1. Introduction
Fibroblast growth factors (FGFs) form a family of signaling proteins involved in diverse cellular processes such as cell proliferation, migration, angiogenesis, embryonic and fetal development [
1,
2]. FGFs interact with specific transmembrane receptor tyrosine kinases (RTKs), FGF receptors (FGFRs), which include four prevalent types (FGFR1–4) and one lacking an intracellular kinase domain (FGFR5) [
3]. FGF-FGFR interaction stimulates receptor dimerization and activation of receptor tyrosine kinases, initiating multiple signal transduction pathways. The complexity of FGF-induced signaling requires precise spatial and temporal regulation, as well as the control of the signal strength [
4,
5]. Regulatory mechanisms of FGFR signaling are not yet fully understood, nevertheless a growing number of proteins specifically affecting the FGF ligands and FGF-induced signaling cascades have been identified [
2]. One of them is integrin α
vβ
3 reported to bind directly to fibroblast growth factor 1 (FGF1) and influence its downstream signals [
6,
7].
Many reports point to the importance of integrin-growth factor receptors crosstalk [
8,
9]. Integrins are able to stimulate many types of RTKs, leading to the promotion of cancer growth and progression [
10]. Accordingly, Takada and coworkers demonstrated that integrin α
vβ
3 may affect the cellular response induced by FGF1 [
6]. The integrin α
vβ
3 binding site on FGF1 was identified as distinct from the receptor binding region and close to the heparin-binding site, with mostly positively charged residues responsible for the interaction [
6]. FGF1 mutant (R35E) deficient in integrin binding exhibits impaired biological function, as shown both in vitro and in vivo [
7,
11]. R35E variant was found to be unable to stimulate cell proliferation and migration [
7], and to suppress FGF1-dependent tumorigenesis in animal models [
11].
On the other hand, even a single amino acid substitution, especially a charge-reversing mutation, as in the case of R35E in FGF1, can lead to a change in protein stability [
12,
13]. Thus, the lack of prolonged cellular response (mitogenesis), coupled with the unchanged affinity for FGF receptors and the ability to induce downstream signaling (short-term response typical for unstable FGFs), suggested to us that R35E FGF1 mutant may exhibit reduced stability. Since the stabilization of FGF ligands with heparan sulfates or specific mutations [
14], as well as the decreasing temperature of cell culture experiments restores the ability of unstable FGFs to induce cellular response [
15], we have decided to verify if protein stability plays a role in the reported effects of R35E FGF1 variant on the cellular outcome. To this end, we took advantage of previously described stabilizing mutations, Q40P, S47I, and H93G, which increase the thermodynamic stability and proteolytic resistance of FGF1, but have no impact on its binding to FGFRs or any other properties [
14,
16]. Here, we present a detailed analysis of integrin-binding deficient mutants of FGF1 with diverse stability and biological activities.
2. Materials and Methods
2.1. Construction, Expression and Purification of Recombinant Proteins
The coding sequence of truncated form of human FGF1 (Ala-FGF1
21–154) cloned in the pET-3c vector was used. R35E mutation [
6] and stabilizing mutations (Q40P, S47I, H93G) [
14] were introduced with QuikChange™ site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using mutagenic primers (Genomed, Warsaw, Poland). Recombinant FGF1 variants were expressed and purified as described before [
17]. Since we used the truncated form of FGF1, we applied a numbering system according to Gimenez-Gallego et al. (1–140) [
18]. The full length extracellular fragment of FGFR1-IIIc fused to the Fc (FGFR1c-Fc) was produced in CHO cells and purified using Protein A Sepharose (GE Healthcare, Piscataway, NJ, USA), as described previously by our group [
19]. Purity and identity of protein samples were confirmed by SDS-PAGE and mass spectrometry.
2.2. Thermal Stability Measurements
To determine the stability of FGF1 variants, thermal denaturation curves were acquired following the changes in the ellipticity signal at 227 nm. Measurements were performed at a protein concentration 4 × 10
−6 M in the presence of 0.7 M GdmCl in 25 mM sodium phosphate buffer, pH 7.3 in a cuvette of 10 mm path length, using a scan rate of 0.25 °C/min, as described previously [
16]. Thermodynamic data were analyzed using PeakFit software (Jandel Scientific Software, San Jose, CA, USA), assuming a two-state reversible equilibrium transition.
