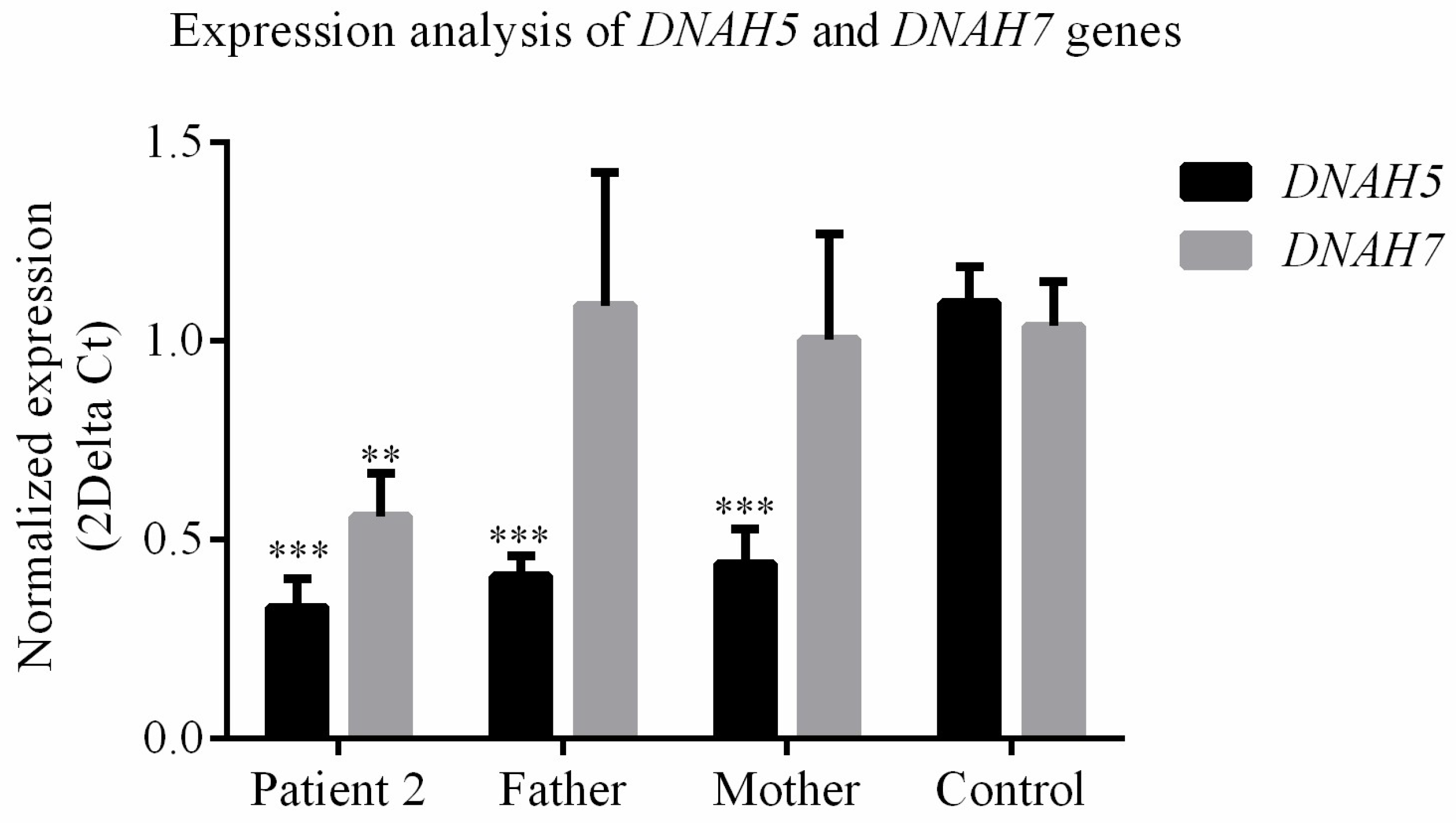
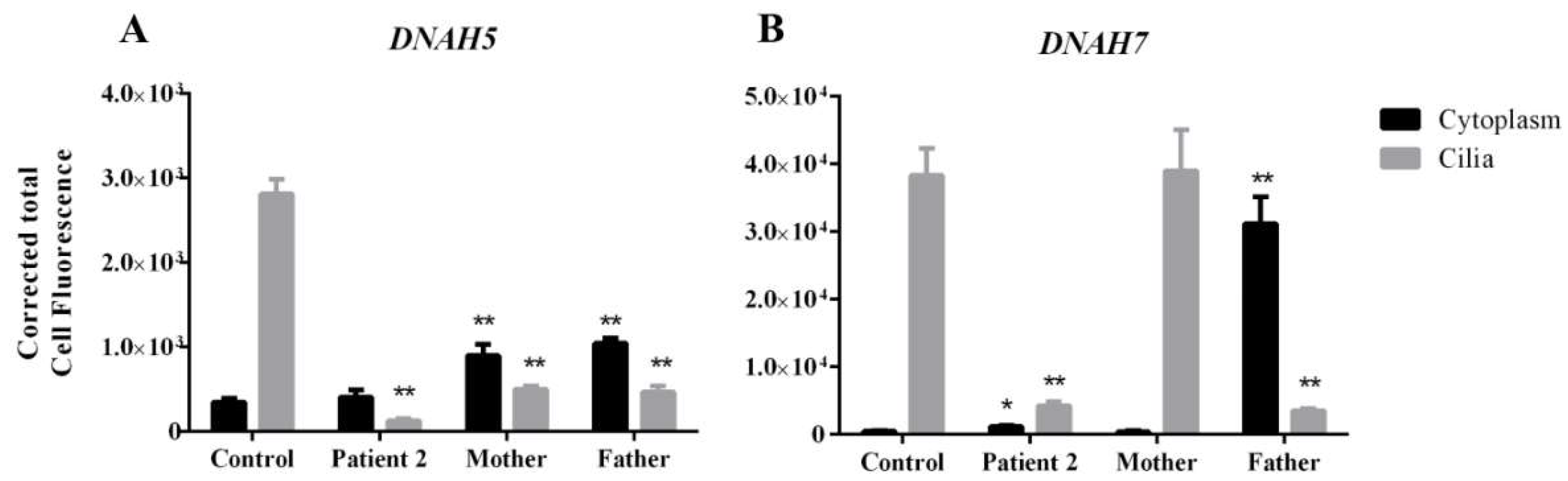
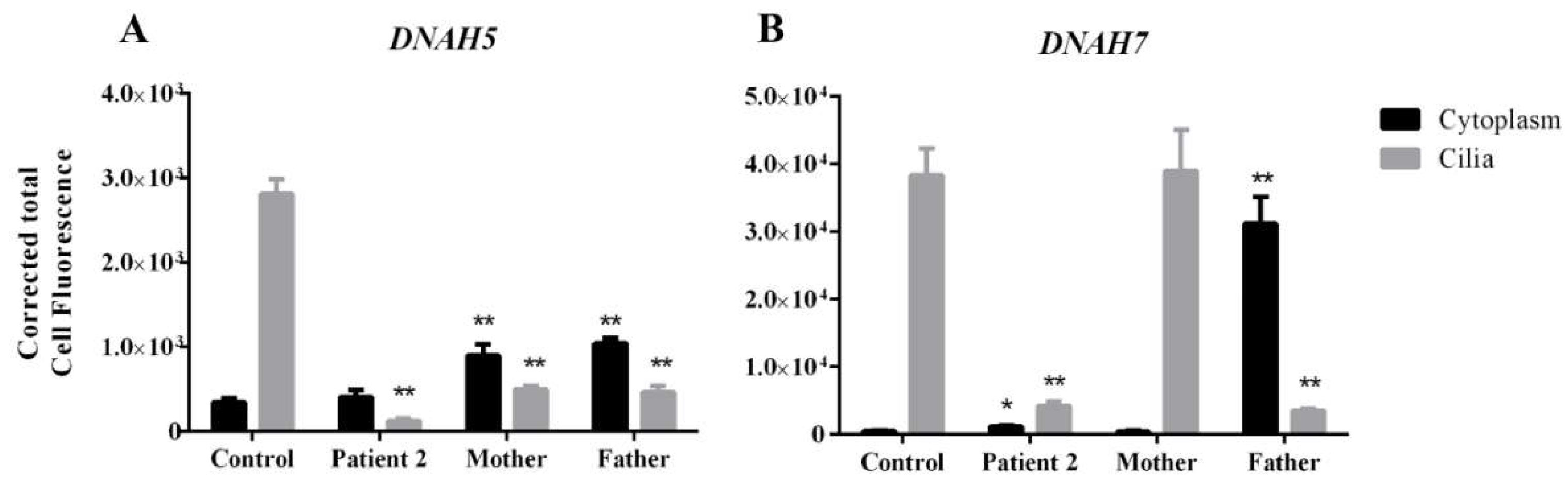
3.4. DNAH5 and DNAH7 mRNA Relative Expression
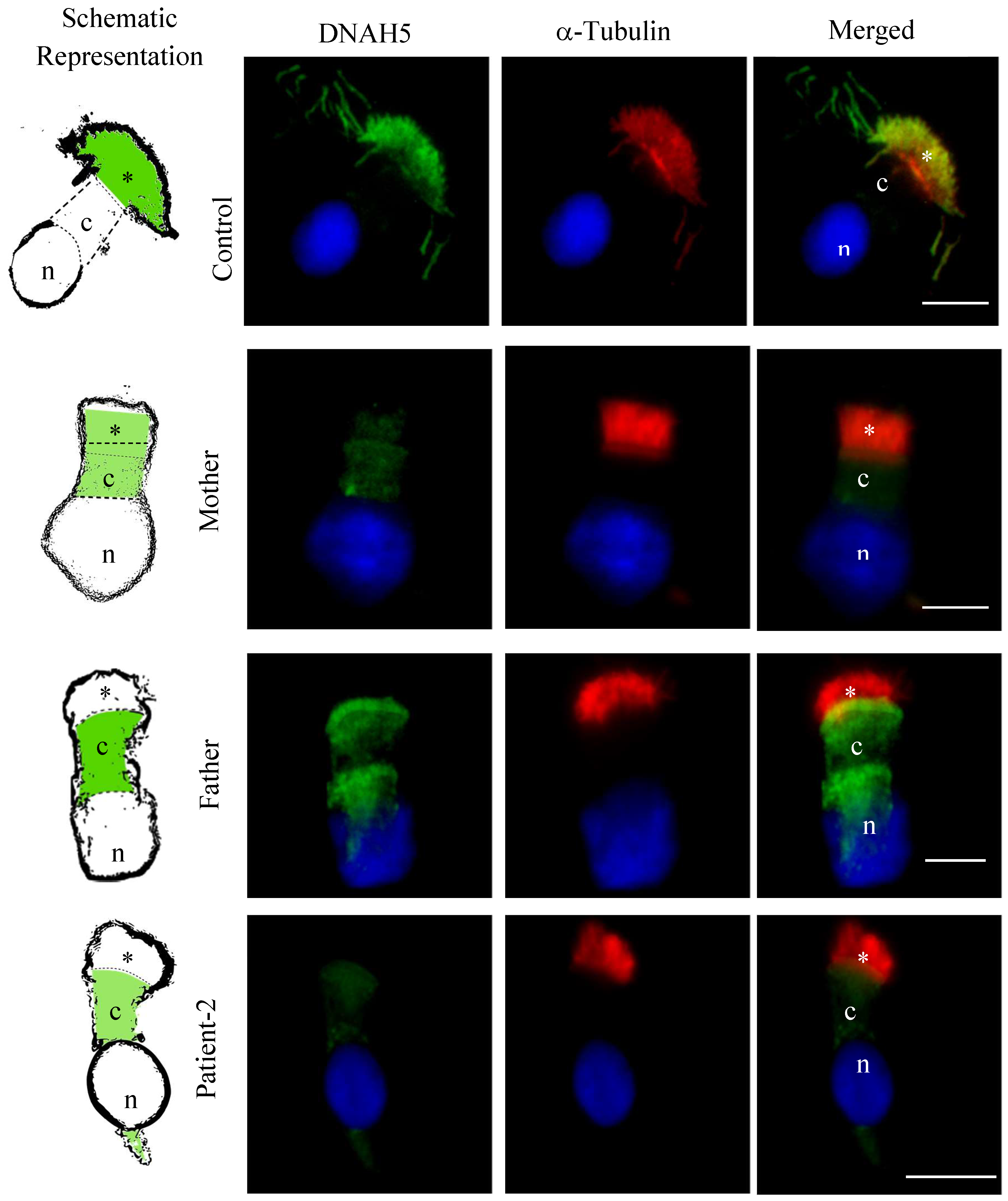
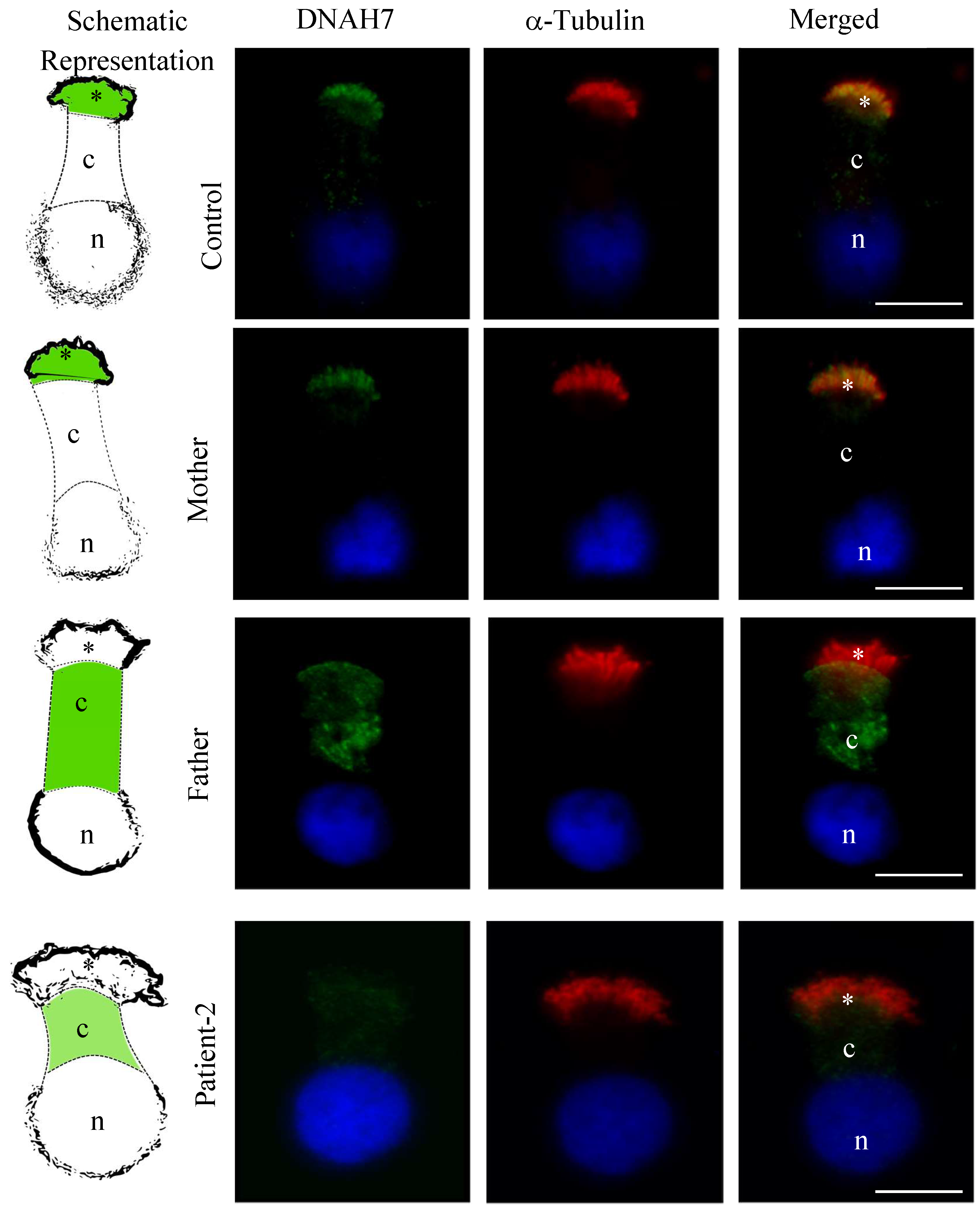
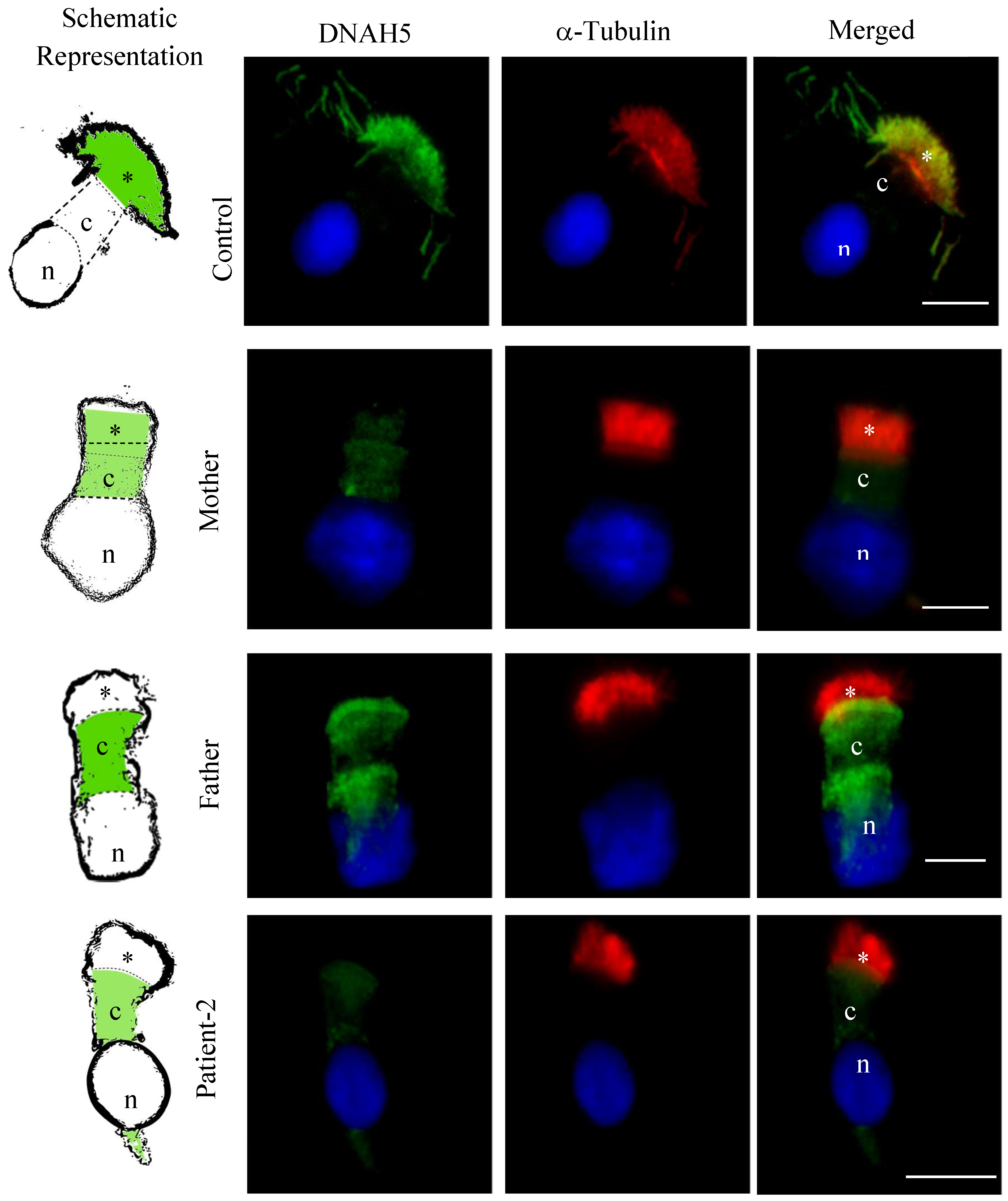
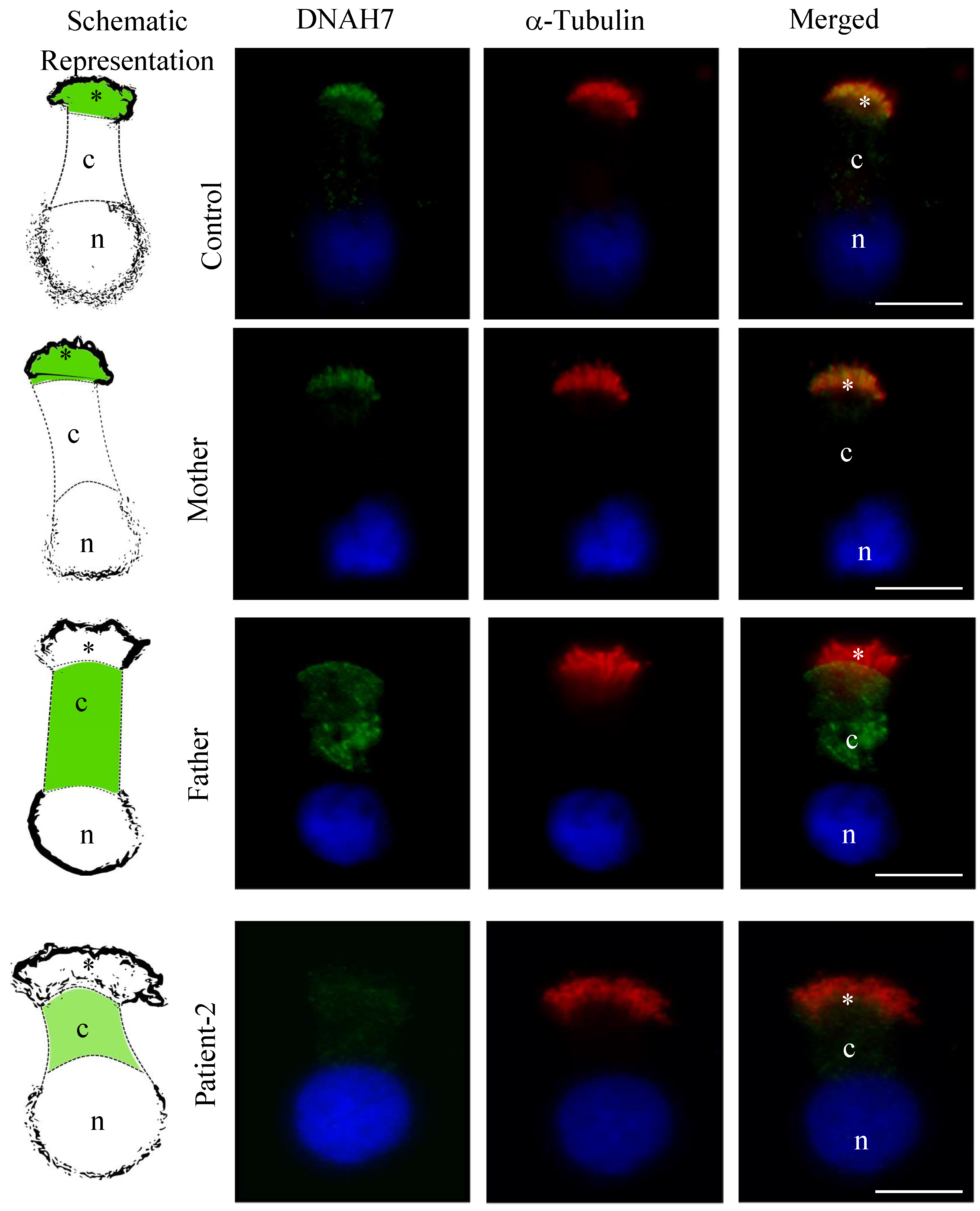
3.5. DNAH5 and DNAH7 Immunofluorescence Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef] [PubMed]
- Faubel, R.; Westendorf, C.; Bodenschatz, E.; Eichele, G. Cilia-based flow network in the brain ventricles. Science 2016, 353, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Sá, R.; Barros, A.; Sousa, M. Major regulatory mechanisms involved in sperm motility. Asian J. Androl. 2017, 19, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kölle, S.; Dubielzig, S.; Reese, S.; Wehrend, A.; König, P.; Kummer, W. Ciliary Transport, Gamete Interaction, and Effects of the Early Embryo in the Oviduct: Ex Vivo Analyses Using a New Digital Videomicroscopic System in the Cow1. Biol. Reprod. 2009, 81, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzelius, B.A.; Srurgess, J.M. The immotile-cilia syndrome: A microtubule-associated defect. Crit. Rev. Biochem. Mol. Biol. 1985, 19, 63–87. [Google Scholar] [CrossRef]
- Lucas, J.S.; Burgess, A.; Mitchison, H.M.; Moya, E.; Williamson, M.; Hogg, C. Diagnosis and management of primary ciliary dyskinesia. Arch. Dis. Child. 2014, 99, 850–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbo, B.; Lucas, J.S. Clinical care for primary ciliary dyskinesia: Current challenges and future directions. Eur. Respir. Rev. 2017, 26, 170023–170034. [Google Scholar] [CrossRef]
- Munro, N.C.; Currie, D.C.; Lindsay, K.S.; Ryder, T.A.; Rutman, A.; Dewar, A.; Greenstone, M.A.; Hendry, W.F.; Cole, P.J. Fertility in men with primary ciliary dyskinesia presenting with respiratory infection. Thorax 1994, 49, 684–687. [Google Scholar] [CrossRef]
