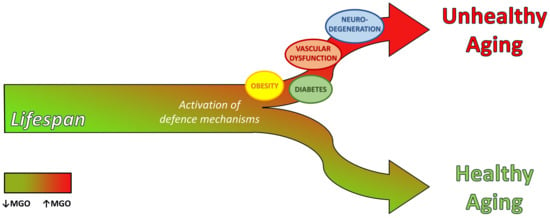
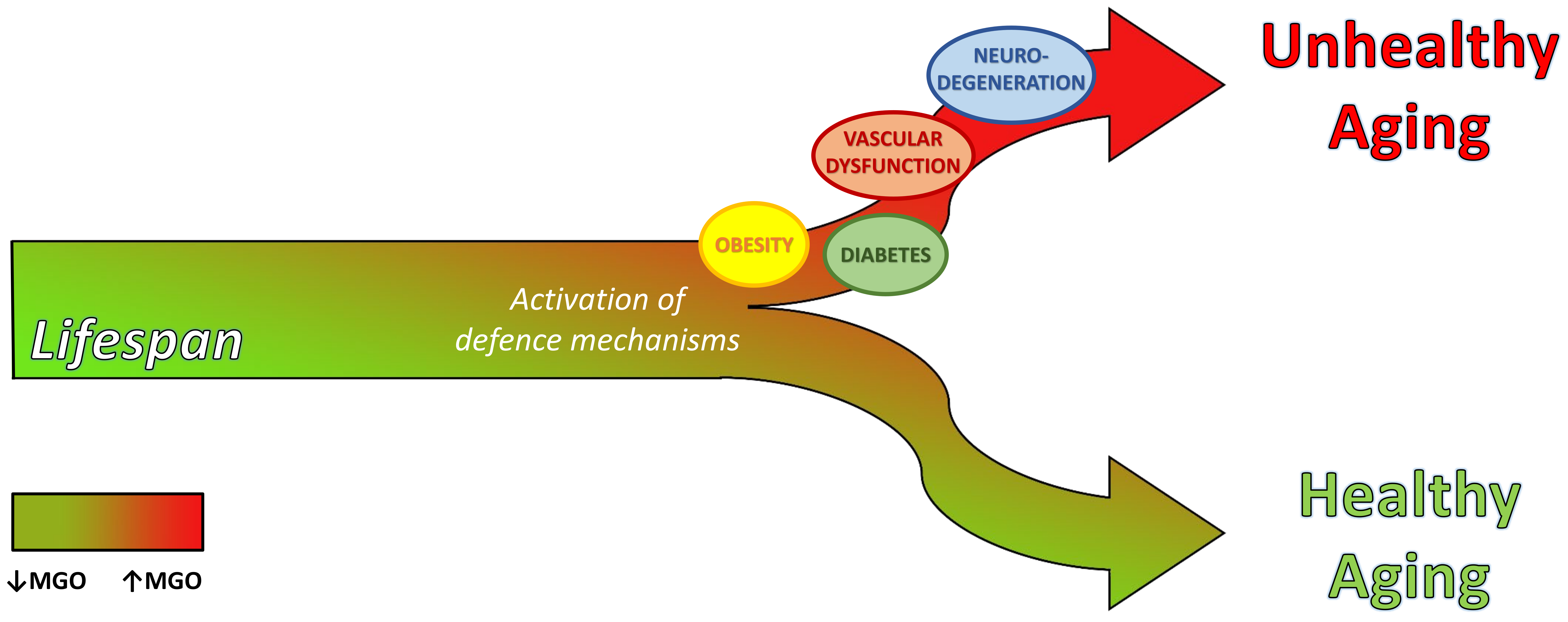
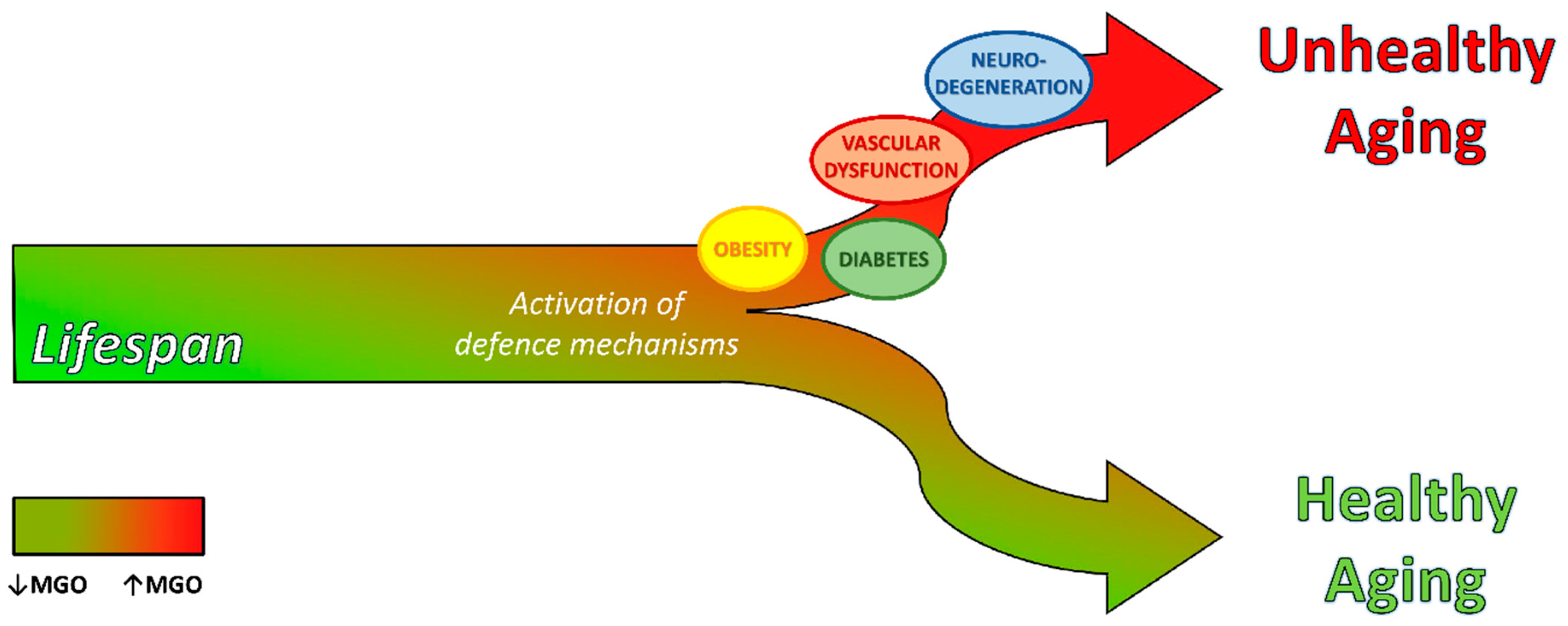
Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging

Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction—Dicarbonyl Stress and Glycation
2. Hormesis: A First Line of Cellular Defense to Counteract Dicarbonyl Stress
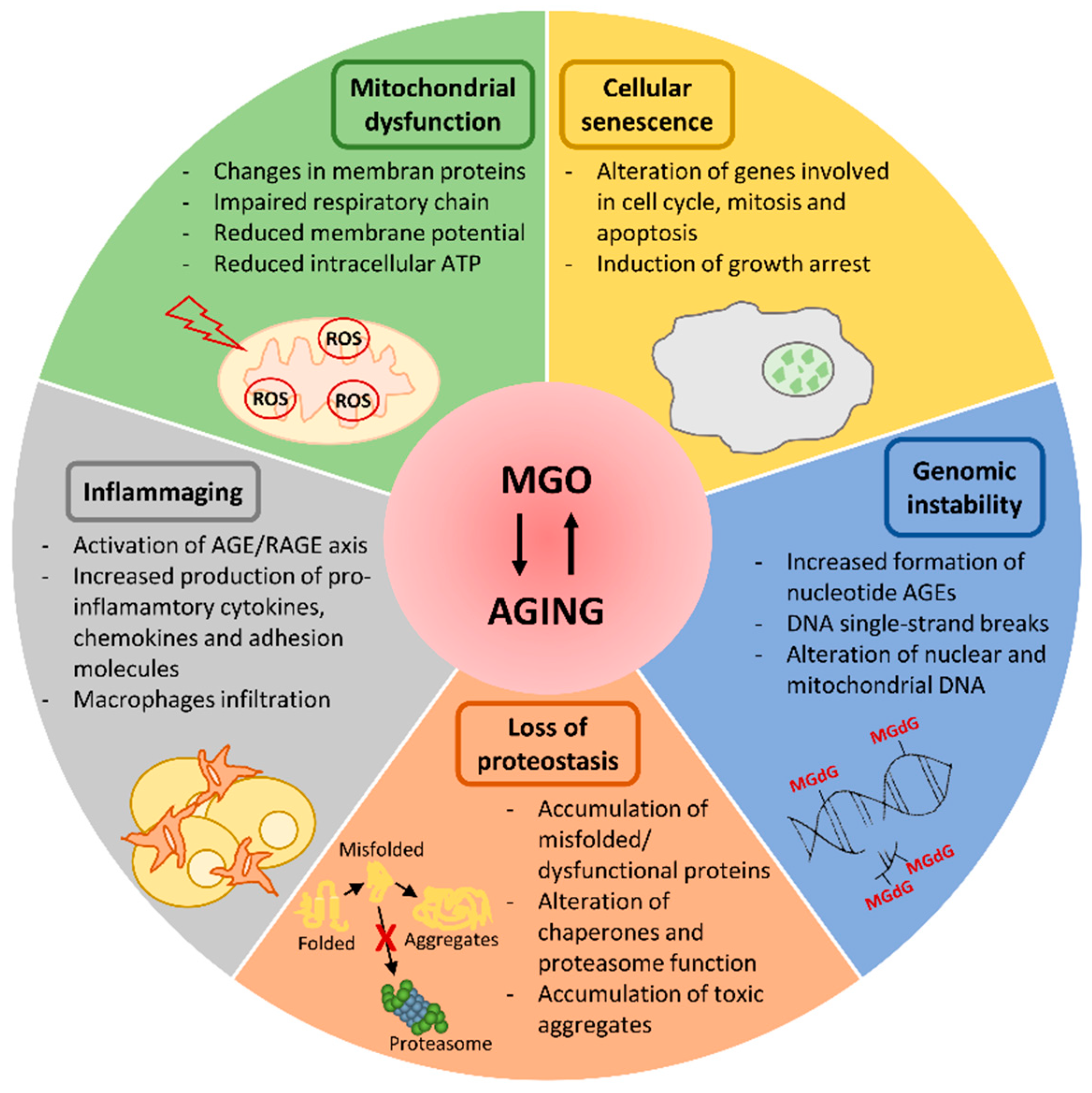
3. Dicarbonyl Stress in Aging
3.1. Mitochondrial Dysfunction
3.2. Loss of Proteostasis
3.3. Cellular Senescence
3.4. Inflammaging
3.5. Genomic Instability
4. Dicarbonyl Stress in Aging-Related Diseases
4.1. Metabolic Disease
4.1.1. Type 2 Diabetes (T2D)
4.1.2. Obesity
4.2. Vascular Dysfunction
4.2.1. Microvascular Dysfunction
4.2.2. Macrovascular Dysfunction
4.2.3. Acute Disease
4.3. Neurodegeneration
4.3.1. Glycation Targets in Parkinson’s (PD) and Alzheimer’s Disease (AD)
4.3.2. Dicarbonyl Stress and Cognitive Decline
4.3.3. Methylglyoxal (MGO) Detoxification and Neurodegeneration
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Rabbani, N.; Xue, M.; Thornalley, P.J. Methylglyoxal-induced dicarbonyl stress in aging and disease: First steps towards glyoxalase 1-based treatments. Clin. Sci. 2016, 130, 1677–1696. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Glyoxalase 1 Modulation in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 30, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates: Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344 Pt 1, 109–116. [Google Scholar] [CrossRef]
- Kazachkov, M.; Yu, P.H. A novel HPLC procedure for detection and quantification of aminoacetone, a precursor of methylglyoxal, in biological samples. J. Chromatogr. B 2005, 824, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Reichard, G.A., Jr.; Skutches, C.L.; Hoeldtke, R.D.; Owen, O.E. Acetone metabolism in humans during diabetic ketoacidosis. Diabetes 1986, 35, 668–674. [Google Scholar] [CrossRef]
- Degen, J.; Vogel, M.; Richter, D.; Hellwig, M.; Henle, T. Metabolic transit of dietary methylglyoxal. J. Agric. Food Chem. 2013, 61, 10253–10260. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem. Soc. Trans. 2014, 42, 425–432. [Google Scholar] [CrossRef]
- Rae, C.; Berners-Price, S.J.; Bulliman, B.T.; Kuchel, P.W. Kinetic analysis of the human erythrocyte glyoxalase system using 1H NMR and a computer model. Eur. J. Biochem. 1990, 193, 83–90. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Yurek-George, A.; Argirov, O.K. Kinetics and mechanism of the reaction of aminoguanidine with the alpha-oxoaldehydes glyoxal, methylglyoxal, and 3-deoxyglucosone under physiological conditions. Biochem. Pharmacol. 2000, 60, 55–65. [Google Scholar] [CrossRef]