2.3. Analysis of FGF1 Variants Interaction with FGFR1
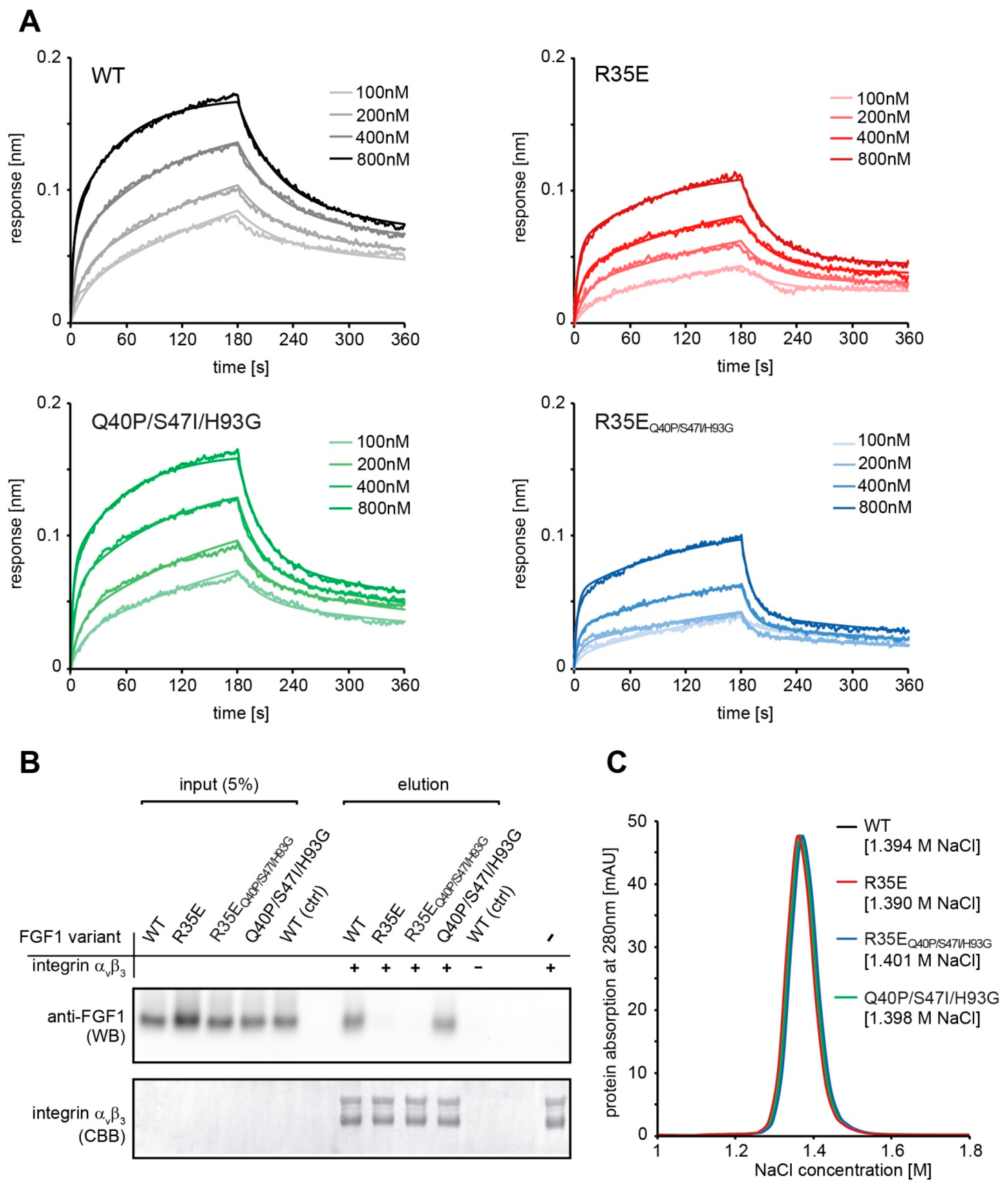
Binding kinetics were measured using bio-layer interferometry (BLI) techniques with ForteBio Octet K2 (Pall ForteBio, San Jose, CA, USA). All steps of interaction studies were performed at 25 °C with shaking at 1000 rpm in PBS buffer. The 96-microwell plates were filled with 200 μL of buffer or samples and incubated for 10 min at 25 °C prior measurements for system stabilization. Protein A biosensors were hydrated in PBS buffer for 10 min and then loaded with FGFR1c-Fc (20 μg/mL for 600 s). Association and dissociation phases (180 s each) were monitored at various concentrations of FGF1 proteins ranging from 100 to 800 nM. As a control a reference sensor without FGFR1c-Fc was used. Kinetic parameters were determined by global fitting with the 2:1 “heterogeneous ligand” model and steady state analysis (averaged response values from 170 to 180 s) from three independent experiments with ForteBio Data Analysis 11.0 software (Pall ForteBio, San Jose, CA, USA).
