1. Introduction
Copines make up a family of calcium-dependent phospholipid-binding proteins found in many eukaryotic organisms [
1,
2]. Copine proteins contain two C2 domains in the N-terminal half of the protein followed by an A domain similar to the von Willebrand A (VWA) domain in the C-terminal half of the protein [
1]. The C2 domain is a calcium-dependent phospholipid-binding motif originally identified in protein kinase C. Most proteins containing a single C2 domain are involved in signaling pathways, while most proteins that have multiple C2 domains are involved in membrane trafficking [
3]. The VWA domain is named from the von Willebrand Factor, a plasma and extracellular matrix protein. VWA domains have been studied in integrins and several extracellular matrix proteins, and appear to function as protein-binding domains [
4]. Although the exact function of copines is not known, a growing body of evidence suggests that copines may mediate an array of cellular processes by conferring calcium regulation to various signaling pathways [
5,
6,
7,
8].
A general hypothesis proposed by Tomsig et al. [
9] for how copines may regulate signaling pathways is that specific copines interact with other cellular proteins through their A domains and then either deliver soluble target proteins to membranes or regulate the function of membrane proteins through the action of the C2 domains in response to a rise in intracellular calcium. Tomsig et al. [
9] identified more than 20 distinct potential targets of A domains of human copines I, II, and IV using a yeast two-hybrid screen. Among the proteins that were found to associate with human copine A domains were various regulators of phosphorylation, transcription, ubiquitination, cytoskeleton, exocytosis, and mitosis, suggesting that copines carry out many diverse functions.
We are studying the function of copines using the model organism
Dictyostelium discoideum. We have identified six copine genes in
Dictyostelium discoideum and have focused our studies on one of the copine proteins, CpnA [
10,
11,
12,
13]. Cells lacking the
cpnA gene (
cpnA−) were previously shown to have defects in cytokinesis, contractile vacuole function, and development [
11].
Dictyostelium discoideum lives as unicellular haploid amoeba feeding on bacteria. However, when starved, the amoeba will secrete and respond to periodic waves of cAMP to aggregate into a mound. A tip is formed on the mound that elongates into a finger-like structure that falls over to form a slug. The slug is capable of moving toward light and heat in processes called phototaxis and thermotaxis, respectively. When conditions are favorable, slug movement will arrest, and the slug will culminate into a fruiting body consisting of a mass of spores on top of a long thin stalk made up of vacuolated cells [
14]. When
cpnA− cells were starved, they were delayed in aggregation to form the mound and then arrested at the slug stage [
11]. The slugs formed by
cpnA− cells were bigger than normal slugs, and they were not able to carry out normal phototaxis and thermotaxis [
13].
Previous studies in our lab have shown that GFP-tagged CpnA localized to the cytosol in live
Dictyostelium cells [
10,
15]. However, when cells were treated with a calcium ionophore in the presence of calcium, GFP-tagged CpnA was found associated with the plasma membrane and intracellular organelles. In addition, in cells primed for aggregation, GFP-tagged CpnA quickly translocated to the plasma membrane, and then back to the cytosol in response to cAMP stimulation, suggesting that CpnA may have a role in cAMP signaling during chemotaxis [
15]. To investigate the specific role of CpnA in these processes, we used column chromatography and immunoprecipitation to identify potential binding partners of CpnA. One protein identified by both techniques was actin. Because several of the defects observed in
cpnA− cells are consistent with a defect in the actin cytoskeleton, we explored this interaction further. We found that CpnA binds to actin filaments in a calcium-dependent manner in vitro. Furthermore, cells lacking CpnA exhibited increased adhesion, were defective in their actin polymerization response to cAMP stimulation, and in their ability to sense and move towards a cAMP gradient.
2. Materials and Methods
2.1. Dictyostelium Strains and Cell Culture
The
Dictyostelium discoideum strain used was NC4A2, an axenic strain derived from the wild-type NC4 strain [
16]. NC4A2 cells are referred to as the parental strain hereafter. Cells were grown at 20 °C on plastic culture dishes in HL-5 media (0.75% proteose peptone, 0.75% thiotone E peptone, 0.5% Oxoid yeast extract, 1% glucose, 2.5 mM Na
2HPO
4, and 8.8 mM KH
2PO
4, pH 6.5) supplemented with penicillin-streptomycin at 60 U/mL. Plasmid transformed cells were cultured in HL-5 media supplemented with 7.5 μg/mL G418. The full-length coding sequence of
cpnA and the A domain of
cpnA (bases 1-1000) were amplified by PCR from the cDNA clone, SLI-395 [
17]. The PCR fragments were subcloned into the
Dictyostelium extrachromosomal plasmid, pTX-GFP [
18], containing a gene for a variant of green fluorescent protein (GFP, S65A, V68L, and S72A mutations) to produce a fusion protein with a HIS-tag and GFP at the N-terminus of CpnA (GFP-CpnA) and the A domain of CpnA (GFP-Ado). As a control,
Dictyostelium cells were also transformed with the pTX-GFP plasmid without a
cpnA cDNA insertion; these cells express a HIS-tagged GFP. The
cpnA cDNA was also subcloned into the pDXA-GST plasmid [
19] to produce a fusion protein with glutathione-S-transferase (GST) at the N-terminus and a HIS-tag at the C-terminus of CpnA.
Dictyostelium cells were transformed with plasmids by electroporation.
Previously, a
cpnA knockout (KO) strain (
cpnA−) was created by replacing the
cpnA gene with the blasticidin S resistance gene (
bsr) in the axenic strain, NC4A2 [
11]. The
cpnA knockout DNA construct included PCR fragments of approximately 1 kb upstream (5’) and downstream (3’) of the
cpnA gene that were ligated into the pBSIIbsr plasmid to flank the
bsr gene. Another
cpnA knockout strain (
cpnA−cre) was made using the pLPBLP plasmid, which contains the
bsr cassette bookended by loxP sites [
20]. The 5’ and 3’ flanking regions of the
cpnA gene were removed from the pBSIIbsr plasmid, and ligated into the pLPBLP plasmid at the KpnI and HindIII, and BamHI and NotI restrictions sites, respectively. The plasmid DNA was linearized and electroporated into NC4A2 cells. Clonal populations were selected by resistance to blasticidin (10 µg/mL) and screened for expression of CpnA by western blot with rabbit polyclonal antisera raised against a bacterially expressed protein fragment of CpnA. Cell lines that did not express CpnA were also screened by PCR using primers designed to amplify the middle of the
cpnA gene. A digoxigenin labeling and detection kit (Roche Diagnostics, Indianapolis, IN, USA) was used in a Southern blot to verify the
bsr gene was found once in the genome and had replaced the
cpnA gene in the genome (
Figure S1).