- van Bussel, B.C.; van de Poll, M.C.; Schalkwijk, C.G.; Bergmans, D.C. Increased Dicarbonyl Stress as a Novel Mechanism of Multi-Organ Failure in Critical Illness. Int. J. Mol. Sci. 2017, 18, 346. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Gallet, X.; Charloteaux, B.; Thomas, A.; Brasseur, R. A fast method to predict protein interaction sites from sequences. J. Mol. Biol. 2000, 302, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Dobler, D.; Dean, M.; Thornalley, P.J. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 2005, 280, 5724–5732. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Waris, S.; Fleming, T.; Santarius, T.; Larkin, S.J.; Winklhofer-Roob, B.M.; Stratton, M.R.; Rabbani, N. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 2010, 38, 5432–5442. [Google Scholar] [CrossRef]
- Bilova, T.; Paudel, G.; Shilyaev, N.; Schmidt, R.; Brauch, D.; Tarakhovskaya, E.; Milrud, S.; Smolikova, G.; Tissier, A.; Vogt, T.; et al. Global proteomic analysis of advanced glycation end products in the Arabidopsis proteome provides evidence for age-related glycation hot spots. J. Biol. Chem. 2017, 292, 15758–15776. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef]
- Collard, F.; Vertommen, D.; Fortpied, J.; Duester, G.; Van Schaftingen, E. Identification of 3-deoxyglucosone dehydrogenase as aldehyde dehydrogenase 1A1 (retinaldehyde dehydrogenase 1). Biochimie 2007, 89, 369–373. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Asp. Med. 1993, 14, 287–371. [Google Scholar] [CrossRef]
- Abordo, E.A.; Minhas, H.S.; Thornalley, P.J. Accumulation of alpha-oxoaldehydes during oxidative stress: A role in cytotoxicity. Biochem. Pharmacol. 1999, 58, 641–648. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Momiji, H.; Rabbani, N.; Barker, G.; Bretschneider, T.; Shmygol, A.; Rand, D.A.; Thornalley, P.J. Frequency Modulated Translocational Oscillations of Nrf2 Mediate the Antioxidant Response Element Cytoprotective Transcriptional Response. Antioxid. Redox Signal. 2015, 23, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Dhawan, G.; Kapoor, R.; Iavicoli, I.; Calabrese, V. HORMESIS: A Fundamental Concept with Widespread Biological and Biomedical Applications. Gerontology 2016, 62, 530–535. [Google Scholar] [CrossRef]
- Pennisi, M.; Crupi, R.; Di Paola, R.; Ontario, M.L.; Bella, R.; Calabrese, E.J.; Crea, R.; Cuzzocrea, S.; Calabrese, V. Inflammasomes, hormesis, and antioxidants in neuroinflammation: Role of NRLP3 in Alzheimer disease. J. Neurosci. Res. 2017, 95, 1360–1372. [Google Scholar] [CrossRef]