2.4. Pull Down Assay
To confirm the interaction of FGF1 variants with integrin αvβ3 1 µg of His-tagged integrin αvβ3 (R&D Systems, Minneapolis, MN, USA) was bound to Co2+-NTA resin (Thermo Fisher Scientific, Waltham, MA, USA) (50 µL) for 30 min at ambient temperature and washed three times with HBS buffer (10 mM HEPES, 150 mM NaCl, 1 mM Mn2+, pH 7.4). Next, FGF1 variants in HBS buffer were added at concentration of 50 ng/mL (200 µL) and incubated at 4 °C overnight. Resin was washed three times with HBS buffer and bound proteins were eluted with SDS-loading buffer. Eluates were analyzed by western blotting with anti-FGF1 antibody. The presence of equal amounts of integrin αvβ3 in each sample was confirmed by Coomassie Brilliant Blue staining. As a control for unspecific FGF1 interaction, a resin sample without immobilized integrin αvβ3 was used.
2.5. Heparin Affinity Measurements
FGF1 affinity for heparin was evaluated using a HiTrap Heparin-Sepharose column (GE Healthcare) and a linear NaCl gradient (in 25 mM Tris-HCl, pH 7.4) in a range from 0.1 M to 2 M using AKTA Explorer FPLC system (GE Healthcare).
2.6. Cell Culture
Mouse embryo fibroblast cells (NIH 3T3) obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) were cultivated in DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin). Murine pro B cell line (BaF3) transfected with FGFR1-IIIc (BaF3-R1c), was a kind gift from Dr. David Ornitz from the Department of Developmental Biology, Washington University School of Medicine. The cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific) supplemented with 10% newborn bovine calf serum (Thermo Fisher Scientific), antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin), β-mercaptoethanol (50 nM) and mouse interleukin 3 (IL-3, PeproTech, Hamburg, Germany). Human osteosarcoma cell line, U2OS, stably transfected with FGFR1-IIIc (U2OS-R1) was provided by Dr. Ellen M. Haugsten from the Department of Molecular Cell Biology, Institute for Cancer Research (Oslo University Hospital). U2OS-R1 cells were grown in DMEM (Biowest) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin and 1 mg/mL geneticin).
2.7. Analysis of Signaling Pathways
NIH 3T3 cells were serum-starved for 6 h and stimulated for 15 min with 10 ng/mL of FGF1 variants in the presence of 10 U/mL heparin (Sigma-Aldrich, St. Louis, MO, USA). Cells were then washed with PBS, lysed with SDS sample buffer and sonicated. Total cell lysates were subjected to SDS-PAGE separation and western blotting analysis with mouse anti-phospho-FGFR (Tyr653/Tyr654) antibody (p-FGFR) (#3476), mouse anti-phospho-p44/42 (Thr202/Tyr204) MAP kinase antibody (p-Erk1/2) (#9106) and rabbit anti-p44/42 MAP kinase antibody (Erk1/2) (#9102) from Cell Signaling Technology (Danvers, MA, USA), anti-phospho-PLCγ (Tyr783) antibody (sc-12943) (p-PLCγ) from Santa Cruz Biotechnology (Dallas, TX, USA). Mouse anti-Hsp90 antibody (610418) from BD Transduction Laboratory (San Jose, CA, USA) was used as a control of equal sample loading. Horseradish peroxidase-conjugated secondary antibodies from Jackson ImmunoResearch (West Grove, PA, USA) and chemiluminescent substrate (Thermo Fisher Scientific) were used for visualization in the ChemiDoc station (BioRad, Hercules, CA, USA). ImageLab software (version 6.0.1) from Biorad was used to quantify the intensities of the bands of interest. The intensity of bands corresponding to phospho-proteins (p-FGFR, p-Erk1/2, p-PLCγ) was normalized to the intensity of Hsp90 bands and then expressed as a fraction of the response observed for the wild-type FGF1.