- Lyons, R.A.; Saridogan, E.; Djahanbakhch, O. The reproductive significance of human Fallopian tube cilia. Hum. Reprod. Update 2006, 12, 363–372. [Google Scholar] [CrossRef]
- Inaba, K.; Mizuno, K. Sperm dysfunction and ciliopathy. Reprod. Med. Biol. 2016, 15, 77–94. [Google Scholar] [CrossRef]
- Pereira, R.; Oliveira, J.; Ferraz, L.; Barros, A.; Santos, R.; Sousa, M. Mutation analysis in patients with total sperm immotility. J. Assist. Reprod. Genet. 2015, 32, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.W.; Ferkol, T.W.; Davis, S.D.; Lee, H.-S.; Rosenfeld, M.; Dell, S.D.; Sagel, S.D.; Milla, C.; Olivier, K.N.; Sullivan, K.M.; et al. Clinical Features and Associated Likelihood of Primary Ciliary Dyskinesia in Children and Adolescents. Ann. Am. Thorac. Soc. 2016, 13, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Rutland, J.; Dewar, A.; Cox, T.; Cole, P. Nasal brushing for the study of ciliary ultrastructure. J. Clin. Pathol. 1982, 35, 357–359. [Google Scholar] [CrossRef] [PubMed]
- De Iongh, R.; Rutland, J. Orientation of respiratory tract cilia in patients with primary ciliary dyskinesia, bronchiectasis, and in normal subjects. J. Clin. Pathol. 1989, 42, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Negrao, L.; Fineza, I.; Taipa, R.; Melo-Pires, M.; Fortuna, A.M.; Goncalves, A.R.; Froufe, H.; Egas, C.; Santos, R.; et al. New splicing mutation in the choline kinase beta (CHKB) gene causing a muscular dystrophy detected by whole-exome sequencing. J. Hum. Genet. 2015, 60, 305–312. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Pongor, L.; Kormos, M.; Hatzis, C.; Pusztai, L.; Szabó, A.; Győrffy, B. A genome-wide approach to link genotype to clinical outcome by utilizing next generation sequencing and gene chip data of 6697 breast cancer patients. Genome Med. 2015, 7, 104–115. [Google Scholar] [CrossRef]
