Parkin-Dependent Mitophagy Is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents and Antibodies
2.3. Stereotaxic Injection of AAV Vectors
2.4. Intracerebroventricular Injection of siRNA and Mdivi-1 Administration
2.5. MCAO Model in Rats
2.6. Cerebral Infarct Volume Evaluation
2.7. Assessment of Neurological Score
2.8. Western Blotting
2.9. Immunofluorescence and Confocal Microscopy
2.10. Electron Microscopy
2.11. HE Staining and Nissl Staining
2.12. Determination of Total Reactive Oxygen Species (ROS) in Tissue Levels
2.13. ELISA of IL-1β and IL-18 Activity
2.14. Statistical Analysis
3. Results
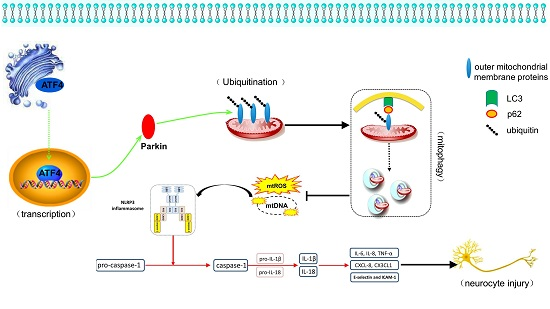
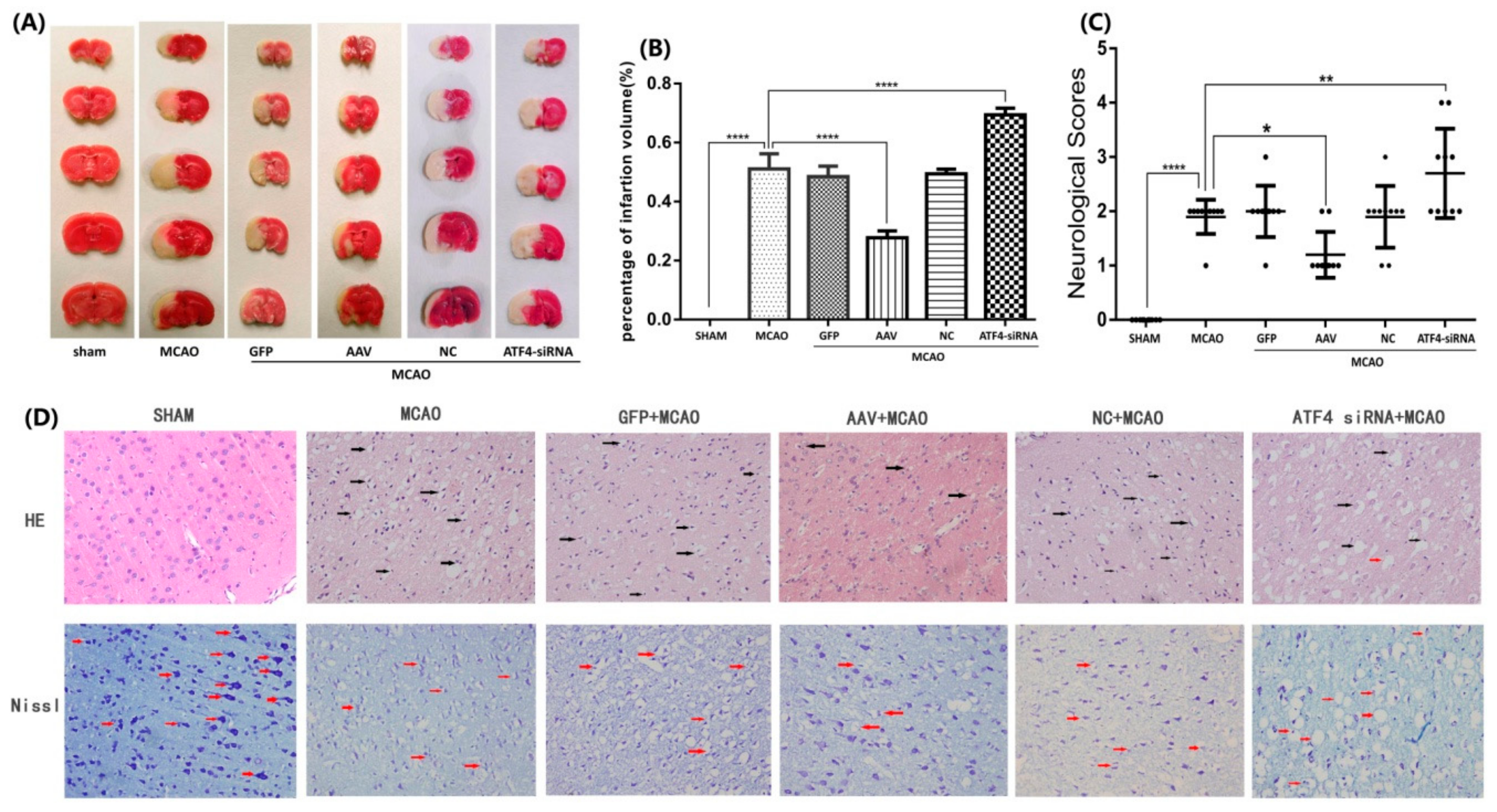
3.1. ATF4 Protects Against Cerebral Ischemia-Reperfusion Injury in Rats
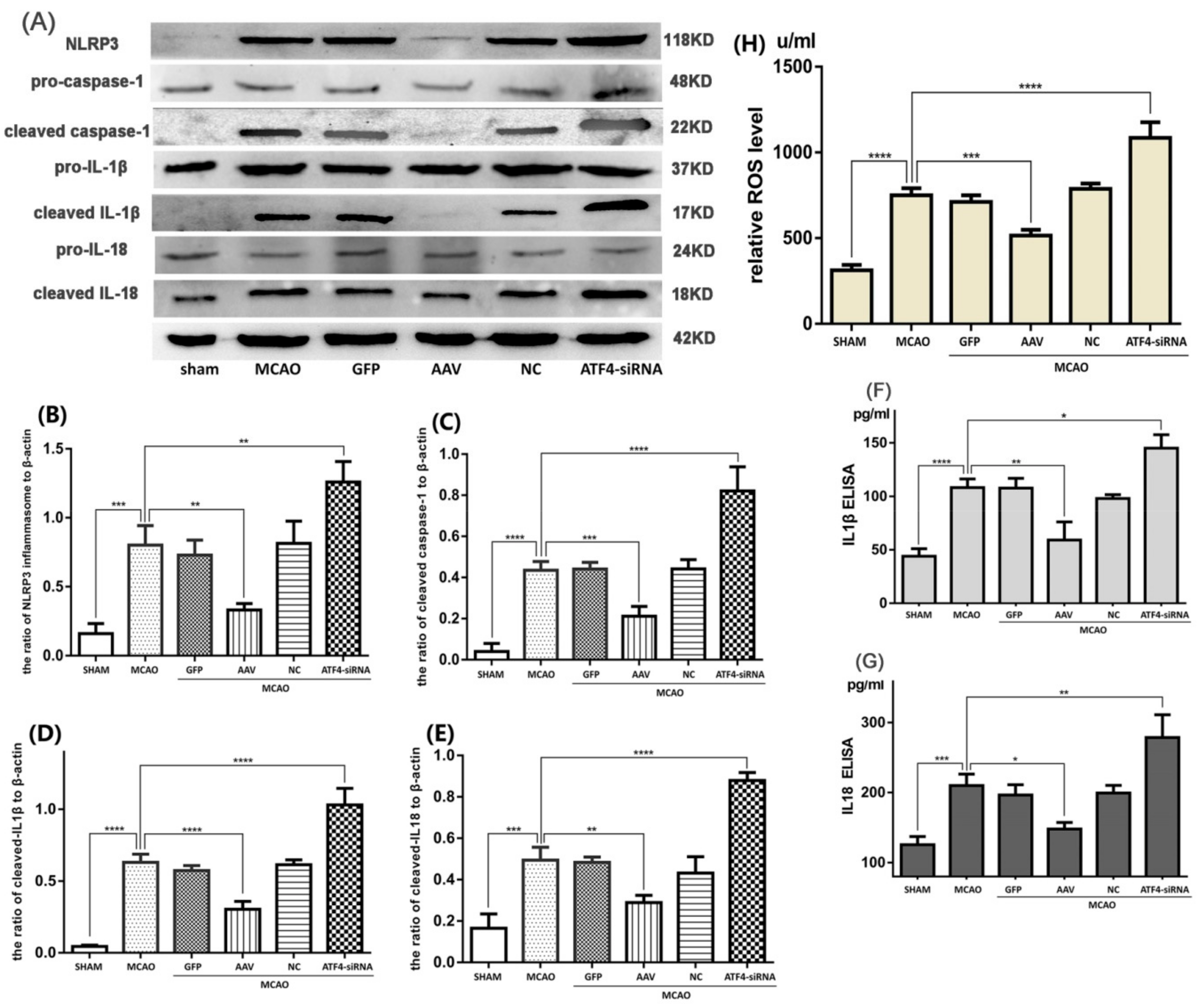
3.2. ATF4 Restrains NLRP3 Inflammasome-Mediated Inflammatory Response during Cerebral I/R Injury in Rats
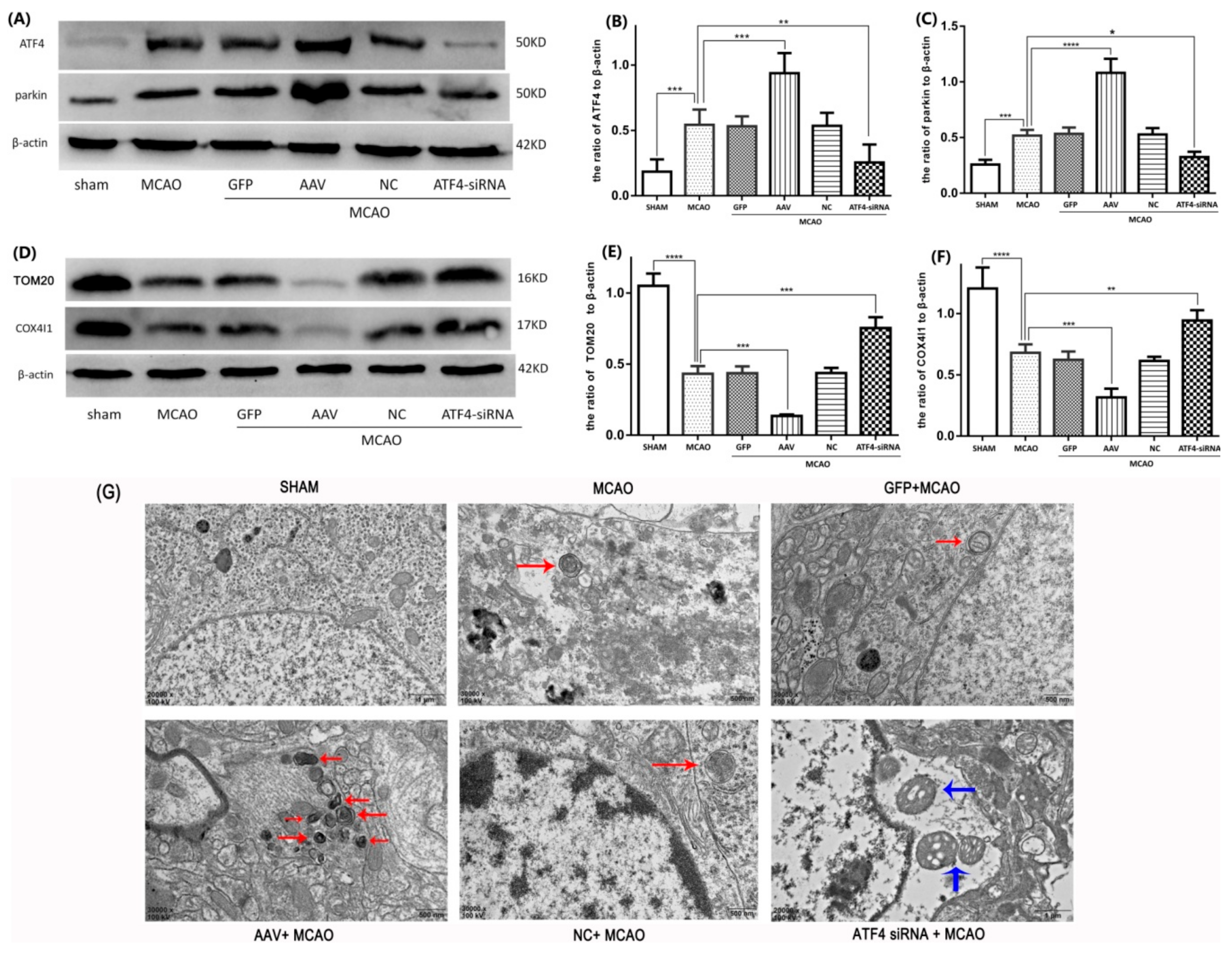
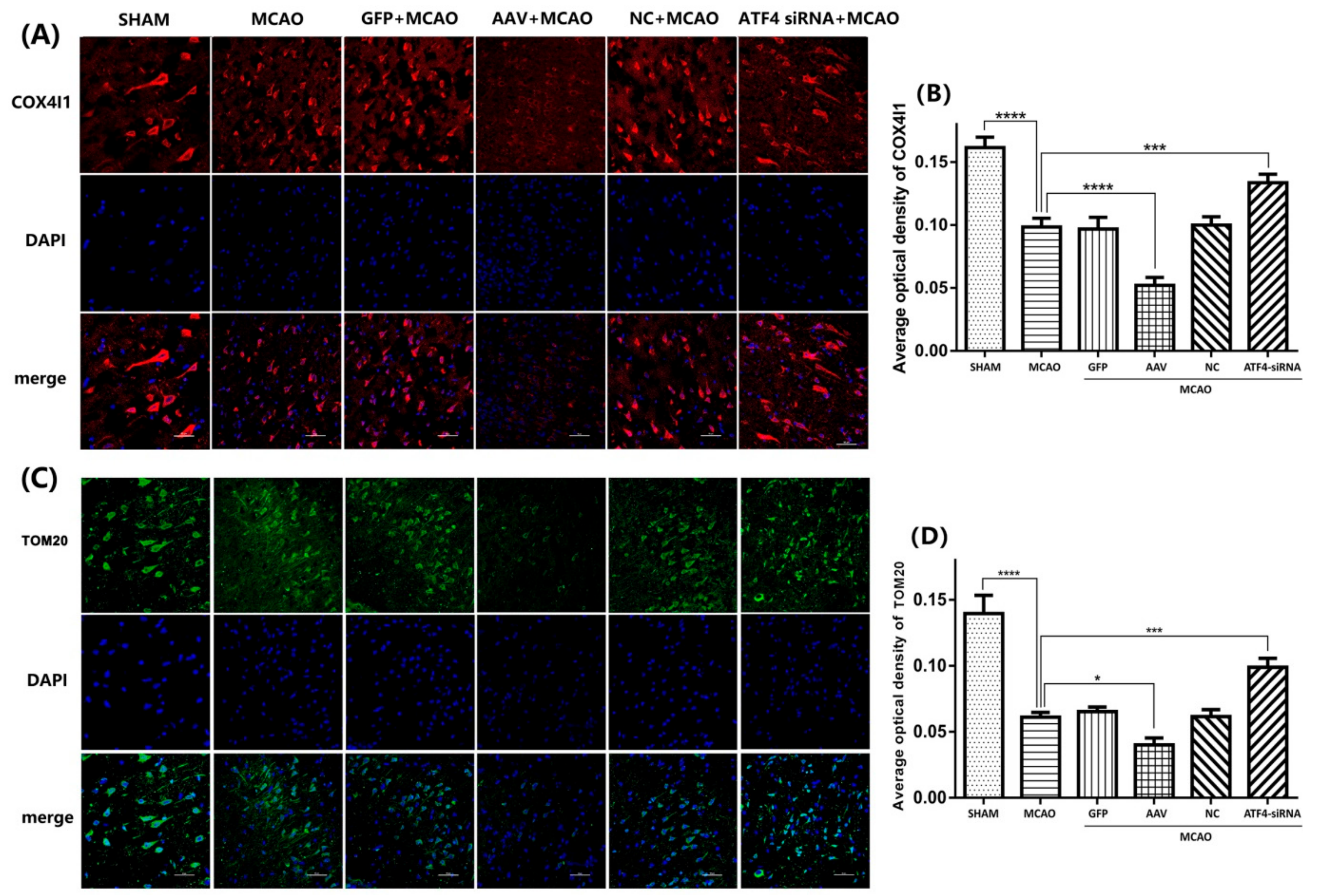
3.3. ATF4 Increases Parkin Expression and Mitophagy Activity
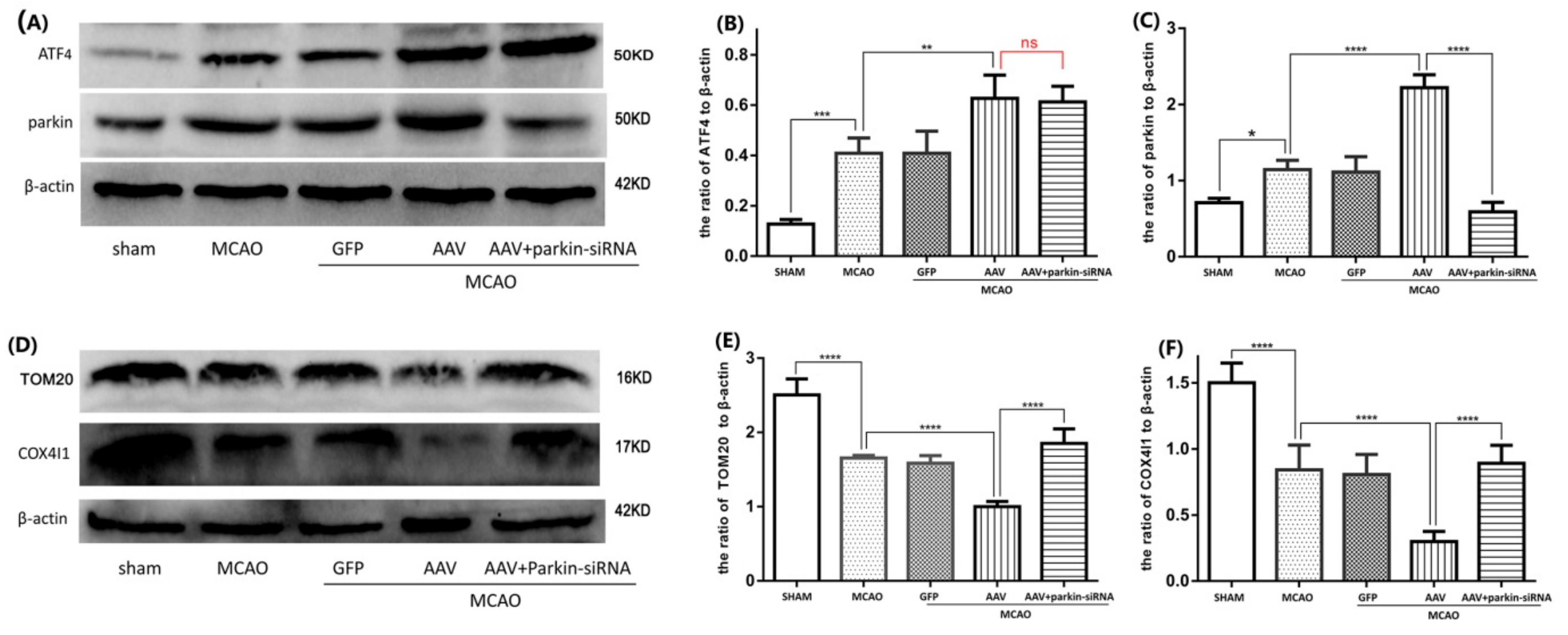
3.4. The ATF4-Mediated Regulation of Mitophagy Relies on Parkin Modulation
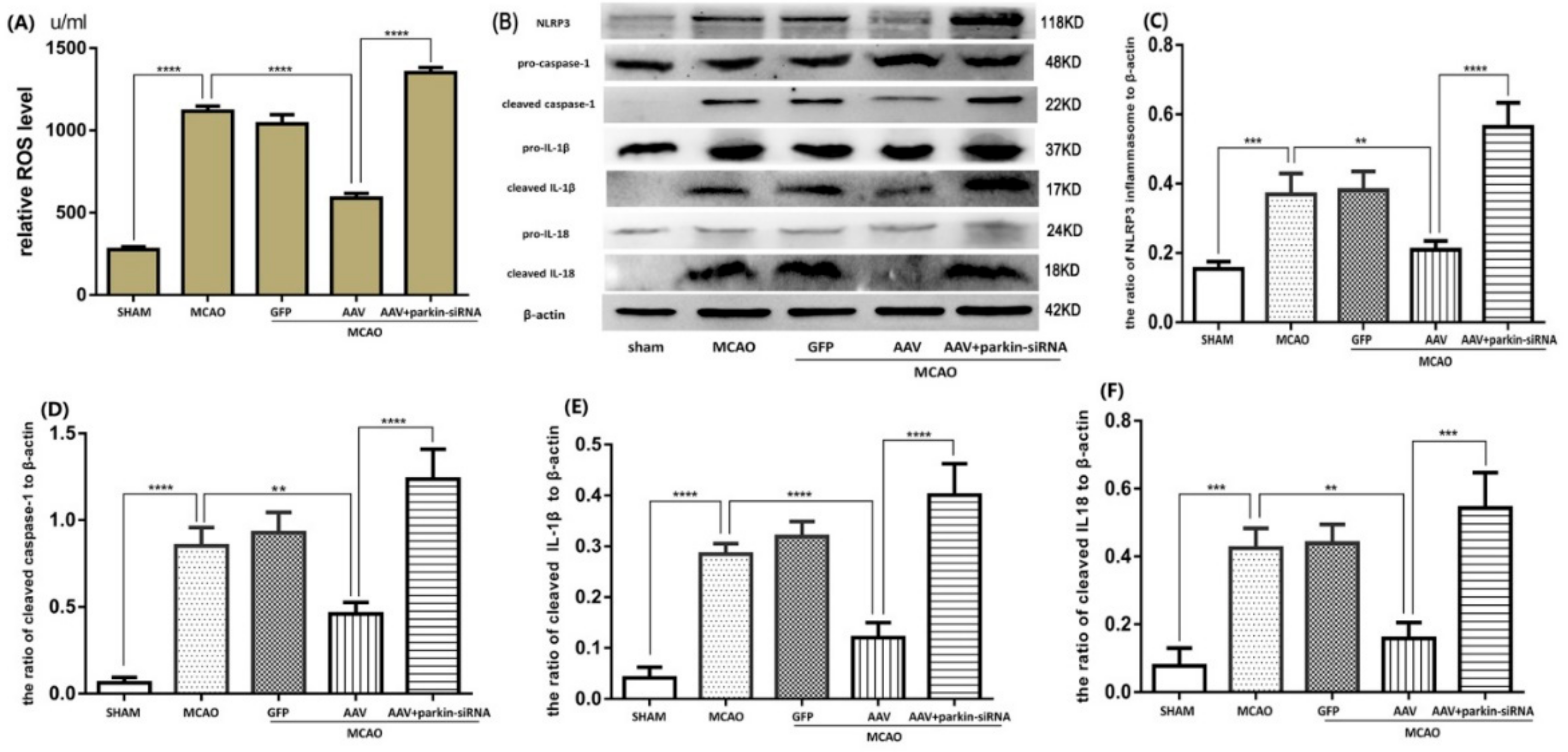
3.5. Crucial Role of Parkin in the ATF4-Mediated Inhibition of NLRP3 Inflammasome Activation