2.8. Microscopy
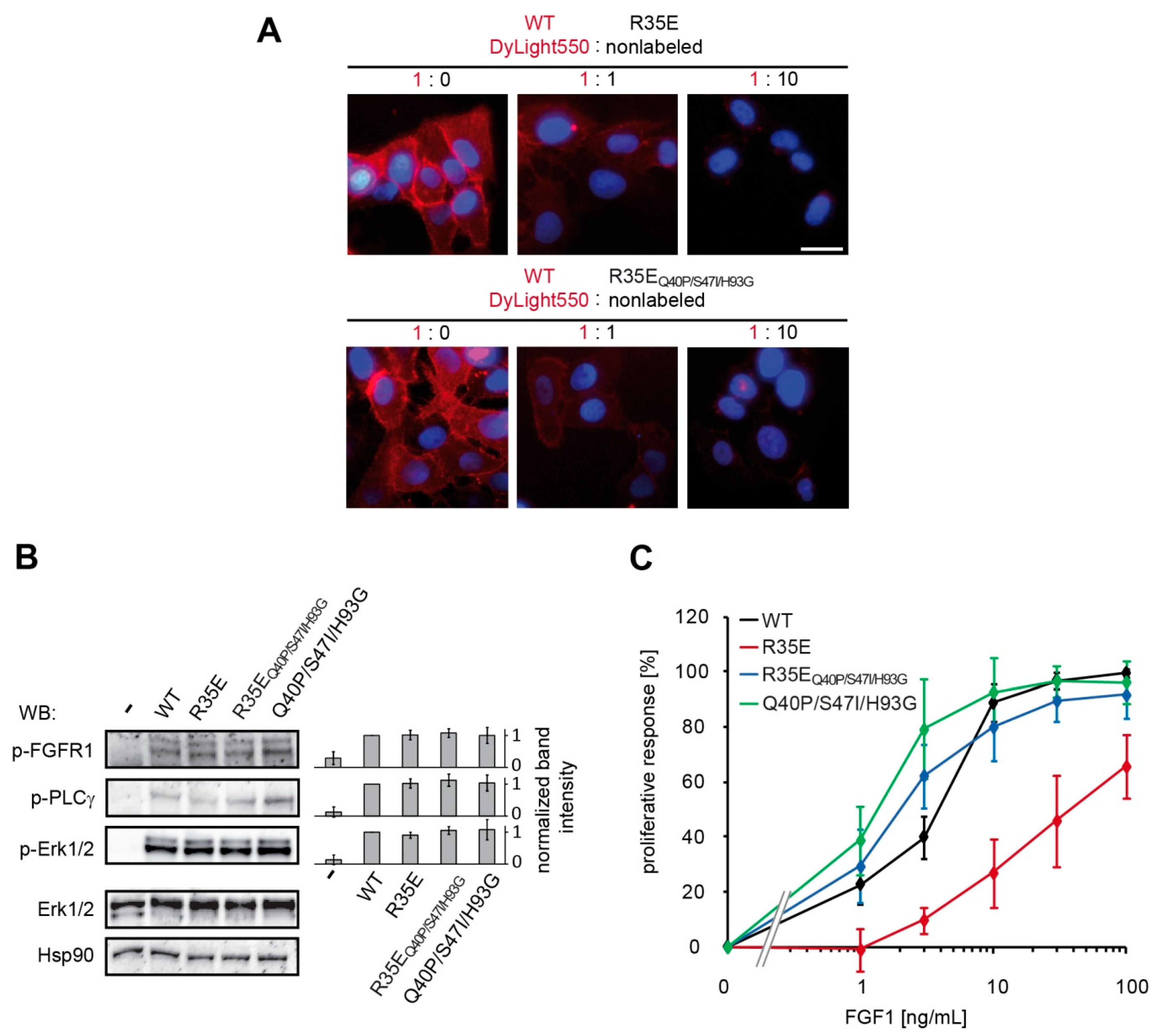
U2OS-R1 cells were incubated with 10 µg/mL of DyLight550-labeled wild type FGF1 alone or in combination with non-labeled FGF1 R35E mutant in 1:1 and 1:10 molar ratio, for 1 h on ice. The cells were subsequently washed with PBS and fixed with 4% paraformaldehyde (20 min, at 4 °C). Next, the cells were washed with PBS and nuclei were stained for 10 min with NucBlue Live dye (Thermo Fisher Scientific). Wide-field fluorescence microscopy was carried out using a Zeiss Axio Observer Z1 microscope (Zeiss, Oberkochen, Germany). Images were taken using LD-Plan-Neofluar 40×/0.6 Korr M27 objective and Axiocam 503 camera. FGF1-DyLight550 signal was visualized with 540/552-nm bandpass excitation filter and 575/640-nm bandpass emission filter. NucBlue Live signal was visualized with 335/383-nm bandpass excitation filter and 420/470-nm emission filter. Image analysis was carried out using ZEN 2.3 (Zeiss, Oberkochen, Germany) and ImageJ (NIH, Bethesda, MD, USA).