2.2. Isolation of CpnA Binding Proteins using Column Chromatography
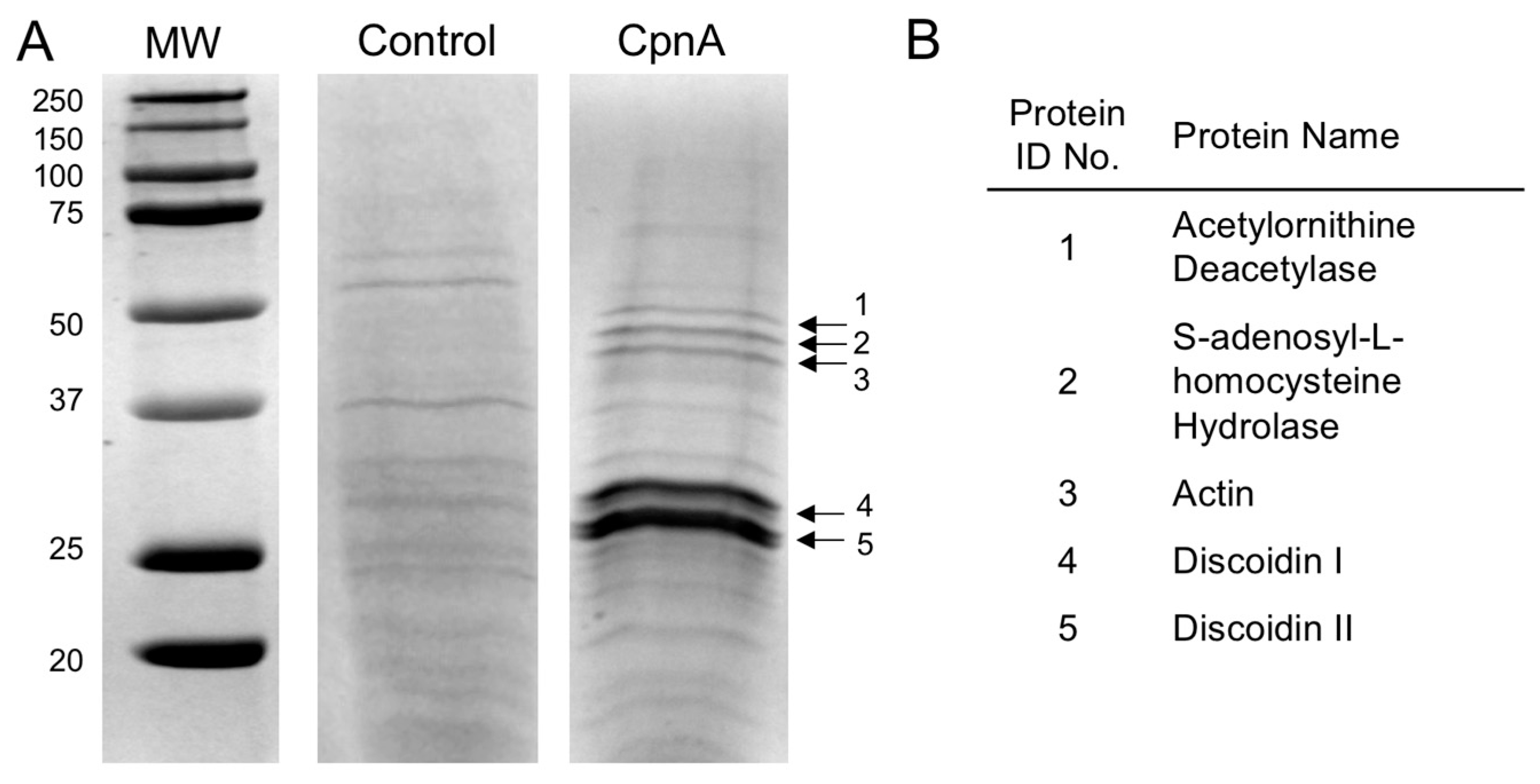
Dictyostelium NC4A2 cells expressing GFP-CpnA were grown to 2 × 106 cells/mL in 2 L of HL-5 media in a shaking incubator at 20 °C. Cells were centrifuged at 7000 RPM in a GSA Beckman rotor. Pellets were resuspended in 5 mL of cold homogenization buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, with protease inhibitors). Cells were disrupted with two passes through a French Press. After adding 5 mM EGTA, the cell lysate was centrifuged at 27,000× g for 15 min at 4 °C. The supernatant containing the GFP-CpnA protein was removed. Liposomes were made by dissolving 200 mg of bovine brain lipids (cat#B1502, Sigma-Aldrich, St. Louis, MO, USA) in 20 mL of homogenization buffer. The lipids were sonicated, then pelleted at 50,000 RPM for one hour in an ultracentrifuge. The liposomes were resuspended in the cell lysate supernatant containing the GFP-CpnA protein, and CaCl2 was added to a final concentration of 8 mM CaCl2. The liposomes were centrifuged again at 50,000 RPM for one hour. The supernatant was discarded, and the pelleted liposomes were washed in Wash Buffer (25 mM HEPES, pH 7.4, 2 mM CaCl2) twice. After the last spin, the liposome pellet was resuspended in Extracting Buffer (25 mM HEPES, pH 7.4, 10 mM EGTA) and the liposomes were pelleted again. The supernatant containing the GFP-CpnA was removed, and the lipids were pelleted and resuspended in Extracting Buffer two more times. The supernatants containing the GFP-CpnA were run over a PD-10 Sephadex G-25M desalting column (Amersham Biosciences, Piscataway, NJ, USA) to remove the EGTA. The desalted protein mixture was then loaded onto a 1 mL HisTrap FF column (GE Healthcare, Waukesha, WI, USA), equilibrated with binding buffer (20 mM sodium phosphate, 500 mM NaCl, 45 mM imidazole, pH 7.4). The column was washed with binding buffer and proteins were eluted with elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4). The eluted protein mixture was desalted using a PD-10 desalting column and analyzed by SDS-PAGE.