- Pomatto, L.C.D.; Davies, K.J.A. Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 2018, 124, 420–430. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Respon. 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Zemva, J.; Fink, C.A.; Fleming, T.H.; Schmidt, L.; Loft, A.; Herzig, S.; Knieß, R.A.; Mayer, M.; Bukau, B.; Nawroth, P.P.; et al. Hormesis enables cells to handle accumulating toxic metabolites during increased energy flux. Redox Biol. 2017, 13, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, M.; Priebe, S.; Grigolon, G.; Rozanov, L.; Groth, M.; Laube, B.; Guthke, R.; Platzer, M.; Zarse, K.; Ristow, M. Impairing L-Threonine Catabolism Promotes Healthspan through Methylglyoxal-Mediated Proteohormesis. Cell Metab. 2018, 27, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59, 670–678. [Google Scholar] [CrossRef]
- Govindan, S.; Amirthalingam, M.; Duraisamy, K.; Govindhan, T.; Sundararaj, N.; Palanisamy, S. Phytochemicals-induced hormesis protects Caenorhabditis elegans against alpha-synuclein protein aggregation and stress through modulating HSF-1 and SKN-1/Nrf2 signaling pathways. Biomed. Pharmacother. 2018, 102, 812–822. [Google Scholar] [CrossRef]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27, 926–934. [Google Scholar] [CrossRef]
- Fabre, N.T.; Thieme, K.; Silva, K.S.; Catanozi, S.; Cavaleiro, A.M.; Pinto, D.A., Jr.; Okamoto, M.M.; Morais, M.R.; Falquetto, B.; Zorn, T.M.; et al. Hormetic modulation of hepatic insulin sensitivity by advanced glycation end products. Mol. Cell. Endocrinol. 2017, 447, 116–124. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef]
- Thao, M.T.; Gaillard, E.R. The glycation of fibronectin by glycolaldehyde and methylglyoxal as a model for aging in Bruch’s membrane. Amino Acids 2016, 48, 1631–1639. [Google Scholar] [CrossRef]
- Trellu, S.; Courties, A.; Jaisson, S.; Gorisse, L.; Gillery, P.; Kerdine-Römer, S.; Vaamonde-Garcia, C.; Houard, X.; Ekhirch, F.P.; Sautet, A.; et al. Impairment of glyoxalase-1, an advanced glycation end-product detoxifying enzyme, induced by inflammation in age-related osteoarthritis. Arthritis Res. Ther. 2019. [Google Scholar] [CrossRef]
- Tasatargil, A.; Tanriover, G.; Barutcigil, A. Turkmen, E. Protective effect of resveratrol on methylglyoxal-induced endothelial dysfunction in aged rats. Aging Clin. Exp. Res. 2019, 31, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Castro, J.P.; Hugo, M.; Braune, S.; Weber, D.; Pignitter, M.; Somoza, V.; Bornhorst, J.; Schwerdtle, T.; Grune, T. Oxidants produced by methylglyoxal-modified collagen trigger ER stress and apoptosis in skin fibroblasts. Free Radic. Biol. Med. 2018, 120, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Narda, M.; Peno-Mazzarino, L.; Krutmann, J.; Trullas, C.; Granger, C. Novel Facial Cream Containing Carnosine Inhibits Formation of Advanced Glycation End-Products in Human Skin. Skin Pharmacol. Physiol. 2018, 31, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyls linked to damage in the powerhouse: Glycation of mitochondrial proteins and oxidative stress. Biochem. Soc. Trans. 2008, 36, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.M.; Chang, T.; Wang, H.; Banigesh, A.; Dhar, A.; Liu, J.; Untereiner, A.; Wu, L. Oxidative stress and aging: Is methylglyoxal the hidden enemy? Can. J. Physiol. Pharmacol. 2010, 88, 273–284. [Google Scholar] [CrossRef]