3.6. ATF4 Inhibits NLRP3 Inflammasome Activation through Mitophagy Induction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| I/R | Ischemia/reperfusion |
| MCAO | middle cerebral artery occlusion-reperfusion |
| siRNA | small interfering RNA |
| AAV | adeno-associated virus |
| GFP | Green fluorescent protein |
| NC | negative control siRNA |
| BSA | bovine serum albumin |
| ATF4 | Activating Transcription Factor 4 |
| COX4I1 | cytochrome c oxidase IV isoform 1 |
| TOM20 | translocase of outer mitochondrial membrane 20 |
| ROS | reactive oxygen species |
| NLRP3 | Nod-like receptor protein 3 |
| Mdivi-1 | mitochondrial division inhibitor-1 |
References
- David, Y.W.; Fann, S.Y.; Leea, S.; Manzaneroa, P.; Chunduri, C.G.; Sobey, C.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966. [Google Scholar]
- Wijitra, C.; Sarinthorn, T.; Piyarat, G.; Chainarong, T.; Jinatta, J.; Jiraporn, T. Neuroprotection of agomelatine against cerebral ischemia/reperfusion injury through an antiapoptotic pathway in rat. Neurochem. Int. 2017, 102, 114–122. [Google Scholar]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.; Wang, X.; Hua, W. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, P.; Wan, Y.; Geng, S.; He, Y.; Feng, B.; Ye, Z.; Zhou, D.; Li, D.; Wei, H.; Li, H. Salvianolic Acids for Injection (SAFI) suppresses inflammatory responses in activated microglia to attenuate brain damage in focal cerebral ischemia. J. Ethnopharmacol. 