- Zhang, K.; Foster, H.E.; Rondelet, A.; Lacey, S.E.; Bahi-Buisson, N.; Bird, A.W.; Carter, A.P. Cryo-EM reveals how human cytoplasmic dynein is auto-inhibited and activated. Cell 2017, 169, 1303–1314. [Google Scholar] [CrossRef]
- Schmidt, H.; Zalyte, R.; Urnavicius, L.; Carter, A.P. Structure of human cytoplasmic dynein-2 primed for its power stroke. Nature 2015, 518, 435. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Oyama, T.; Shimo-Kon, R.; Imamula, K.; Shima, T.; Sutoh, K.; Kurisu, G. The 2.8 Å crystal structure of the dynein motor domain. Nature 2012, 484, 345. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Gleave, E.S.; Carter, A.P. Insights into dynein motor domain function from a 3.3-Å crystal structure. Nat. Struct. Mol. Biol. 2012, 19, 492. [Google Scholar] [CrossRef] [PubMed]
- Seelow, D.; Schuelke, M.; Hildebrandt, F.; Nürnberg, P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009, 37, W593–W599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]
- Fliegauf, M.; Olbrich, H.; Horvath, J.; Wildhaber, J.H.; Zariwala, M.A.; Kennedy, M.; Knowles, M.R.; Omran, H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005, 171, 1343–1349. [Google Scholar] [CrossRef]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef]
- Burgess, S.A.; Walker, M.L.; Sakakibara, H.; Knight, P.J.; Oiwa, K. Dynein structure and power stroke. Nature 2003, 421, 715–718. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.; Pereira, R.; Santos, R.; Sousa, M. Homozygosity Mapping using Whole-Exome Sequencing: A Valuable Approach for Pathogenic Variant Identification in Genetic Diseases. In Biomedical Engineering Systems and Technologies, Proceedings of the 10th International Joint Conference, Porto, Portugal, 21–23 February 2017; Springer International Publishing: Cham, Switzerland, 2017; pp. 210–216. [Google Scholar] [CrossRef]