- Seo, K.; Seo, S.; Han, J.Y.; Ki, S.H.; Shin, S.M. Resveratrol attenuates methylglyoxal-induced mitochondrial dysfunction and apoptosis by Sestrin2 induction. Toxicol. Appl. Pharmacol. 2014, 280, 314–322. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Padilla-Tejeda, D.E.; Carrillo-Ibarra, S.; Miranda-Díaz, A.G. Oxidative Stress, Apoptosis, and Mitochondrial Function in Diabetic Nephropathy. Int. J. Endocrinol. 2018. [Google Scholar] [CrossRef]
- Bellahcene, A.; Nokin, M.J.; Castronovo, V.; Schalkwijk, C. Methylglyoxal-derived stress: An emerging biological factor involved in the onset and progression of cancer. Semin. Cancer Biol. 2018, 49, 64–74. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; Wu, L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem. Pharmacol. 2009, 77, 1709–1716. [Google Scholar] [CrossRef]
- de Arriba, S.G.; Stuchbury, G.; Yarin, J.; Burnell, J.; Loske, C.; Münch, G. Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells—Protection by carbonyl scavengers. Neurobiol. Aging 2007, 28, 1044–1050. [Google Scholar] [CrossRef]
- Chang, Y.C.; Hsieh, M.C.; Wu, H.J.; Wu, W.C.; Kao, Y.H. Methylglyoxal, a reactive glucose metabolite, enhances autophagy flux and suppresses proliferation of human retinal pigment epithelial ARPE-19 cells. Toxicol. In Vitro 2015, 29, 1358–1368. [Google Scholar] [CrossRef]
- Suh, K.S.; Choi, E.M.; Rhee, S.Y.; Kim, Y.S. Methylglyoxal induces oxidative stress and mitochondrial dysfunction in osteoblastic MC3T3-E1 cells. Free Radic. Res. 2014, 48, 206–217. [Google Scholar] [CrossRef]
- Suh, K.S.; Chon, S.; Choi, E.M. Protective effects of piceatannol on methylglyoxal-induced cytotoxicity in MC3T3-E1 osteoblastic cells. Free Radic. Res. 2018, 52, 712–723. [Google Scholar] [CrossRef]
- Breyer, V.; Becker, C.M.; Pischetsrieder, M. Intracellular glycation of nuclear DNA, mitochondrial DNA, and cytosolic proteins during senescence-like growth arrest. DNA Cell Biol. 2011, 30, 681–689. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef]
- Rabbani, N.; Shaheen, F.; Anwar, A.; Masania, J.; Thornalley, P.J. Assay of methylglyoxal-derived protein and nucleotide AGEs. Biochem. Soc. Trans. 2014, 42, 511–517. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; van Bezu, J.; van der Schors, R.C.; Uchida, K.; Stehouwer, C.D.; van Hinsbergh, V.W. Heat-shock protein 27 is a major methylglyoxal-modified protein in endothelial cells. FEBS Lett. 2006, 580, 1565–1570. [Google Scholar] [CrossRef]
- Oya-Ito, T.; Liu, B.F.; Nagaraj, R.H. Effect of methylglyoxal modification and phosphorylation on the chaperone and anti-apoptotic properties of heat shock protein 27. J. Cell. Biochem. 2006, 99, 279–291. [Google Scholar] [CrossRef]