The resulting purified GFP-CpnA was coupled to agarose beads using an Aminolink Plus Immobilization kit according to the manufacturer’s instructions (cat#44894, ThermoFisher Scientific, Waltham, MA, USA). Dictyostelium NC4A2 cells were grown in 1 L of HL-5 media in a shaking incubator at 20 °C. Cells were centrifuged at 7000 RPM in a GSA Beckman rotor. Pellets were resuspended in 5 mL of cold homogenization buffer. EGTA was added to a final concentration of 2 mM and cells were disrupted with two passes through a French Press. The cell lysate was centrifuged at 50,000 RPM in an ultracentrifuge for 45 min at 4 °C. CaCl2 was added to the supernatant to a final concentration of 10 mM, and the pH was adjusted to 7.4. The supernatant containing soluble proteins was applied to the GFP-CpnA-linked agarose column and a control column containing agarose beads. Both columns were washed twice in homogenization buffer containing 2 mM CaCl2. Proteins were eluted from the columns with a low pH buffer (0.2 M glycine, pH 2.5) and these column fractions were neutralized with 1 M Tris, pH 9. Column fractions were analyzed by SDS-PAGE and stained with Coomassie Blue.
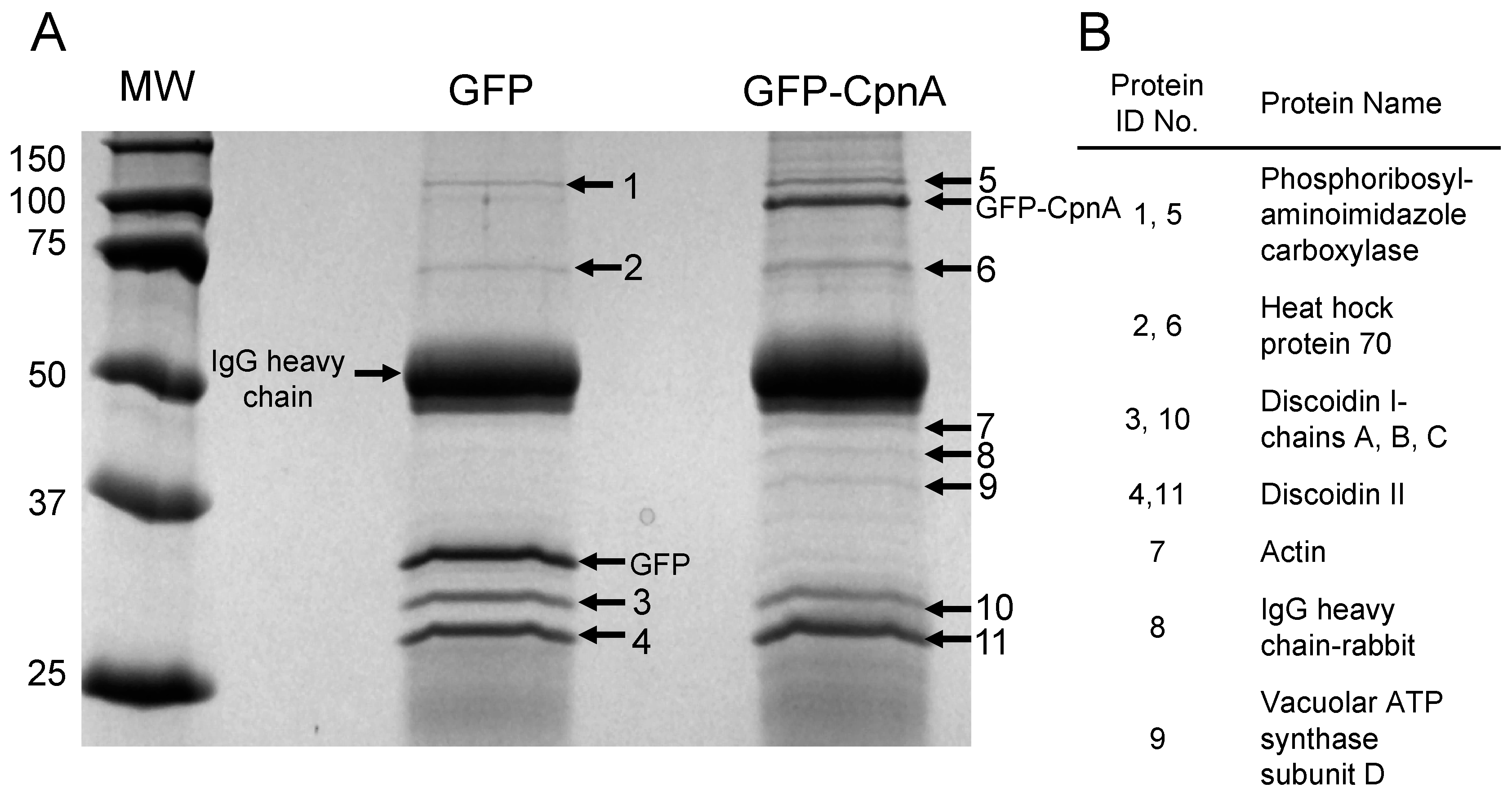
2.3. Isolation of CpnA Binding Proteins using Immunoprecipitation
Dictyostelium NC4A2 cells (2 × 108) expressing GFP, GFP-Ado, or GFP-CpnA were harvested from plates and pelleted by centrifugation at 1500 RPM for 5 min. Cells were washed twice with 5 mL of HEPES Buffer (50 mM, pH 7.4) at 4 °C. Cells were then resuspended in 2 mL of Lysis Buffer (50 mM HEPES, 100 mM NaCl, 2 mM CaCl2, 10% Sucrose, 0.3% NP-40, pH 7.4 with protease inhibitors). Cell lysates were centrifuged at 14,000 RPM for 15 min. Supernatants (2 mL) and 0.5 mL HEPES buffer were applied to HEPES buffer equilibrated PD-10 desalting columns, then 3.5 mL of HEPES buffer was applied to each column, and the flow-through was collected. Protein A agarose beads (Roche Diagnostics) were washed four times with 4 mL of HEPES Buffer and then resuspended in HEPES Buffer to create a 50% v/v bead slurry. Cell supernatants were pre-cleared twice by adding 50 µL of 50% v/v protein A agarose beads, incubating at 4°C with rocking for 1 hour, and then centrifuging at 3000× g for 10 min. The pre-cleared supernatant was then incubated with 3 µL of anti-GFP antibody (ab-290, Abcam, Cambridge, UK) for 1 h with rocking, then incubated with protein A agarose beads for an additional hour. The beads were pelleted by centrifugation at 4500 RPM for 30–45 sec and washed four times with 1 mL of HEPES Buffer. After the last wash, the supernatant was discarded. The pelleted beads were incubated with 50 µL of sample buffer at 95 °C for 5 min. The beads were pelleted again with centrifugation at 10,000 × g for 5 min. Supernatant samples were analyzed by SDS-PAGE and stained with Coomassie Blue.