2017, 198, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Arjun, S.; Bhakta, P.G.; Kyu, S.C.; Se, J.J.; Oh, W.K.; Dae, S.J.; Sun, Y.K.; Jong, H.R.; Ji, W.C. Eupatilin exerts neuroprotective effects in mice with transient focal cerebral ischemia by reducing microglial activation. PLoS ONE 2017, 12, e0171479. [Google Scholar]
- Schwartz, M. Macrophages and microglia in central nervous system injury: Are they helpful or harmful? J. Cereb. Blood Flow Metab. 2003, 23, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Kenichi, S.; Timothy, R.C.; Justin, K.; Jargalsaikhan, D.; Norika, C.; Chen, S.V.; Krishnan, R.; Andrea, J.W.; Laurent, V.; David, M.O. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Justine, M.A.; Xia, M.; Zhang, Y.; Krishna, M.B.; Li, P. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar]
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef]
- Amna, A.; Khadija, E.; Elodie, B.; Bertrand, B.; Mohamed-Naceur, S.; Berthold, B.; Thomas, S.; Dominique, C.; Mustapha, R. Inhibition of the Inflammasome NLRP3 by Arglabin Attenuates Inflammation, Protects Pancreatic beta-Cells from Apoptosis, and Prevents Type 2 Diabetes Mellitus Development in ApoE2Ki Mice on a Chronic High-Fat Diet. J. Pharmacol. Exp. Ther. 2016, 357, 487–494. [Google Scholar]
