Autoantibodies Specific to ERα are Involved in Tamoxifen Resistance in Hormone Receptor Positive Breast Cancer

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Patients and Methods
2.1. Purification of Specific Autoantibodies from Patients’ Sera
2.2. Cell Culture and Treatment
2.3. Flow Cytometry
2.3.1. Apoptosis, Proliferation and Cell Cycle
2.3.2. Quantitative Fluorescence Resonance Energy Transfer (FRET)
2.4. Fluorescence Microscopy
2.5. Western Blot
2.6. Activity of Anti-ERα Abs in Tumor-Bearing Severe Combined Immunodeficient (SCID) Mice
2.7. Immunofluorescence Analysis on Formalin-Fixed, Paraffin-Embedded (FFPE) Tumor Tissues
2.8. Data Analysis and Statistic
3. Results and Discussion
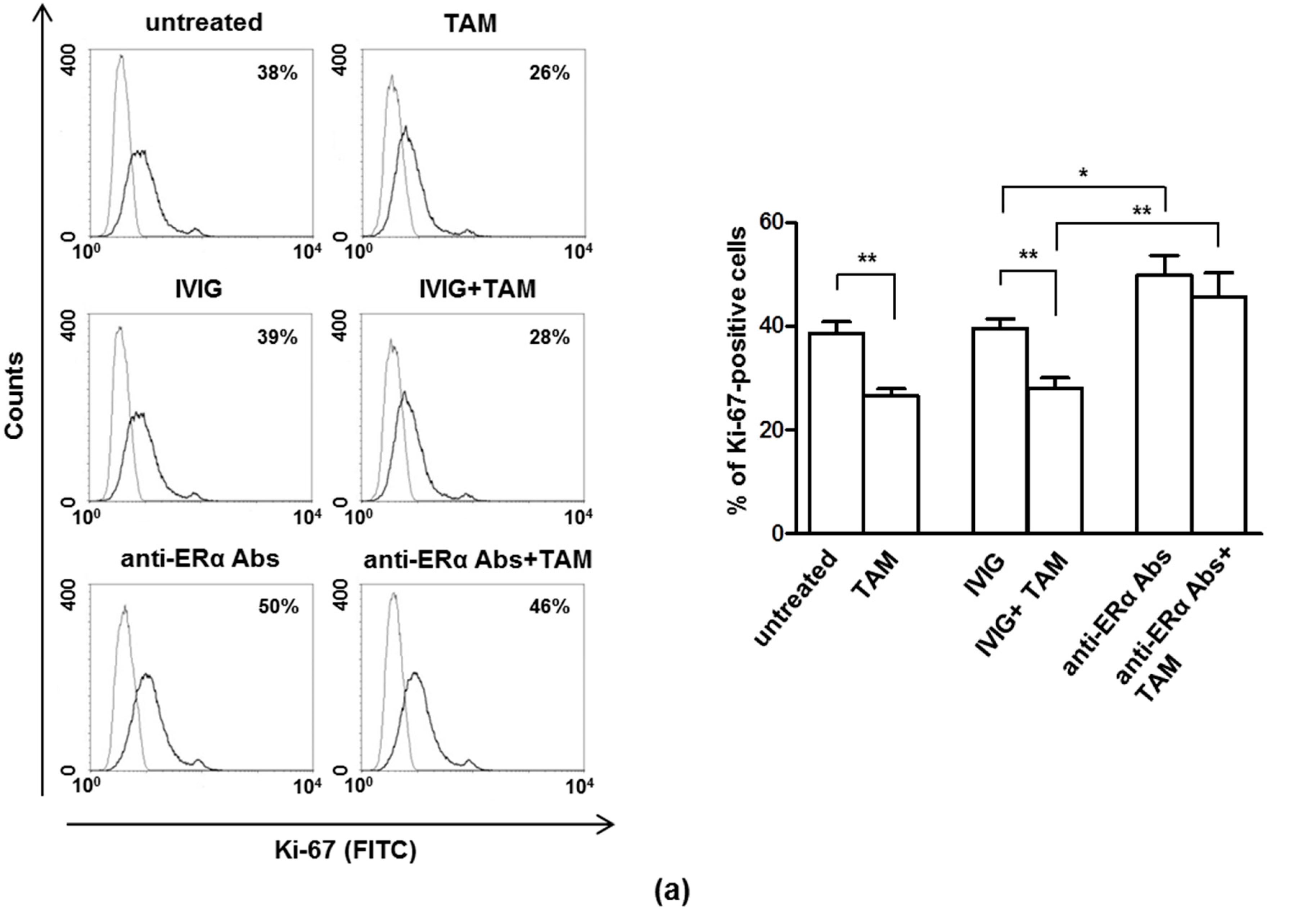
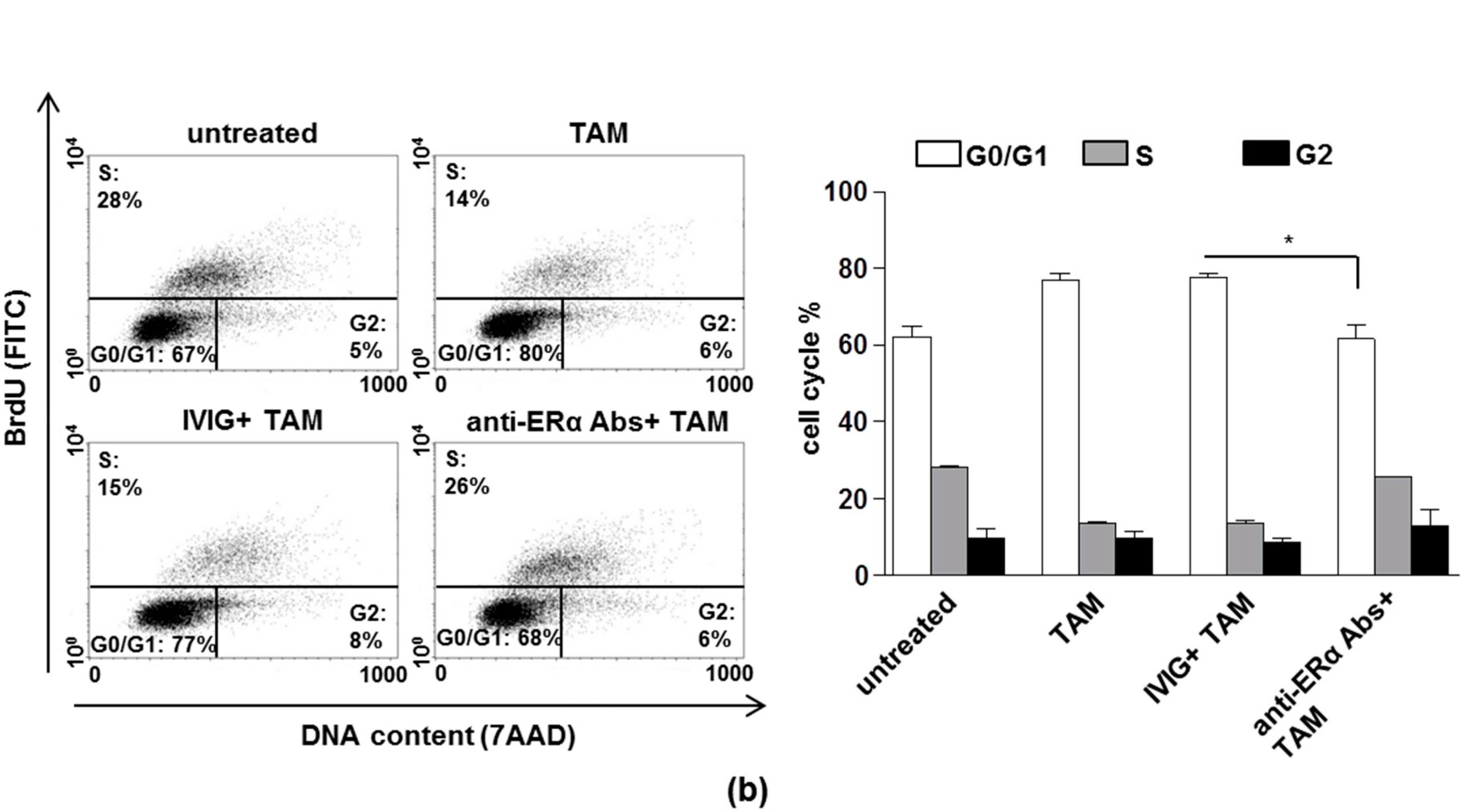
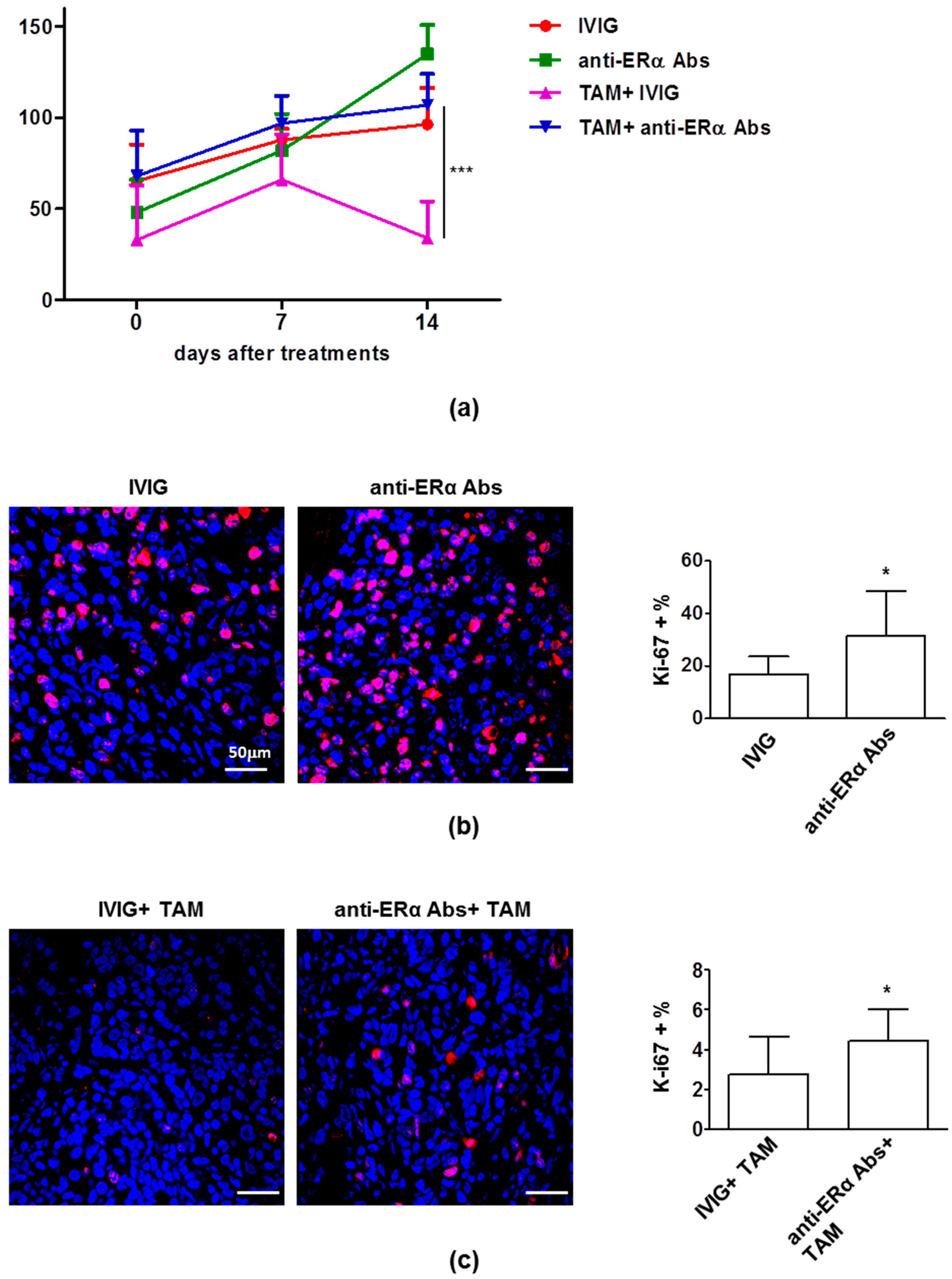
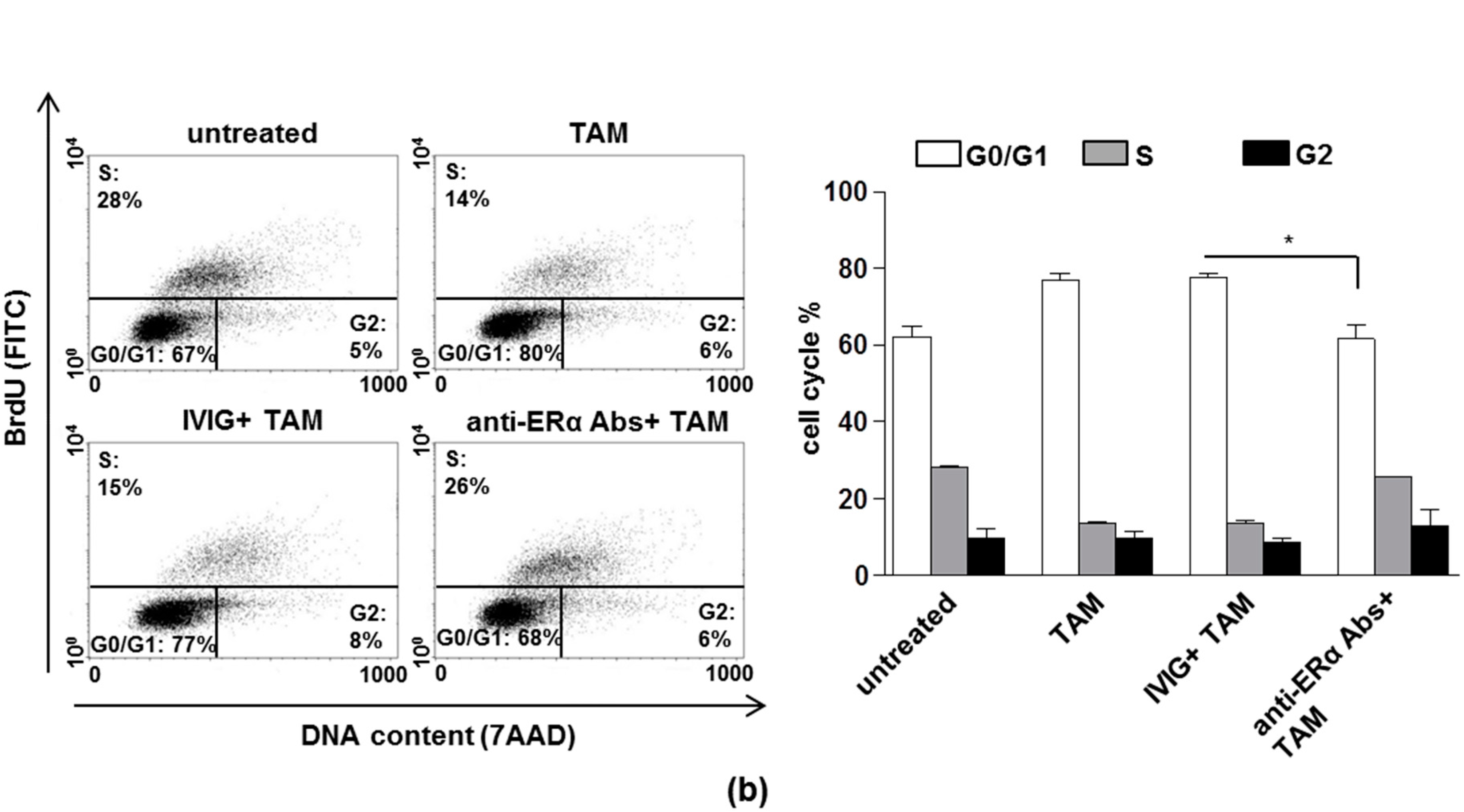
3.1. The Anti-Proliferative Effects of Tamoxifen is Inhibited by Anti-ERα Abs
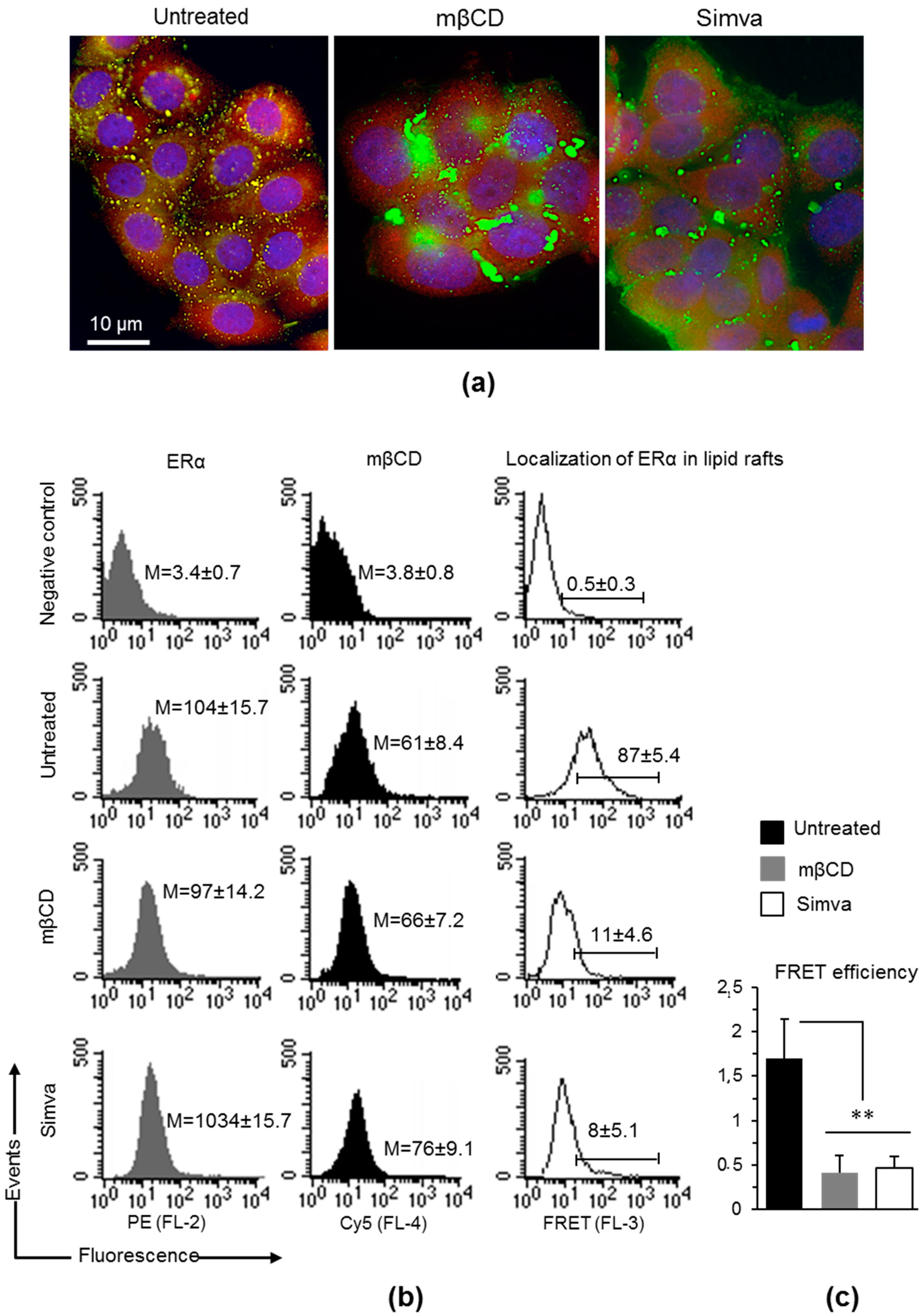
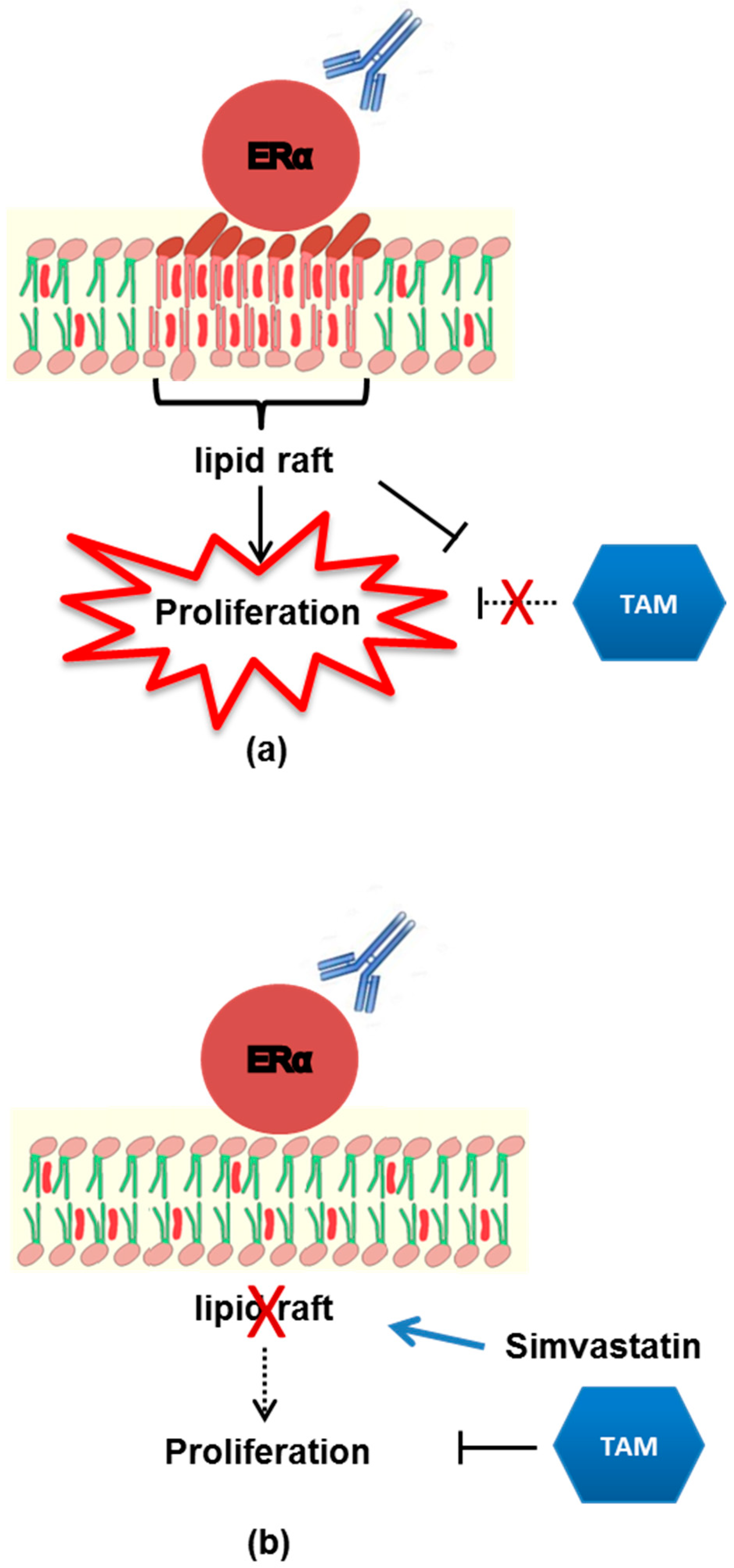
3.2. Localization of mERα in Lipid Rafts
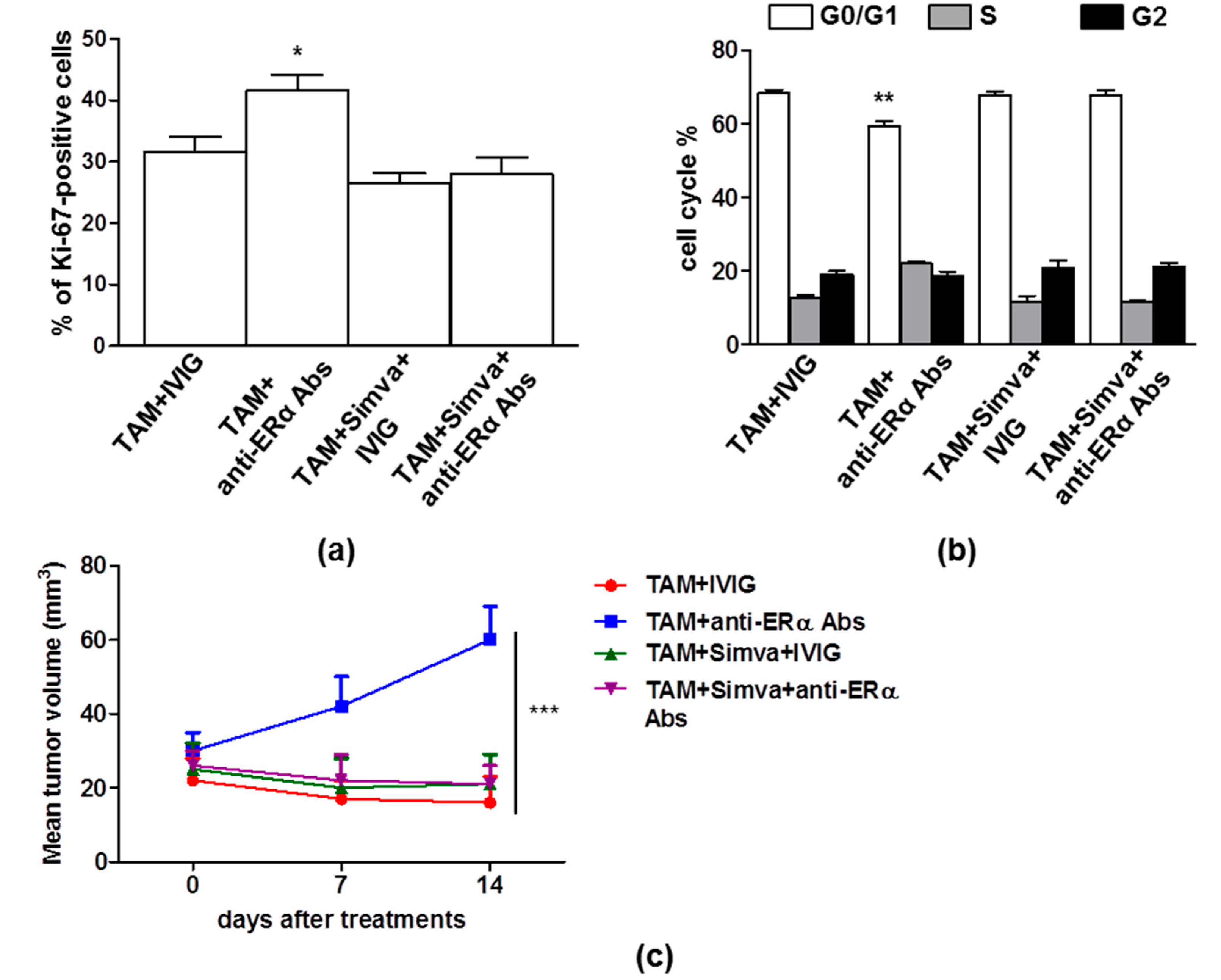
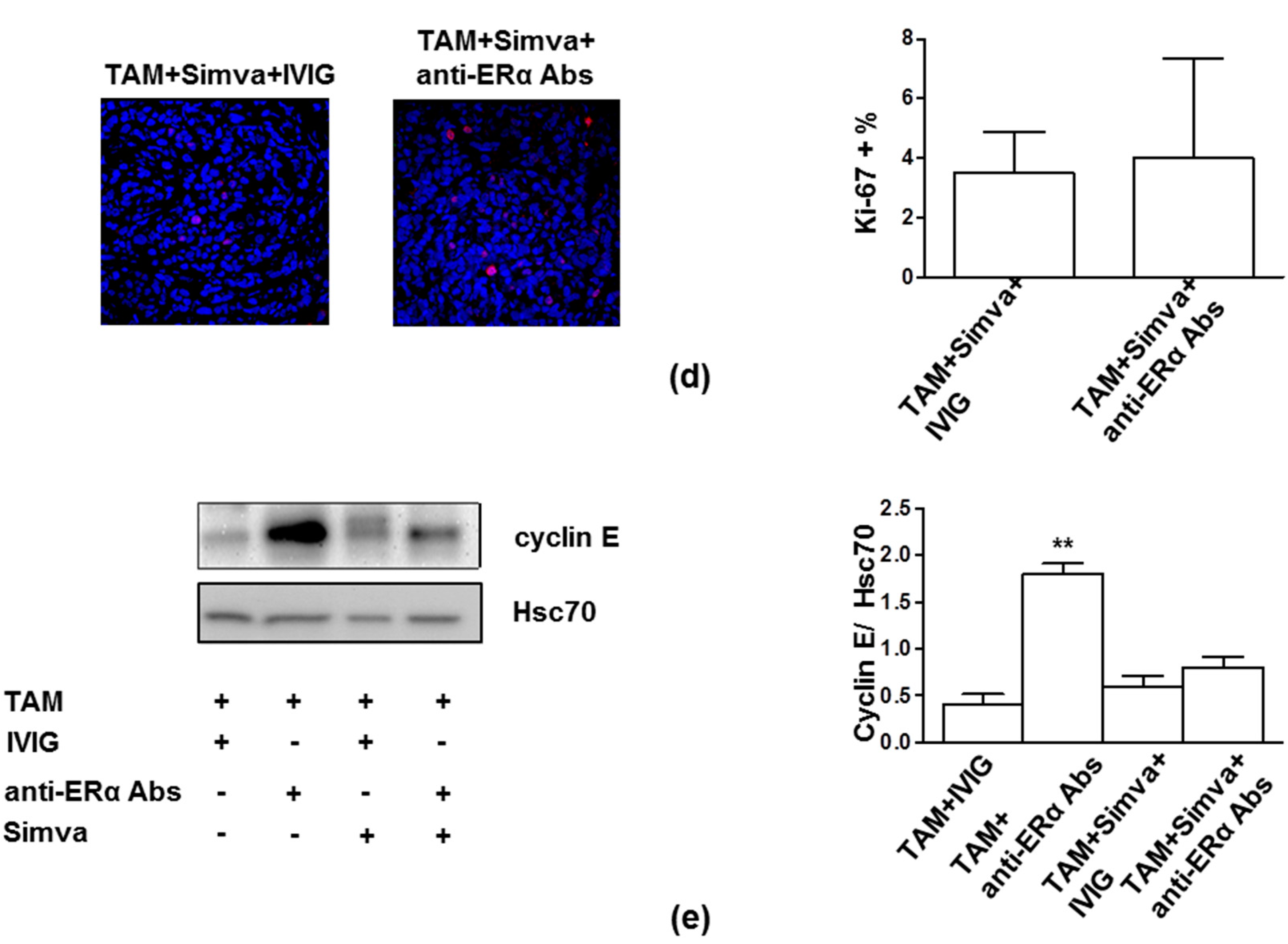
3.3. Perturbation of Lipid Rafts is a Potential Approach to Restore TAM Effects in Presence of Anti-ERα Abs
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; Wang, Y.C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [PubMed]