2.9. FGF1-Induced Proliferative Response
24-h-starved NIH 3T3 and BaF3-R1c cells grown on the 96-well plates were treated with increasing concentrations (1, 3, 10, 30, and 100 ng/mL) of FGF1 variants in the presence of heparin (10 U/mL or 0.36 U/mL). After 48 h, cell viability was determined by addition of AlamarBlue Cell Viability Reagent (Thermo Fisher Scientific) for 4 h. The emission of fluorescent reduced form of the dye was measured at 590 nm upon excitation at 560 nm on Infinite M1000 PRO plate reader (Tecan, Männedorf, Switzerland). The data were normalized and expressed as a percentage of maximal response observed for the wild-type.
2.10. Protein Stability in Cell-Conditioned Medium
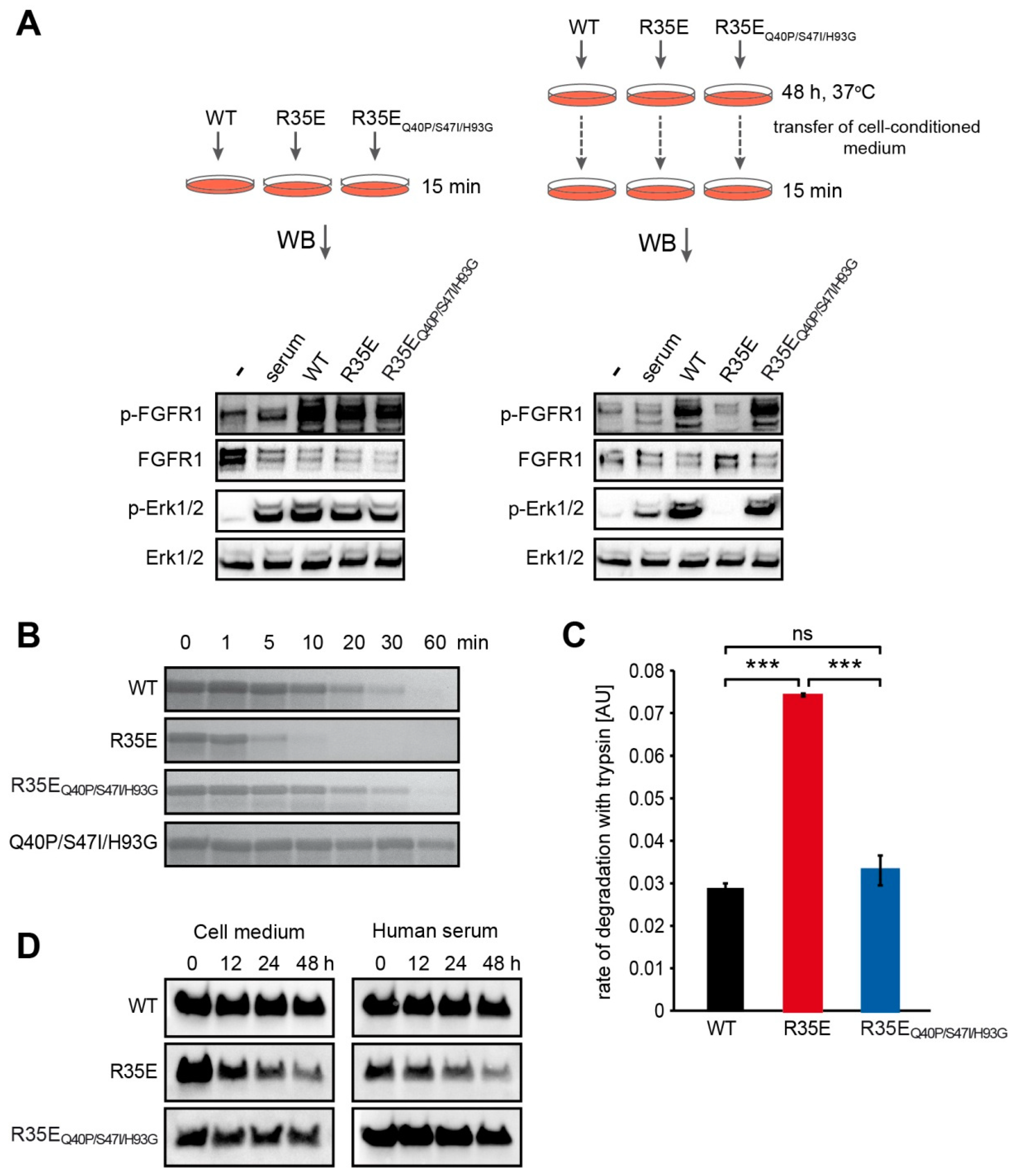
NIH 3T3 cells were starved in a serum-free medium for 24 h. FGF1 variants were added to the medium to a final concentration of 50 ng/mL, in the presence of 10 U/mL heparin, and incubated with cells for 48 h at 37 °C. Then, conditioned medium was aspirated and added to the new set of 24-h-starved NIH 3T3 cells. Activation of cell signaling cascades was used as a sensitive readout of FGF1 proteins degradation. Freshly prepared 50 ng/mL FGF1 solutions served as positive controls. Cells were incubated for 15 min at 37 °C and lysed with SDS sample buffer. The total cell lysate was separated by SDS-PAGE and analyzed by western blotting.