- Oliveira, J.; Pereira, R.; Santos, R.; Sousa, M. Evaluating Runs of Homozygosity in Exome Sequencing Data - Utility in Disease Inheritance Model Selection and Variant Filtering. In Communications in Computer and Information Science; Barbosa, S.D.J., Chen, P., Filipe, J., Kotenko, I., Sivalingam, K.M., Washio, T., Yuan, J., Zhou, L., Eds.; Springer: Cham, Switzerland; Berlin, Germany, 2018; Volume 881, pp. 268–288. [Google Scholar] [CrossRef]
- Oda, T.; Yanagisawa, H.; Kamiya, R.; Kikkawa, M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346, 857–860. [Google Scholar] [CrossRef]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389. [Google Scholar] [CrossRef] [PubMed]
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations in CCDC39 and CCDC40 are the Major Cause of Primary Ciliary Dyskinesia with Axonemal Disorganization and Absent Inner Dynein Arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Blanchon, S.; Legendre, M.; Copin, B.; Duquesnoy, P.; Montantin, G.; Kott, E.; Dastot, F.; Jeanson, L.; Cachanado, M.; Rousseau, A. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J. Med. Genet. 2012, 49, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 Is Mutated in Primary Ciliary Dyskinesia and Interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakhleh, N.; Francis, R.; Giese, R.A.; Tian, X.; Li, Y.; Zariwala, M.A.; Yagi, H.; Khalifa, O.; Kureshi, S.; Chatterjee, B. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation 2012, 125, 2232–2242. [Google Scholar] [CrossRef]
- Sui, W.; Hou, X.; Che, W.; Ou, M.; Sun, G.; Huang, S.; Liu, F.; Chen, P.; Wei, X.; Dai, Y. CCDC40 mutation as a cause of primary ciliary dyskinesia: A case report and review of literature. Clin. Respir. J. 2016, 10, 614–621. [Google Scholar] [CrossRef]