- Mélanie, B.; Grégoire, M.; Valentin, D.; Fanny, C.; Angélique, C.; Frédérique, V.; Wilfrid, B.; Benoit, S.; Bernhard, R.; Jean, L.C. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar]
- Zahra, H.; Fatemeh, S.; Bahman, R.; Amirhossein, S.; Aria, M.; Hamed, M. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar]
- Yin, J.; Zhao, F.; Jeremy, E.C.; Jacob, F.; William, L.K.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.; Lee, S.-Y.; Manzanero, S.; Tang, S.-C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.-L.; Thundyil, J. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wan, M.; Zhang, J.; Cai, Q.; Lu, D.; Li, Y.; Dong, Y.; Zhao, T.; Chen, H. The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. Int. Immunopharmacol. 2016, 40, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Moyzis, A.G.; Sadoshima, J.; Gustafsson, A.B. Mending a broken heart: The role of mitophagy in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H183–H192. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, Y.; La, S. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis. 2011, 43, 46–51. [Google Scholar] [CrossRef]
- Danielle, A.S.; Jennifer, M.; Hao, L.; Chen, X.; Sun, N.; Tara, D.F.; Jonathon, L.B.; Li, Y.; Zhang, Z.; Derek, P.N. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar]
- Zhao, Y.; Huang, S.; Liu, J.; Wu, X.; Zhou, S.; Dai, K.; Kou, Y. Mitophagy Contributes to the Pathogenesis of Inflammatory Diseases. Inflammation 2018, 41, 1590–1600. [Google Scholar] [CrossRef]
- Cui, C.; Chen, S.; Qiao, J.; Qing, L.; Wang, L.; He, T.; Wang, C.; Liu, F.; Gong, L.; Chen, L. PINK1-Parkin alleviates metabolic stress induced by obesity in adipose tissue and in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2018, 498, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.Z.; Macleod, K.F. In Brief: Mitophagy: Mechanisms and role in human disease. J. Pathol. 2016, 240, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Atsushi, U.; Elsa, S.L.; Liang, S.; Shabnam, S.; Jerry, W.; He, F.; Daniela, B.; Guy, P.; Syed, R.A. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liu, L.; Zhu, Y.; Chen, Q. Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 2013, 319, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, J.; John, F.C.; Cristina, M.; David, S.; Lloyd, A.G.; Oren, A.L. ATF4 protects against neuronal death in cellular Parkinson’s disease models by maintaining levels of parkin. J. Neurosci. 2013, 33, 2398–2407. [Google Scholar] [CrossRef]
- Bouman, L.; Schlierf, A.K.; Shan, J.; Deinlein, J.K.; Galehdar, V.P.; Patenge, D.B. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef]