2.11. Limited Proteolysis with Trypsin
FGF1 variants (200 µg/mL) were subjected to the limited proteolysis with trypsin (15 µg/mL) (Worthington Biochemical Corporation, Lakewood, NJ, USA), in reaction buffer (100 mM TrisHCl, 20 mM CaCl2, pH 8.3) at 37 °C. Proteolysis was terminated by the addition of SDS sample buffer at different time points. Proteolytic fragments were visualized by SDS-PAGE and Coomassie Brilliant Blue staining.
For the quantitative measurement of FGF1 variants susceptibility to proteolysis, the fluorescence of tryptophan was monitored as a readout of degraded protein fraction. Proteolytic reactions were carried out with 80 µg/mL of FGF1, 6 µg/mL of trypsin in the reaction buffer at 37 °C. Fluorescence emission measurements at 353 nm upon excitation at 280 nm were performed using FP-750 spectrofluorimeter (Jasco, Easton, MD, USA). The rate of proteolysis was determined as a slope of the linear regression of tryptophane fluorescence signal versus time during the first 300 s.
2.12. Half-Life Analysis
There were 24-h serum starved NIH 3T3 cells treated with FGF1 variants (100 ng/mL) in the presence of 10 U/mL heparin and incubated at 37 °C. At different time points (0, 12, 24 and 48 h), medium was aspirated and then incubated with 30 μL of Heparin-Sepharose resin (GE Healthcare) for 2 h at 4 °C. In addition, FGF1 variants (500 ng/mL) were incubated at 37 °C in human serum (H4522, Sigma-Aldrich) in the presence of 10 U/mL heparin. At different time points (0, 12, 24 and 48 h), 200 μL of sample was aspirated and then incubated with 30 μL of Heparin-Sepharose resin for 2 h at 4 °C.
In each case, after washing twice with PBS, bound proteins were eluted from the resin with SDS sample buffer and analyzed by western blotting with rabbit anti-FGF1 antibody (#3139) from Cell Signaling Technology (Danvers). ImageLab software (version 6.0.1) from Biorad was used to quantify the intensities of the bands of interest and the half-life values were derived from single exponential decay equation with SigmaPlot 12 software (Systat Software).