- Takeuchi, K.; Kitano, M.; Kiyotoshi, H.; Ikegami, K.; Ogawa, S.; Ikejiri, M.; Nagao, M.; Fujisawa, T.; Nakatani, K. A targeted next-generation sequencing panel reveals novel mutations in Japanese patients with primary ciliary dyskinesia. Auris Nasus Larynx 2018, 45, 585–591. [Google Scholar] [CrossRef]
- Yang, L.; Banerjee, S.; Cao, J.; Bai, X.; Peng, Z.; Chen, H.; Huang, H.; Han, P.; Feng, S.; Yi, N.; et al. Compound Heterozygous Variants in the Coiled-Coil Domain Containing 40 Gene in a Chinese Family with Primary Ciliary Dyskinesia Cause Extreme Phenotypic Diversity in Cilia Ultrastructure. Front. Genet. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Djakow, J.; Svobodová, T.; Hrach, K.; Uhlík, J.; Cinek, O.; Pohunek, P. Effectiveness of sequencing selected exons of DNAH5 and DNAI1 in diagnosis of primary ciliary dyskinesia. Pediatr. Pulmonol. 2012, 47, 864–875. [Google Scholar] [CrossRef]
- Failly, M.; Bartoloni, L.; Letourneau, A.; Munoz, A.; Falconnet, E.; Rossier, C.; De Santi, M.M.; Santamaria, F.; Sacco, O.; DeLozier-Blanchet, C.D. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J. Med. Genet. 2009, 46, 281–286. [Google Scholar] [CrossRef]
- Hornef, N.; Olbrich, H.; Horvath, J.; Zariwala, M.a.; Fliegauf, M.; Loges, N.T.; Wildhaber, J.; Noone, P.G.; Kennedy, M.; Antonarakis, S.E.; et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 2006, 174, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Ferkol, T.W.; Puffenberger, E.G.; Lie, H.; Helms, C.; Strauss, K.A.; Bowcock, A.; Carson, J.L.; Hazucha, M.; Morton, D.H.; Patel, A.C. Primary ciliary dyskinesia-causing mutations in Amish and Mennonite communities. J. Pediatr. 