2.4. Identification of Proteins using Mass Spectrometry
Proteins were analyzed by SDS-PAGE and stained with Coomassie Blue. Visible bands were excised from the gel manually and cut into small pieces approximately 1 mm × 1 mm. These gel pieces were washed three times with water, then washed twice with 1:1 acetonitrile: 100 mM ammonium bicarbonate. To further prepare the gel pieces for digestion, the gel pieces were then dehydrated in 100% acetonitrile. After removing all acetonitrile, 25 µL of porcine trypsin (Promega, Madison, WI, USA) dissolved in 25 mM ammonium bicarbonate at a concentration of 4 µg/mL was added to the gel pieces. The gel pieces were then kept at room temperature (RT) overnight (approximately 12–16 h). Following digestion, the supernatant was transferred to a second tube, and acetonitrile was added to the gel pieces to complete the extraction of digested peptides. This extract was added to the first supernatant, and this combined solution, containing the extracted peptides was frozen and lyophilized. The peptides were resuspended in 5 µL of 100:99:1 acetonitrile: water: trifluoroacetic acid immediately prior to spotting on the MALDI target.
For matrix-assisted laser desorption/ionization (MALDI) analysis, the matrix solution consisted of alpha-cyano-4-hydroxycinnamic acid (Aldrich Chemical Co. Milwaukee, WI, USA) saturating a solution of 1:1:0.01 acetonitrile: 25 mM ammonium citrate: trifluoroacetic acid. Approximately 0.15 µL of peptide solution was spotted on the MALDI target immediately followed by 0.15 µL of the matrix solution. This combined solution was allowed to dry at RT. MALDI MS and MS/MS data were then acquired using the ABSCIEX 5800 TOFTOF Mass Spectrometer. Resultant peptide mass fingerprint and peptide sequence data were submitted to the UniProt database using the Mascot search engine to which relevance is calculated, and scores are displayed (
Tables S1 and S2).
2.5. Glutathione Agarose Chromatography
Dictyostelium NC4A2 cells expressing GST-tagged CpnA were harvested from plates and counted with a hemocytometer. The cells were pelleted at 1500 RPM for 5 min at 4 °C, and the supernatant was discarded. Lysis buffer containing 5 mM EGTA, 1% Triton X-100, 1 mM DTT and 1× protease inhibitor in PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na
2HPO
4, 1.47 mM KH
2PO
4, pH 7.4) was used to resuspend the pelleted cells, and the cell suspension was incubated on ice for 30 min. Glutathione agarose beads (Sigma, 50 mg) were suspended in 5 mL of deionized water, placed on the rotator, and incubated at RT for 30 min. The beads were washed three times in PBS by pelleting at 3000 RPM for 5 min at 4 °C. The beads were resuspended in 150 µL of PBS to make a 50% bead slurry. The cells in lysis buffer were spun in a microcentrifuge at 14,000 RPM for 5 min at 4 °C. The bead slurry was added to the cell lysate supernatant and placed on the rotator at RT for 30 min. The beads were washed by pelleting at 3000 RPM for 5 min at 4 °C four times in PBS, and two times using lysis buffer. The beads were washed with 5 mL of 10 mM Tris (pH 8.0), then incubated for 10 min on a rotator at RT in elution buffer (10 mM reduced glutathione in 50 mM Tris-HCl, pH 8.0). The beads were washed three times with elution buffer, and the supernatants were analyzed by SDS-PAGE (
Figure S2) and stored at −20 °C. The concentration of the purified GST-CpnA was determined using a BIO-RAD protein assay kit.
2.6. F-actin Binding Assay
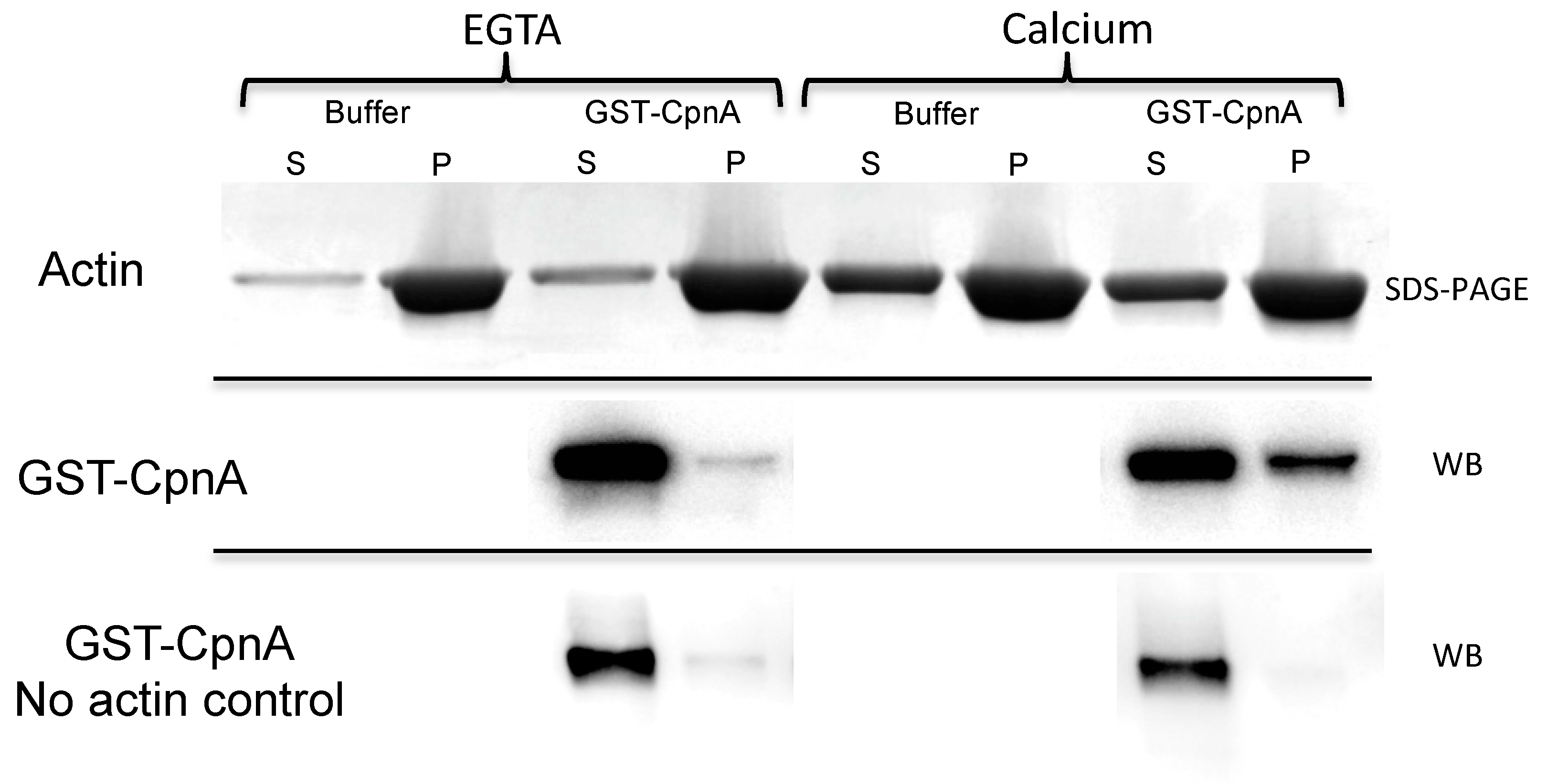
Purified GST-CpnA samples were thawed on ice and spun at 150,000× g (50,000 RPM) for 1 h at 4 °C. The supernatant was removed and kept on ice. An F-actin stock was prepared as described in the published protocol (Cytoskeleton, Inc.). Three samples were prepared for the F-actin binding assay: (1) An F-actin control sample made by mixing 40 µL F-actin stock (1 mg/mL) and 10 µL of glutathione elution buffer; (2) a no actin protein control made by mixing 10 μL GST-CpnA protein (20 μg/mL) and 40 μL F-actin buffer; and (3) an experimental sample made by mixing 40 μL F-actin stock (1 mg/mL) and 10 μL GST-CpnA protein (24 μg/mL). In addition, the same