- EBCTCTG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: Extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef]
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids 2008, 73, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer--An overview and update. Mol. Cell Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Gu, W.; Dong, N.; Wang, P.; Shi, C.; Yang, J.; Wang, J. Tamoxifen resistance and metastasis of human breast cancer cells were mediated by the membrane-associated estrogen receptor ER-alpha36 signaling in vitro. Cell Biol. Toxicol. 2017, 33, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Yin, L. Estrogen receptor alpha-36 (ER-alpha36): A new player in human breast cancer. Mol. Cell Endocrinol. 2015, 418, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Maselli, A.; Capoccia, S.; Pugliese, P.; Raggi, C.; Cirulli, F.; Fabi, A.; Malorni, W.; Pierdominici, M.; Ortona, E. Autoantibodies specific to estrogen receptor alpha act as estrogen agonists and their levels correlate with breast cancer cell proliferation. Oncoimmunology 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Pierdominici, M.; Maselli, A.; Locatelli, S.L.; Ciarlo, L.; Careddu, G.; Patrizio, M.; Ascione, W.; Matarrese, P.; Oetona, E. Estrogen receptor beta ligation inhibits Hodgkin lymphoma growth by inducing autophagy. Oncotarget 2017, 8, 8522–8535. [Google Scholar] [CrossRef] [PubMed]