2013, 163, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Kano, G.; Tsujii, H.; Takeuchi, K.; Nakatani, K.; Ikejiri, M.; Ogawa, S.; Kubo, H.; Nagao, M.; Fujisawa, T. Whole-exome sequencing identification of novel DNAH5 mutations in a young patient with primary ciliary dyskinesia. Mol. Med. Report. 2016, 14, 5077–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, M.R.; Leigh, M.W.; Ostrowski, L.E.; Huang, L.; Carson, J.L.; Hazucha, M.J.; Yin, W.; Berg, J.S.; Davis, S.D.; Dell, S.D. Exome Sequencing Identifies Mutations in CCDC114 as a Cause of Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2013, 92, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Olbrich, H.; Fenske, L.; Mussaffi, H.; Horvath, J.; Fliegauf, M.; Kuhl, H.; Baktai, G.; Peterffy, E.; Chodhari, R.; et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008, 83, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gong, P.; Wen, J. Clinical and genetic analysis of a family with Kartagener syndrome caused by novel DNAH5 mutations. J. Assist. Reprod. Genet. 2017, 34, 275–281. [Google Scholar] [CrossRef]
- Davis, S.D.; Ferkol, T.W.; Rosenfeld, M.; Lee, H.-S.; Dell, S.D.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Pittman, J.E.; Shapiro, A.J. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am. J. Respir. Crit. Care Med. 2015, 191, 316–324. [Google Scholar] [CrossRef]
- Mocz, G.; Gibbons, I.R. Model for the motor component of dynein heavy chain based on homology to the AAA family of oligomeric ATPases. Structure 2001, 9, 93–103. [Google Scholar] [CrossRef]
- King, S.M. Composition and Assembly of Axonemal Dyneins. In Dyneins; King, S.M., Ed.; Academic Press: Boston, MA, USA, 2012; pp. 208–243. [Google Scholar] [CrossRef]
- Hung, M.-C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [Green Version]