2.13. Anti-Apoptotic Assay
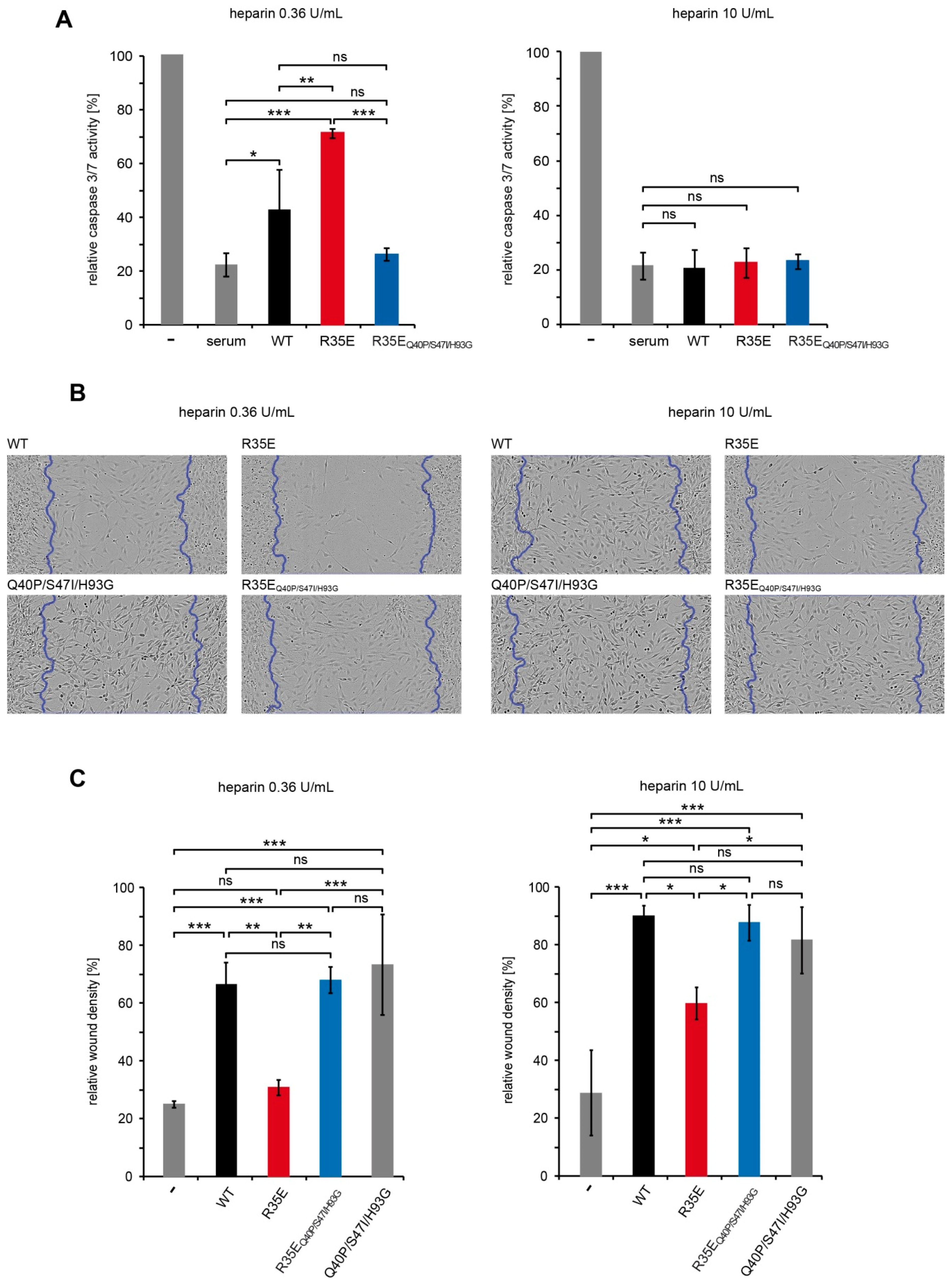
There were 24-h serum starved NIH 3T3 cells treated for 16 h with FGF1 variants (50 ng/mL) or fetal calf serum (10%) in the presence of 0.36 U/mL or 10 U/mL heparin. Next, the caspase-3/7 activity and the cell viability were determined using ApoLive-Glo Multiplex Assay (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The ratio of the caspase-3/7 activity to the cell viability was normalized towards the untreated cells, and denoted as relative caspase-3/7 activity.
2.14. Scratch Wound Assay
Cell migration was measured with the IncuCyte Scratch Wound Assay (Essen BioScience, Ann Arbor, MI, USA). NIH 3T3 cells seeded on a 96-well ImageLock plate were serum-starved for 24 h and scratched with WoundMaker (Essen BioScience). Then, the cells were stimulated with FGF1 variants (10 ng/mL) in the presence of heparin (10 U/mL or 0.36 U/mL) for 48 h. Every 2 h, images of the wounds were automatically acquired. The data were analyzed with respect to the spatial cell density in the wound area using the IncuCyte software package.
2.15. Statistical Analysis
For statistical analyses, one-way analysis of variance (ANOVA) with Tukey’s posttest was applied using SigmaPlot 12 software (Systat Software). p < 0.05 was considered statistically significant.
4. Discussion
The R35E (R50E in full-length numbering system) variant of FGF1 designed and described by Takada and colleagues is unable to stimulate cell proliferation, even though it initiates FGFR-dependent signaling [
6]. This mutant was found to be defective in integrin α
vβ
3 binding and therefore FGF1-integrin interaction was considered to be critical for FGF1 mitogenic response [
6,
7].