- Riemann, D.; Tcherkes, A.; Hansen, G.H.; Wulfaenger, J.; Blosz, T.; Danielsen, E.M. Functional co-localization of monocytic aminopeptidase N/CD13 with the Fc gamma receptors CD32 and CD64. Biochem. Biophys. Res. Commun. 2005, 331, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Wojewodzka, U.; Gajkowska, B.; Orzechowski, A. Lipid rafts in anticancer therapy: Theory and practice (Review). Mol. Med. Rep. 2008, 1, 167–172. [Google Scholar] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Marquez, D.C.; Chen, H.W.; Curran, E.M.; Welshons, W.V.; Pietras, R.J. Estrogen receptors in membrane lipid rafts and signal transduction in breast cancer. Mol. Cell Endocrinol. 2006, 246, 91–100. [Google Scholar] [CrossRef]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.W.; Wang, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2003, 95, 353–361. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.; Levin, E.R. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J. Biol. Chem. 2002, 277, 50768–50775. [Google Scholar] [CrossRef]
- Mahammad, S.; Parmryd, I. Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol. Biol. 2015, 1232, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Beckwitt, C.H.; Brufsky, A.; Oltvai, Z.N.; Wells, A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Novel Molecular Markers for Breast Cancer. Biomark. Cancer 2016, 8, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.L.; Musgrove, E.A. Cyclins and breast cancer. J. Mammary Gland Biol. Neoplasia 2004, 9, 95–104. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maselli, A.; Parlato, S.; Puglisi, R.; Raggi, C.; Spada, M.; Macchia, D.; Pontecorvi, G.; Iessi, E.; Pagano, M.T.; Cirulli, F.; et al. Autoantibodies Specific to ERα are Involved in Tamoxifen Resistance in Hormone Receptor Positive Breast Cancer. Cells 2019, 8, 750. https://doi.org/10.3390/cells8070750
Maselli A, Parlato S, Puglisi R, Raggi C, Spada M, Macchia D, Pontecorvi G, Iessi E, Pagano MT, Cirulli F, et al. Autoantibodies Specific to ERα are Involved in Tamoxifen Resistance in Hormone Receptor Positive Breast Cancer. Cells. 2019; 8(7):750. https://doi.org/10.3390/cells8070750
Chicago/Turabian StyleMaselli, Angela, Stefania Parlato, Rossella Puglisi, Carla Raggi, Massimo Spada, Daniele Macchia, Giada Pontecorvi, Elisabetta Iessi, Maria Teresa Pagano, Francesca Cirulli, and et al. 2019. "Autoantibodies Specific to ERα are Involved in Tamoxifen Resistance in Hormone Receptor Positive Breast Cancer" Cells 8, no. 7: 750. https://doi.org/10.3390/cells8070750
APA StyleMaselli, A., Parlato, S., Puglisi, R., Raggi, C., Spada, M., Macchia, D., Pontecorvi, G., Iessi, E., Pagano, M. T., Cirulli, F., Gabriele, L., Carè, A., Vici, P., Pizzuti, L., Barba, M., Matarrese, P., Pierdominici, M., & Ortona, E. (2019). Autoantibodies Specific to ERα are Involved in Tamoxifen Resistance in Hormone Receptor Positive Breast Cancer. Cells, 8(7), 750. https://doi.org/10.3390/cells8070750