- Ibañez-Tallon, I.; Gorokhova, S.; Heintz, N. Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum. Mol. Genet. 2002, 11, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, P.; Ferreira, R.R.; Guerrero, A.; Pintado, P.; Tavares, B.; Amaro, J.; Smith, A.A.; Montenegro-Johnson, T.; Smith, D.J.; Lopes, S.S. Left-Right Organizer Flow Dynamics: How Much Cilia Activity Reliably Yields Laterality? Dev. Cell 2014, 29, 716–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; O’Neal, W.K.; Randell, S.H.; Blackburn, K.; Moyer, M.B.; Boucher, R.C.; Ostrowski, L.E. Identification of Dynein Heavy Chain 7 as an Inner Arm Component of Human Cilia That Is Synthesized but Not Assembled in a Case of Primary Ciliary Dyskinesia. J. Biol. Chem. 2002, 277, 17906–17915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redwine, W.B.; Hernandez-Lopez, R.; Zou, S.; Huang, J.; Reck-Peterson, S.L.; Leschziner, A.E. Structural Basis for Microtubule Binding and Release by Dynein. Science 2012, 337, 1532–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonce, M.P.; Tikhonenko, I. Functional elements within the dynein microtubule-binding domain. Mol. Biol. Cell 2000, 11, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, I.R.; Garbarino, J.E.; Tan, C.E.; Reck-Peterson, S.L.; Vale, R.D.; Carter, A.P. The Affinity of the Dynein Microtubule-binding Domain Is Modulated by the Conformation of Its Coiled-coil Stalk. J. Biol. Chem. 2005, 280, 23960–23965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, M.J.; Russell, R.B. Amino Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; Barnes, M.R., Gray, I.C., Eds.; John Wiley and Sons, Ltd.: New York, NY, USA, 2003; pp. 289–316. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Castel, S.E.; Cervera, A.; Mohammadi, P.; Aguet, F.; Reverter, F.; Wolman, A.; Guigo, R.; Iossifov, I.; Vasileva, A.; Lappalainen, T. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat. Genet. 2018, 50, 1327. [Google Scholar] [CrossRef]