three samples were prepared again, but 2 mM EGTA was added to the samples. All samples were incubated at RT for 30 min then spun at 150,000× g in an Airfuge for 1.5 h at 24 °C. The supernatants were removed, and 10 μL 6× sample buffer was added into each supernatant. The pellets were resuspended with 30 µL 2× sample buffer. All the samples were analyzed by SDS-PAGE and stained with Coomassie Blue. A western Blot was performed for the supernatants and pellets from the samples that included GST-CpnA using an HRP conjugated anti-GST antibody (1:5000). An ECL chemiluminescence kit (Amersham Biosciences) and the Kodak Gel Logic 2200 were used to image the blot.
2.7. Dynabead Immunoprecipitations
Dictyostelium NC4A2 cells (5 × 107) expressing GFP, GFP-Ado, and GFP-CpnA were harvested from plates in HL-5 media. Cells were then centrifuged at 1500 RPM for 5 min, the supernatant was removed, and the cells were washed with 5 mL of 50 mM HEPES buffer, pH 7.4. The cells were centrifuged again, the supernatant was removed, and the cells were resuspended in 1.5 mL of Lysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 2mM EGTA, 10% w/v sucrose, and 0.3% NP-40). Cell lysates were centrifuged in a microcentrifuge at 14,000 RPM for 10 min. Magnetic Protein G Dynabeads (Life Technologies, Carlsbad, CA, USA) were washed in PBS and incubated with 2 μL of rabbit polyclonal anti-GFP antibody (Abcam, ab290) for 10 min. The beads were resuspended with the cell lysate and incubated for 30 min. The beads were washed three times with 200 μL of 50 mM HEPES, pH 7.4. After the final wash, the beads were resuspended with 100 μL 50 mM HEPES, pH 7.4 and transferred to new tubes.
G-actin and F-actin stocks were prepared as described in the published protocol (Cytoskeleton, Inc., Denver, CO, USA). The F-actin or G-actin stock (40 μL) was added to the beads-antibody-protein complex and incubated for 10 min at RT; then the beads were washed once with 200 μL of 50 mM HEPES buffer. The same protocol was followed again, but EGTA (2mM) was added to the actin stocks. The beads were precipitated out of solution using the magnet and then resuspended in 40 μL of 2× sample buffer and heated at 95 °C for 5 min. Samples were analyzed by western blotting with a mouse monoclonal anti-actin antibody (cat# sc-47778, Santa Cruz Biotechnology, Inc) diluted 1:2000 and anti-mouse HRP-conjugated antibody diluted 1:15,000. An ECL chemiluminescence kit (Amersham Biosciences, ) and the Kodak Gel Logic 2200 was used to image the blot. Blots were re-probed with a mouse monoclonal anti-GFP antibody (cat# sc-9996, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) diluted 1:2000 and the same secondary antibody diluted 1:15,000.
2.8. Folate and cAMP Chemotaxis Assays
Dictyostelium NC4A2 and cpnA KO cells were grown in HL-5 media in a shaking incubator at 20 °C at 180 RPM. Small wells were made in agar plates containing Development Buffer ((DB) 5 mM Na2HPO4, 5 mM KH2PO4). DB alone or with 100 µM folate or 100 µM cAMP was placed in the wells. For folate chemotaxis assays, cells were washed once in DB and then resuspended in DB at a concentration of 2.5 × 108 cells/mL. Cells were placed as 1 µL drops 5 mm away from each well. For cAMP chemotaxis assays, cells were washed in DB and resuspended at 1 × 107 cells/mL. Cells were placed in a shaking incubator at 180 RPM for eight hours at 20 °C. Starved cells were pelleted, washed once in DB, and then resuspended in DB containing 2 mM caffeine so that the final cell concentration was 2.5 × 108 cells/mL. The cells were placed as 1 µL drops 5 mm from each well. The plates were placed in a humidity chamber for 3 h at 20 °C. Cell drops were imaged using a Leica dissecting microscope at 12.5× magnification. The distance moved by cells, away from the edge of the cell drop, was measured in the direction towards the chemoattractant well, as well as in the opposite direction, using ImageJ. Two trials (three replicates per trial) for each cell type and chemoattractant were performed. Distance data towards and away from chemoattractant were analyzed separately by a 2-way ANOVA with post-hoc comparisons using the Tukey method.