- Wu, L.; Luo, N.; Zhao, H.; Gao, Q.; Lu, J.; Pan, Y.; Shi, J.; Tian, Y.; Zhang, Y. Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 2014, 1549, 52–62. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef]
- Wang, P.; Liang, J.; Li, Y.; Li, J.; Yang, X.; Zhang, X.; Han, S.; Li, S.; Li, J. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem. Res. 2014, 39, 1279–1291. [Google Scholar] [CrossRef]
- Wang, P.; Xu, T.; Guan, Y.; Tian, W.; Benoit, V.; Rui, Y.; Zhai, Q.; Su, D.; Miao, C. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Wang, Y.; Hou, Y.; LI, L.; ZHAO, J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 2017, 50, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Denise, P.A.; Juan, P.R.V.; Lozano, J.D.; George, L.; Robert, W.K.; W, D.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544. [Google Scholar]
- Guo, H.; Bi, X.; Zhou, P.; Zhu, S.; Ding, W. NLRP3 Deficiency Attenuates Renal Fibrosis and Ameliorates Mitochondrial Dysfunction in a Mouse Unilateral Ureteral Obstruction Model of Chronic Kidney Disease. Mediat. Inflamm. 2017, 2017, 8316560. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, R.; Wang, X.; Fu, Q.; Ma, S. Umbelliferone ameliorates cerebral ischemia-reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci. Lett. 2015, 600, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; He, Q.; Zheng, J.; Li, L.; Hou, Y.; Song, F. Sulforaphane improves outcomes and slows cerebral ischemic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int. Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Zohreh, G.; Patrick, S.; Fuerth, B.; Steven, M.C.; David, S.P.; Sean, P.C. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar]
- Kumar, A.; Sushmita, D.; Ashish, K.; Ajay, K.; Vinod, K.; Savita, S.; Abhishek, M.; Sudha, V.; Manjay, K.; Pradeep, D. Leishmania donovani induced Unfolded Protein Response delays host cell apoptosis in PERK dependent manner. PLoS Negl. Trop. Dis. 2018, 12, e0006646. [Google Scholar]
- Hyo, J.K.; Yeonsoo, J.; Seul-Ki, K.; Se-Ung, P.; Jeongmin, P.; Chen, Y.; Jin, K.; Jinhyun, R.; Gyeong, J.C.; Young-Joon, S.; et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2alpha-ATF4 pathway. Free Radic. Biol. Med. 2017, 110, 81–91. [Google Scholar]
- YU, W.L.; TSUNG, Y.C.; CHIA, Y.H.; SHIH, H.T.; SHENG, Y.H.; CHE, C.C.; HSIN, Y.H.; E, J.L. Melatonin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress. Int. J. Mol. Med. 2018, 42, 182–192. [Google Scholar]
- Wu, F.; Qiu, J.; Fan, Y.; Zhang, Q.; Cheng, B.; Wu, Y.; Bai, B. Apelin-13 attenuates ER stress-mediated neuronal apoptosis by activating Galphai/Galphaq-CK2 signaling in ischemic stroke. Exp. Neurol. 