- Ruzycki, P.A.; Tran, N.M.; Kefalov, V.J.; Kolesnikov, A.V.; Chen, S. Graded gene expression changes determine phenotype severity in mouse models of CRX-associated retinopathies. Genome Biol. 2015, 16, 171. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilbault, C.; Saeed, Z.; Downey, G.P.; Radzioch, D. Cystic Fibrosis Mouse Models. Am. J. Respir. Cell Mol. Biol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gene | Variant Description | Freq.* | Variant Origin |
|---|---|---|---|---|
| Patient-1 | CCDC40 | NG_029761.1(NM_017950.3): c.1989 + 1G > A; p.(=) | 0.0008% | P |
| NM_017950.3: c.2824_2825insCTGT; p.(Arg942Thrfs*57) | 0.0042% | M | ||
| Patient-2 | DNAH5 | NM_001369.2: c.4530del; p.(Asn1511Metfs*6) | New | M |
| NM_001369.2: c.6000C > A; p.(Tyr2000*) | New | P | ||
| DNAH7 | NM_018897.3: c.8209G > A; p.(Gly2737Ser) | 0.0008% | P | |
| NM_018897.3: c.11947C > T; p.(Arg3983Trp) | 0.8666% | M |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, R.; Barbosa, T.; Gales, L.; Oliveira, E.; Santos, R.; Oliveira, J.; Sousa, M. Clinical and Genetic Analysis of Children with Kartagener Syndrome. Cells 2019, 8, 900. https://doi.org/10.3390/cells8080900
Pereira R, Barbosa T, Gales L, Oliveira E, Santos R, Oliveira J, Sousa M. Clinical and Genetic Analysis of Children with Kartagener Syndrome. Cells. 2019; 8(8):900. https://doi.org/10.3390/cells8080900
Chicago/Turabian StylePereira, Rute, Telma Barbosa, Luís Gales, Elsa Oliveira, Rosário Santos, Jorge Oliveira, and Mário Sousa. 2019. "Clinical and Genetic Analysis of Children with Kartagener Syndrome" Cells 8, no. 8: 900. https://doi.org/10.3390/cells8080900
APA StylePereira, R., Barbosa, T., Gales, L., Oliveira, E., Santos, R., Oliveira, J., & Sousa, M. (2019). Clinical and Genetic Analysis of Children with Kartagener Syndrome. Cells, 8(8), 900. https://doi.org/10.3390/cells8080900