2.9. cAMP Gradient Chemotaxis Assay
Dictyostelium NC4A2 and cpnA KO cells were harvested from plates, counted with a hemocytometer, centrifuged at 1,500 RPM for 5 min at 4°C, washed twice with KK2 buffer (16.2 mM KH2PO4, 4.0 mM K2HPO4), and resuspended in KK2 buffer at 1 × 107 cells/mL. The cells were starved for 1 hour in shaking suspension at 150 RPM at 20 °C, and subsequently pulsed with 50 nM cAMP every 6 min for 5 h with shaking at 150 RPM. Cells were diluted to 7.5 × 105 cells/mL and treated with 2.5 mM caffeine for 15 min, while shaking at 150 RPM. Cells were placed on coverslips for 10 min and placed on the Dunn Chemotaxis Chamber (DCC100, Hawksley, Sussex, UK) and sealed with a hot wax mixture (Vaseline, paraffin, beeswax, 1:1:1). For control experiments with no cAMP gradient, the inner well was filled with KK2 buffer solution with 2.5 mM caffeine. For cAMP gradient experiments, the inner well was filled with KK2 buffer solution with 2.5 mM caffeine and 5 µM cAMP. Time-lapse images of cells were captured with a Leica DFC7000 T camera and a Leica DMi8 light microscope using a 20× DIC objective every 10 s for 10 min. Trials for each cell line with and without a cAMP gradient were performed at least twice for each cell line on different days, except for the pulsed cpnA− cells in which there was only one trial with and without a cAMP gradient. Time-lapse images were taken from 2–3 different areas of the Dunn chamber within the same trial. The ImageJ manual tracking plug-in was used to track the movement of all cells (~15–20 cells) that appeared viable and stayed within the field of view. To measure directionality, the changes in x and y coordinates every 10 s for 10 min were averaged for each cell. The change in x and y averages from each cell were averaged for each cell type and analyzed for differences among the cell types using t-tests. Individual cells from all trials, which included 2–6 movies, were analyzed for velocity and roundness using ImageJ. An average and standard error were calculated from the averages of velocity and roundness for each movie and these averages were analyzed by a 2-way ANOVA with post-hoc comparisons using the Tukey method.
2.10. Actin Polymerization in Response to cAMP Stimulation Assay
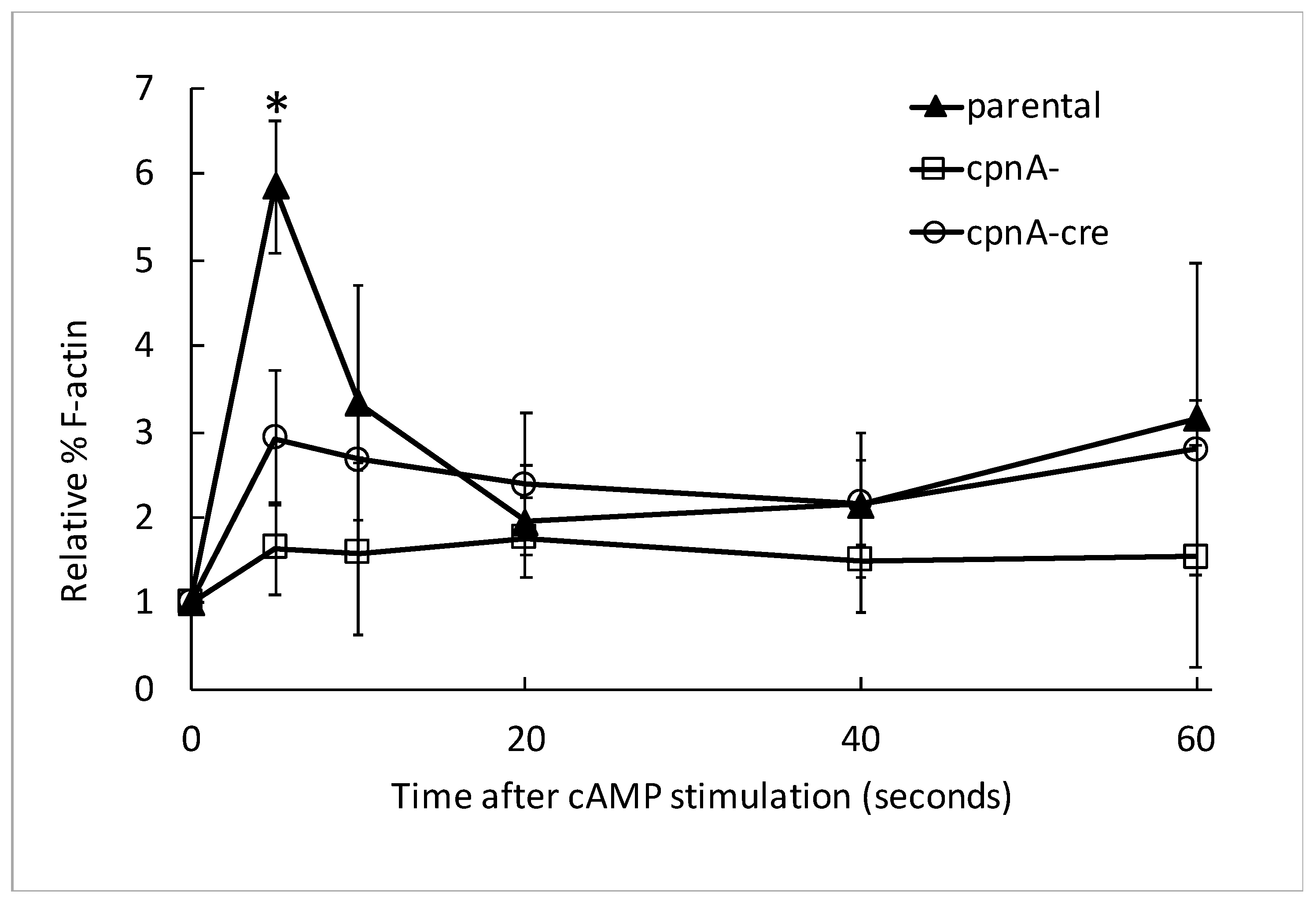
Dictyostelium NC4A2 and cpnA KO cells were harvested from plates, counted with a hemocytometer, centrifuged at 1500 RPM for 5 min at 4 °C, washed twice with KK2 buffer, and resuspended in KK2 at 1 × 107 cells/mL. The cells were starved for 1 h in shaking suspension at 150 RPM at 20 °C, and subsequently pulsed with 50 nM cAMP every 6 min for 5 h with shaking at 150 RPM. Cells were treated with 2.5 mM caffeine for 15 min, while shaking at 150 RPM. Cells (250 µL) were removed from the flask for the 0-s timepoint. Cells were treated with 100 µM cAMP and 100 mM dithiothreitol (DTT), and 250 µL of cells were removed from the flask at 5, 10, 15, 20, 40, and 60 second timepoints and subsequently treated with 250 µL of 2× lysis buffer (1% Triton X-100, 5 mM EGTA, 2 mM MgCl2, 4 mM ATP, 20 mM MES, pH 6.8) on ice. Samples were centrifuged at 13,000 RPM for 2 min at 4 °C. The supernatants were removed, placed in 1 mL of acetone, and stored in -20 °C overnight. Pellets were resuspended in 35 µL of 2× SDS sample buffer and boiled at 95 °C for 5 min. Supernatants were removed from −20 °C, samples were centrifuged at 13,000 RPM for 15 min. The supernatants were discarded, and the pellets were air dried to remove any excess acetone and then resuspended in 35 µL of 2× SDS sample buffer and boiled at 95 °C for 3 min. Pellet and supernatant protein samples were separated by SDS-PAGE using BIO-RAD TGX Stain-Free gels. Gels were imaged with a BIO-RAD ChemiDoc Touch imaging system. Densitometry analysis was performed using ImageJ to measure background-subtracted band intensities. Percent F-actin was calculated by dividing the actin band intensity of the pellet by the sum of the pellet and supernatant actin bands at each timepoint. The data were normalized to the 0-timepoint. These experiments were carried out three times from the same pulsed cell populations for each cell type. The normalized data at each timepoint from the three experiments were averaged, and then those means were averaged from two different pulsed cell populations of cpnA− and cpnA−cre cells and three different pulsed cell populations of parental NC4A2 cells. Differences between the averages for each cell type at timepoints 0 and 5 s were analyzed by t-tests.