We reasoned that the observed lack of long-term cellular response to R35E mutant, together with its full functionality for the short period of time reflected in the FGF receptor activation, could be a result of decreased half-life of the protein. To verify this hypothesis, we determined the thermal stability of this mutant (
Figure 1) and its susceptibility to degradation (
Figure 4). We found that R35E variant is significantly less stable than the wild-type FGF1, with denaturation temperature lowered by almost 6 degrees. The thermodynamic instability promoted FGF1 degradation and reduced half-life, which prevents a long-term response, such as cell proliferation. This is in agreement with previous observation that a maximal mitogenic activity requires the presence of active FGF1 for over 12 h [
25,
26]. Further, we observed a strong correlation between the stability/proteolytic degradation of FGF1 mutants and their biological activity [
14,
17]. Accordingly, here we showed that the increase in the thermal stability of R35E mutant by the introduction of three well characterized stabilizing mutations (Q40P, S47I, H93G) significantly reduced its susceptibility to proteolysis by trypsin and prolonged its half-life (
Figure 4,
Figure S2), resulting in restoration of its mitogenic potential in NIH 3T3 as well as BaF3-R1c cells (
Figure 3,
Figure S1). We also observed the activity rescue for the stabilized variant R35E
Q40P/S47I/H93G in the migration assay and in the anti-apoptotic response (
Figure 5). Thus, these results indicate that the low stability, rather than the defective integrin binding, is a major cause for the lack of mitogenic activity of R35E mutant.
The same effects were observed in the case of STAT3 in autosomal dominant hyper-IgE syndrome, where destabilizing mutations decreased the half-life of the protein leading to its dysfunction [
27]. Similarly, loss of function of tumor suppressor, merlin, causing neurofibromatosis type 2, was directly correlated with a reduction in protein half-life and its increased degradation. Moreover, it was shown that merlin mutants maintain intrinsic functional capacity [
28]. In the same manner, despite the low stability, FGF1 R35E variant was able to evoke short-term response, such as endocytosis, FGFR activation and initiation of downstream signaling. However, the incubation with cells, due to an elevated temperature and extracellular proteases’ action, deactivated the FGF1 R35E variant, while not affecting its stabilized version (R35E
Q40P/S47I/H93G) (
Figure 4).
Importantly, none of the stabilizing mutations affected the interaction of FGF1 with integrin α
vβ
3. The stabilized variant, R35E
Q40P/S47I/H93G, similarly to single R35E mutant, exhibit no affinity for recombinant integrin α
vβ
3. We also verified the ability of these two mutants to bind FGFR1 using BLI technique (
Figure 2). The slight decrease in the receptor affinity observed for the FGF1 variants containing R35E mutation can be explained by the involvement of Arg35 residue in the FGF1-FGFR1 interaction, as seen in the crystal structures of FGF1-FGFR1 complex [
29]. Side-chain of Arg35 forms van der Waals contacts with carbonyl carbon and Cβ of Glu162 of D2 domain of FGFR1.
Since Arg35 residue is positively charged and could participate in the FGF1 interaction with negatively charged heparin or cell-surface heparans, which strongly stabilize the FGF1-FGFR complex and protect FGF1 molecule against degradation [
26], we evaluated the affinity of R35E variant for heparin. We showed that the substitution to Glu did not change the elution profile of FGF1 from heparin column (
Figure 2). However, the presence of sugar moieties on the cell surface affected the proliferative potential of the R35E mutant, as can be seen in the comparative experiment in BaF3-R1c cells negative for heparan sulfate glycosaminoglycans [
22,
30] and NIH 3T3 fibroblasts expressing notable level of HSGAGs. At the low heparin concentration (0.36 U/mL, used previously in R35E variant studies [
6,
7]) in NIH 3T3 cells R35E variant exhibited some mitogenic activity, whereas in BaF3-R1c cells it did not evoke any proliferative response (
Figure S1). This discrepancy suggests that R35E variant can be stabilized and protected against the degradation by the surface heparans to certain extent, partially preserving its activity. Notably, we did not observe any difference in the mitogenic potential of the stabilized R35E
Q40P/S47I/H93G mutant in these two cell lines. We further confirmed that high concentration of heparin may protect the low stable R35E protein, similarly to structural changes causes by stabilizing mutations (
Figure 5). In wound healing experiments we observed higher migration-inducing activity of R35E variant at 10 U/mL heparin concentration than at 0.36 U/mL. Similarly, at low heparin concentration we found significant differences in the caspase activity in serum-starved cells between R35E variant and the wild-type, but when heparin was present at high concentration all tested variants exhibited the same anti-apoptotic properties. Again the stable R35E
Q40P/S47I/H93G variant was similarly active independently of heparin concentration. Thus, we showed that the protective effect of heparin can be substituted by the increase in the protein stability [
14].
Taking together, we have provided the explanation for the lack of mitogenic activity of R35E variant and suggested that the protein stability, rather than the integrin binding, is a driving force for the long-term response of FGF1. In light of our data the biological significance of the interaction between FGF/FGFR signaling system and integrins requires further in depth studies. We are convinced that our findings may facilitate the understanding how the stability of FGF proteins dictates specificity and duration of cellular response.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}