2018, 302, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, R.; Gao, M.; Zhao, G.; Wu, S.; Wu, C.; Du, G. Pinocembrin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress induced apoptosis. Neurosci. Lett. 2013, 546, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Pan, J.; Shen, Q.; LI, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 242. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Min-Ji, K.; Soo, H.B.; Jae-Chan, R.; Younghee, K.; Ji-Hwan, O.; Jeongho, K.; Jong-Seok, M.; Kyubo, K.; Atsushi, M.; Min, G.L.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Li, Z.; Meng, C.; Wu, J.; Zhao, Y.; Zhao, J. Parkin-Dependent Mitophagy Is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells 2019, 8, 897. https://doi.org/10.3390/cells8080897
He Q, Li Z, Meng C, Wu J, Zhao Y, Zhao J. Parkin-Dependent Mitophagy Is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells. 2019; 8(8):897. https://doi.org/10.3390/cells8080897
Chicago/Turabian StyleHe, Qi, Zhenyu Li, Changchang Meng, Jingxian Wu, Yong Zhao, and Jing Zhao. 2019. "Parkin-Dependent Mitophagy Is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats" Cells 8, no. 8: 897. https://doi.org/10.3390/cells8080897
APA StyleHe, Q., Li, Z., Meng, C., Wu, J., Zhao, Y., & Zhao, J. (2019). Parkin-Dependent Mitophagy Is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells, 8(8), 897. https://doi.org/10.3390/cells8080897