2.11. Adhesion Assay
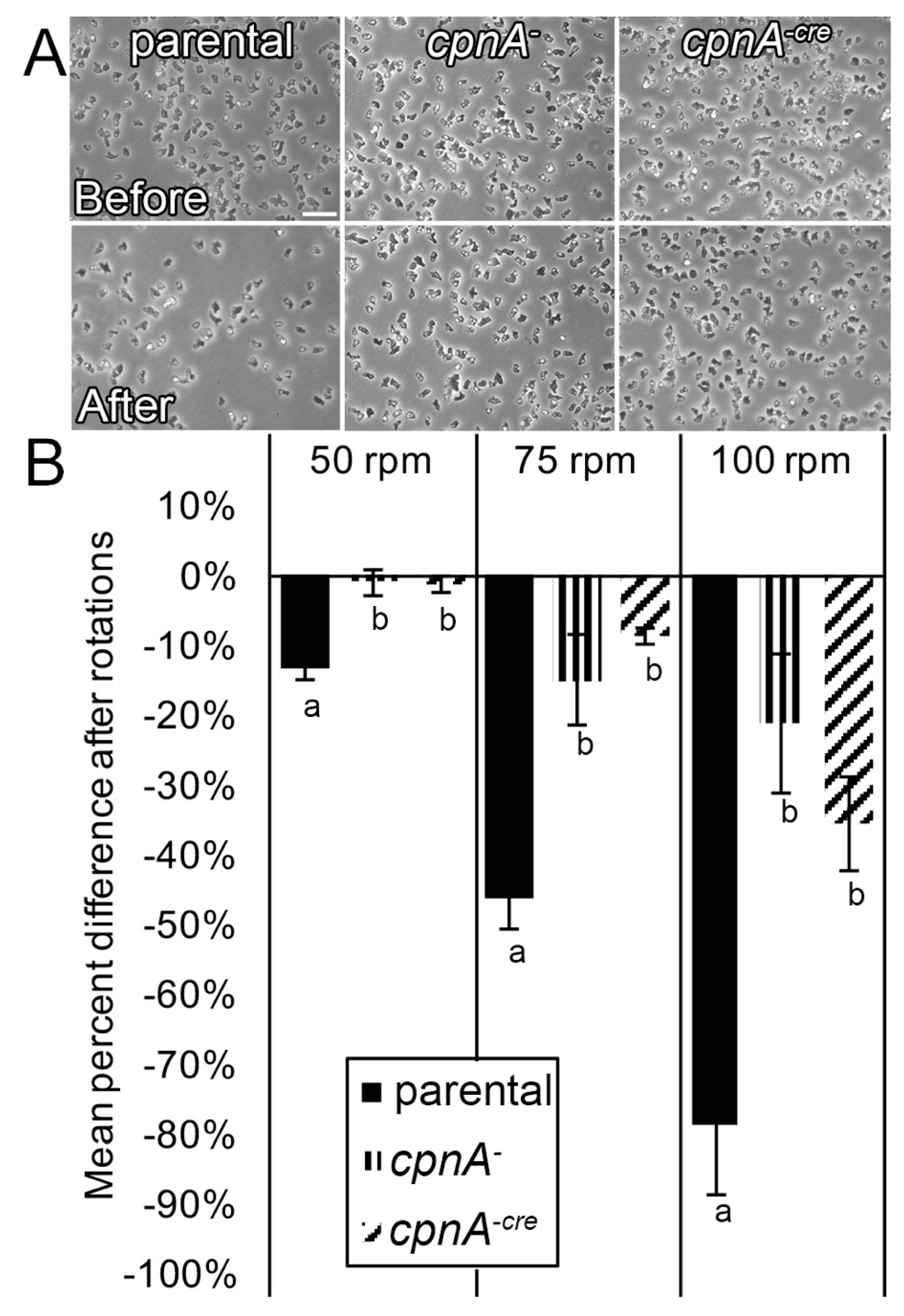
Dictyostelium NC4A2 and cpnA KO cells were harvested from plates and pelleted by centrifugation at 1500 RPM for 5 min. Cells were resuspended in HL-5 at 1 × 106 cells/mL, and 2 mL of cells were allowed to settle on 35 × 10 mm Petri dishes. Cells that had not adhered to the surface of the Petri dish after 20 min were removed by replacing the media with fresh HL-5. Adherent cells were imaged with a Nikon TE2000 microscope with a 20× phase-contrast objective in 5 marked spots on the Petri dishes, one in the middle and four around the periphery of the dish. Plates were placed on a rotator for 15 min at 50, 75, or 100 RPM. Cells that had separated from the bottom surface of the Petri dish while shaking were removed by replacing the media with fresh HL-5. Images were taken at the same marked spots on the Petri dishes after rotation. The number of cells in each image were then counted using the Cell Counter plugin in ImageJ and averaged for each RPM for three trials. The percent difference of cells still adherent after shaking was calculated by subtracting the average number of cells remaining from the average number of cells present before shaking and divided by the number of cells present before shaking. The percent difference was averaged from three trials. Differences between the cell types at each RPM were analyzed by t-tests.