- Sudnitsyna, M.V.; Gusev, N.B. Methylglyoxal and Small Heat Shock Proteins. Biochemistry 2017, 82, 751–759. [Google Scholar] [CrossRef]
- Gawlowski, T.; Stratmann, B.; Stork, I.; Engelbrecht, B.; Brodehl, A.; Niehaus, K.; Körfer, R.; Tschoepe, D.; Milting, H. Heat shock protein 27 modification is increased in the human diabetic failing heart. Horm. Metab. Res. 2009, 41, 594–599. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Aging, Proteotoxicity, Mitochondria, Glycation, NAD and Carnosine: Possible Inter-Relationships and Resolution of the Oxygen Paradox. Front. Aging Neurosci. 2010. [Google Scholar] [CrossRef]
- Bento, C.F.; Marques, F.; Fernandes, R.; Pereira, P. Methylglyoxal alters the function and stability of critical components of the protein quality control. PLoS ONE 2010. [Google Scholar] [CrossRef]
- Navarrete Santos, A.; Jacobs, K.; Simm, A.; Glaubitz, N.; Horstkorte, R.; Hofmann, B. Dicarbonyls induce senescence of human vascular endothelial cells. Mech. Ageing Dev. 2017, 166, 24–32. [Google Scholar] [CrossRef]
- Braun, J.D.; Pastene, D.O.; Breedijk, A.; Rodriguez, A.; Hofmann, B.B.; Sticht, C.; von Ochsenstein, E.; Allgayer, H.; van den Born, J.; Bakker, S.; et al. Methylglyoxal down-regulates the expression of cell cycle associated genes and activates the p53 pathway in human umbilical vein endothelial cells. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Kellow, N.J.; Coughlan, M.T. Effect of diet-derived advanced glycation end products on inflammation. Nutr. Rev. 2015, 73, 737–759. [Google Scholar] [CrossRef]
- Lv, X.; Lv, G.H.; Dai, G.Y.; Sun, H.M.; Xu, H.Q. Food-advanced glycation end products aggravate the diabetic vascular complications via modulating the AGEs/RAGE pathway. Chin. J. Nat. Med. 2016, 14, 844–855. [Google Scholar] [CrossRef]
- Longo, M.; Spinelli, R.; D’sposito, V.; Zatterale, F.; Fiory, F.; Nigro, C.; Raciti, G.A.; Miele, C.; Formisano, P.; Beguinot, F.; et al. Pathologic endoplasmic reticulum stress induced by glucotoxic insults inhibits adipocyte differentiation and induces an inflammatory phenotype. Biochim. Biophys. Acta 2016, 1863, 1146–1156. [Google Scholar] [CrossRef]
- Zhou, Q.; Xu, H.; Yu, W.; Li, E.; Wang, M. Anti-Inflammatory Effect of an Apigenin-Maillard Reaction Product in Macrophages and Macrophage-Endothelial Cocultures. Oxid. Med. Cell. Longev. 2019. [Google Scholar] [CrossRef]
- Yu, W.; Hu, X.; Wang, M. Pterostilbene inhibited advanced glycation end products (AGEs)-induced oxidative stress and inflammation by regulation of RAGE/MAPK/NF-κB in RAW264.7 cells. J. Funct. Foods 2018, 40, 272–279. [Google Scholar] [CrossRef]
- Subramanian, U.; Nagarajan, D. All-Trans Retinoic Acid supplementation prevents cardiac fibrosis and cytokines induced by Methylglyoxal. Glycoconj. J. 2017, 34, 255–265. [Google Scholar] [CrossRef]