4. Discussion
In this study, actin was identified as a binding protein of CpnA using column chromatography and immunoprecipitation. In addition, purified GST-CpnA was shown to bind F-actin, suggesting that CpnA can bind directly to actin filaments. However, we cannot rule out the possibility that another protein that may have been co-purified with the GST-CpnA mediated this interaction. Some of the human copines have also been reported to bind to actin filaments [
9]. In a yeast two-hybrid study, the A domains of human copines I, III, and IV were used as bait and β-actin was identified as a binding partner for all three. In addition, the interaction with β-actin was confirmed in an in vitro binding assay [
9].
To investigate whether CpnA has a role in an actin-dependent process in vivo, we characterized the chemotaxis properties of cpnA KO cells. Chemotaxis to cAMP was assessed for cpnA KO cells in two different types of assays that differed with respect to cAMP concentration, substrate surface, and time period. The results of the chemotaxis assays on an agar substrate over a 3-hour period indicated that populations of cpnA KO cells were able to sense and move in response to a steep cAMP gradient, but were not able to move in the direction of the chemoattractant. This behavior may be indicative of oversensitivity to cAMP in that the cpnA KO cells are overwhelmed by the signal and are not able to sense the direction of the signal.
The results of the Dunn chamber assays where individual cells were imaged moving within a shallower cAMP gradient on glass over a 10-min period indicated that cpnA KO cells were defective in both motility and directional movement. Therefore, in contrast to the agar assays, cpnA KO cells not only had a defect in directional movement, but they also moved more slowly than parental cells. The decreased speed on glass could be attributed to the increased adherence of cpnA KO cells. Both the Dunn chamber and agar assays indicated that cpnA KO cells were impaired in directional movement towards cAMP. In addition to a defect in directional movement, cpnA KO cells within a cAMP gradient did not have the characteristic elongated, polar shape of a chemotactic cell, indicating a defect in cell polarity.
A defect in directional movement or cell polarity could be caused by a multitude of defects that are not directly related to a specific role in regulating the actin cytoskeleton. Therefore, we tested whether cpnA KO cells have a normal actin polymerization response to global cAMP stimulation in the absence of chemotaxis. We found that the fast actin polymerization response is not as robust in cpnA KO cells as observed in the parental cells, suggesting that CpnA’s role in chemotaxis is related to the regulation of the actin cytoskeleton.
In a recent study, we showed that five
Dictyostelium copines, including CpnA, translocated from the cytoplasm to the plasma membrane and back to the cytoplasm in response to cAMP stimulation [
15]. Compared to the other copines, the translocation of CpnA to the plasma membrane in response to cAMP was the most difficult to observe, in that the amount of protein found at the membrane was much lower and appeared more diffuse than the other copines. This diffuseness may be more indicative of an association with actin filaments than the plasma membrane [
28]. The translocation response of CpnA to cAMP stimulation was also the fastest, with CpnA going to the membrane first and coming off the membrane first compared to the other copines [
15]. The translocation timing of CpnA is similar to the fast actin polymerization response to cAMP stimulation [
15,
23,
24,
25,
26], suggesting that CpnA may translocate to the plasma membrane or cell cortex to either activate or inhibit other actin regulating proteins or regulate actin filaments directly.
This rapid translocation of CpnA to the plasma membrane in response to cAMP is similar to what has been observed for several other proteins functioning within chemotactic signaling pathways [
29,
30,
31]. Studies using a tagged Ras-GTP binding domain peptide have shown that upon cAMP stimulation, activated Ras transiently localizes to the plasma membrane and this recruitment is not dependent on actin filaments [
31]. Another protein that is rapidly recruited to the plasma membrane is PI3K; this recruitment is dependent on actin filaments [
31]. As a calcium-dependent membrane-binding protein, CpnA may be responding to the rapid transient increase in intracellular calcium concentration that occurs after cAMP stimulation [
32,
33,
34,
35]. However, other copines (CpnC and CpnF) translocate to the plasma membrane in response to cAMP more slowly than CpnA [
15] and this response is much later than the transient calcium concentration increase observed with cAMP stimulation, suggesting that the translocation of at least some copines to the membrane is not in direct response to a rise in calcium concentration. Furthermore, although cAMP stimulation triggers a transient rise in calcium, the role of calcium in chemotaxis in
Dictyostelium is not clear [
29,
36]. Deletion of the IP3 receptor necessary for cAMP triggered calcium release does not affect chemotaxis, indicating the fast transient rise in calcium is not necessary for chemotaxis [
36].
There are a few known examples of mammalian copines that translocate to the plasma membrane in response to a stimulus to regulate an actin-based process. Human copine 3 was shown to translocate from the cytosol to the plasma membrane in response to a rise in calcium concentration and colocalize with activated ErbB2 receptors in breast cancer cells. Copine 3 also localized to focal adhesions in migrating cells and was required for ErbB2-dependent cell migration [
37]. Copine 6 was shown to translocate to the membranes of dendritic spines in response to synaptic activity in mouse hippocampal neurons. Copine 6 was also shown to interact with and activate Rac1, a Rho GTPase involved in the regulation of the actin cytoskeleton, to mediate changes in dendritic spine morphology important in learning and memory [
8]. Neither of these studies indicated a direct interaction with actin filaments. Although our in vitro data suggests that CpnA interacts with actin filaments directly, CpnA, may function more like Copine 6, and indirectly regulate actin filaments by regulating other proteins, like Rac proteins. In
Dictyostelium, several Rac proteins have been shown to be involved in chemotaxis by regulating actin polymerization [
38,
39].
Our previous studies on
cpnA− cells are consistent with CpnA having a role in regulating the actin cytoskeleton [
11,
12,
13].
cpnA− cells have defects in cytokinesis, contractile vacuole function, and development [
11,
12,
13]. All of these processes depend on the actin cytoskeleton for proper function [
40,
41,
42]. In this study, we showed that
cpnA KO cells had defects in chemotaxis, polarity, and adhesion, which are all dependent on the actin cytoskeleton. Therefore, the assortment of defects observed in
cpnA KO cells may be due to a defect in the regulation of actin filament dynamics.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