- Vulesevic, B.; McNeill, B.; Giacco, F.; Maeda, K.; Blackburn, N.J.; Brownlee, M.; Milne, R.W.; Suuronen, E.J. Methylglyoxal-Induced Endothelial Cell Loss and Inflammation Contribute to the Development of Diabetic Cardiomyopathy. Diabetes 2016, 65, 1699–1713. [Google Scholar] [CrossRef]
- Kim, J.; Kim, N.H.; Sohn, E.; Kim, C.S.; Kim, J.S. Methylglyoxal induces cellular damage by increasing argpyrimidine accumulation and oxidative DNA damage in human lens epithelial cells. Biochem. Biophys. Res. Commun. 2010, 391, 346–351. [Google Scholar] [CrossRef]
- Li, H.; Nakamura, S.; Miyazaki, S.; Morita, T.; Suzuki, M.; Pischetsrieder, M.; Niwa, T. N2-carboxyethyl-2’-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney Int. 2006, 69, 388–392. [Google Scholar] [CrossRef]
- An, S.H.; Kang, J.H. Oxidative damage of DNA induced by the reaction of methylglyoxal with lysine in the presence of ferritin. BMB Rep. 2013, 46, 225–229. [Google Scholar] [CrossRef]
- Waris, S.; Winklhofer-Roob, B.M.; Roob, J.M.; Fuchs, S.; Sourij, H.; Rabbani, N.; Thornalley, P.J. Increased DNA dicarbonyl glycation and oxidation markers in patients with type 2 diabetes and link to diabetic nephropathy. J. Diabetes Res. 2015. [Google Scholar] [CrossRef]
- Sompong, W.; Cheng, H.; Adisakwattana, S. Ferulic acid prevents methylglyoxal-induced protein glycation, DNA damage, and apoptosis in pancreatic beta-cells. J. Physiol. Biochem. 2017, 73, 121–131. [Google Scholar] [CrossRef]
- Meeprom, A.; Sompong, W.; Suantawee, T.; Thilavech, T.; Chan, C.B.; Adisakwattana, S. Isoferulic acid prevents methylglyoxal-induced protein glycation and DNA damage by free radical scavenging activity. BMC Complement. Altern. Med. 2015. [Google Scholar] [CrossRef]
- Suantawee, T.; Cheng, H.; Adisakwattana, S. Protective effect of cyanidin against glucose- and methylglyoxal-induced protein glycation and oxidative DNA damage. Int. J. Biol. Macromol. 2016, 93, 814–821. [Google Scholar] [CrossRef]
- Al-Hussaini, H.; Kilarkaje, N. Trans-resveratrol mitigates type 1 diabetes-induced oxidative DNA damage and accumulation of advanced glycation end products in glomeruli and tubules of rat kidneys. Toxicol. Appl. Pharmacol. 2018, 339, 97–109. [Google Scholar] [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef]
- Risk Factor Collaboration. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef]
- Nigro, C.; Leone, A.; Raciti, G.A.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int. J. Mol. Sci. 2017, 18, 188. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P.H. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. 1994, 87, 21–29. [Google Scholar] [CrossRef]
- Lapolla, A.; Flamini, R.; Dalla Vedova, A.; Senesi, A.; Reitano, R.; Fedele, D.; Basso, E.; Seraglia, R.; Traldi, P. Glyoxal and methylglyoxal levels in diabetic patients: Quantitative determination by a new GC/MS method. Clin. Chem. Lab. Med. 2003, 41, 1166–1173. [Google Scholar] [CrossRef]
- Kold-Christensen, R.; Jensen, K.K.; Smedegård-Holmquist, E.; Sørensen, L.K.; Hansen, J.; Jørgensen, K.A.; Kristensen, P.; Johannsen, M. ReactELISA method for quantifying methylglyoxal levels in plasma and cell cultures. Redox Biol. 2019. [Google Scholar] [CrossRef]
- Kong, X.; Ma, M.Z.; Huang, K.; Qin, L.; Zhang, H.M.; Yang, Z.; Li, X.Y.; Su, Q. Increased plasma levels of the methylglyoxal in patients with newly diagnosed type 2 diabetes 2. J. Diabetes 2014, 6, 535–540. [Google Scholar] [CrossRef]
- Mey, J.T.; Haus, J.M. Dicarbonyl Stress and Glyoxalase-1 in Skeletal Muscle: Implications for Insulin Resistance and Type 2 Diabetes. Front. Cardiovasc. Med. 2018. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef]
- Mey, J.T.; Blackburn, B.K.; Miranda, E.R.; Chaves, A.B.; Briller, J.; Bonini, M.G.; Haus, J.M. Dicarbonyl stress and glyoxalase enzyme system regulation in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef]
- Amicarelli, F.; Ragnelli, A.M.; Aimola, P.; Bonfigli, A.; Colafarina, S.; Di Ilio, C.; Miranda, M. Age-dependent ultrastructural alterations and biochemical response of rat skeletal muscle after hypoxic or hyperoxic treatments. Biochim. Biophys. Acta 1999, 1453, 105–114. [Google Scholar] [CrossRef]
- Stratmann, B.; Goldstein, B.; Thornalley, P.J.; Rabbani, N.; Tschoepe, D. Intracellular Accumulation of Methylglyoxal by Glyoxalase 1 Knock Down Alters Collagen Homoeostasis in L6 Myoblasts. Int. J. Mol. Sci. 2017, 18, 480. [Google Scholar] [CrossRef]
- Lopez-Diez, R.; Shen, X.; Daffu, G.; Khursheed, M.; Hu, J.; Song, F.; Rosario, R.; Xu, Y.; Li, Q.; Xi, X.; et al. Ager Deletion Enhances Ischemic Muscle Inflammation, Angiogenesis, and Blood Flow Recovery in Diabetic Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1536–1547. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef]
- Engelbrecht, B.; Mattern, Y.; Scheibler, S.; Tschoepe, D.; Gawlowski, T.; Stratmann, B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm. Metab. Res. 2014, 46, 77–84. [Google Scholar] [CrossRef]
- Miele, C.; Riboulet, A.; Maitan, M.A.; Oriente, F.; Romano, C.; Formisano, P.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J. Biol. Chem. 2003, 278, 47376–47387. [Google Scholar] [CrossRef]
- Cassese, A.; Esposito, I.; Fiory, F.; Barbagallo, A.P.; Paturzo, F.; Mirra, P.; Ulianich, L.; Giacco, F.; Iadicicco, C.; Lombardi, A.; et al. In skeletal muscle advanced glycation end products (AGEs) inhibit insulin action and induce the formation of multimolecular complexes including the receptor for AGEs. J. Biol. Chem. 2008, 283, 36088–36099. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Jiang, B.; Desai, K.M.; Wu, L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes 2011, 60, 899–908. [Google Scholar] [CrossRef]
- Nigro, C.; Raciti, G.A.; Leone, A.; Fleming, T.H.; Longo, M.; Prevenzano, I.; Fiory, F.; Mirra, P.; D’Esposito, V.; Ulianich, L.; et al. Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia 2014, 57, 1485–1494. [Google Scholar] [CrossRef]
- Cheng, A.S.; Cheng, Y.H.; Lee, C.Y.; Chung, C.Y.; Chang, W.C. Resveratrol protects against methylglyoxal-induced hyperglycemia and pancreatic damage in vivo. Nutrients 2015, 7, 2850–2865. [Google Scholar] [CrossRef]
- Francisco, F.A.; Barella, L.F.; Silveira, S.D.S.; Saavedra, L.P.J.; Prates, K.V.; Alves, V.S.; Franco, C.C.D.S.; Miranda, R.A.; Ribeiro, T.A.; Tófolo, L.P.; et al. Methylglyoxal treatment in lactating mothers leads to type 2 diabetes phenotype in male rat offspring at adulthood. Eur. J. Nutr. 2018, 57, 477–486. [Google Scholar] [CrossRef]
- Sheader, E.A.; Benson, R.S.; Best, L. Cytotoxic action of methylglyoxal on insulin-secreting cells. Biochem. Pharmacol. 2001, 61, 1381–1386. [Google Scholar] [CrossRef]
- Fiory, F.; Lombardi, A.; Miele, C.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 2011, 54, 2941–2952. [Google Scholar] [CrossRef]
- Bo, J.; Xie, S.; Guo, Y.; Zhang, C.; Guan, Y.; Li, C.; Lu, J.; Meng, Q.H. Methylglyoxal Impairs Insulin Secretion of Pancreatic beta-Cells through Increased Production of ROS and Mitochondrial Dysfunction Mediated by Upregulation of UCP2 and MAPKs. J. Diabetes Res. 2016. [Google Scholar] [CrossRef]
- Elmhiri, G.; Barella, L.F.; Vieau, D.; Camous, S.; Mathias, P.C.; Abdennebi-Najar, L. Acute exposure to a precursor of advanced glycation end products induces a dual effect on the rat pancreatic islet function. Int. J. Endocrinol. 2014. [Google Scholar] [CrossRef]
- Cao, D.S.; Zhong, L.; Hsieh, T.H.; Abooj, M.; Bishnoi, M.; Hughes, L.; Premkumar, L.S. Expression of transient receptor potential ankyrin 1 (TRPA1) and its role in insulin release from rat pancreatic beta cells. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Matafome, P.; Rodrigues, T.; Sena, C.; Seiça, R. Methylglyoxal in Metabolic Disorders: Facts, Myths, and Promises. Med. Res. Rev. 2017, 37, 368–403. [Google Scholar] [CrossRef]
- Jia, X.; Olson, D.J.; Ross, A.R.; Wu, L. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 2006, 20, 1555–1557. [Google Scholar] [CrossRef]
- Wilson, A.F.; Elston, R.C.; Tran, L.D.; Siervogel, R.M. Use of the robust sib-pair method to screen for single-locus, multiple-locus, and pleiotropic effects: Application to traits related to hypertension. Am. J. Hum. Genet. 1991, 48, 862–872. [Google Scholar]
- Wuschke, S.; Dahm, S.; Schmidt, C.; Joost, H.G.; Al-Hasani, H. A meta-analysis of quantitative trait loci associated with body weight and adiposity in mice. Int. J. Obes. 2007, 31, 829–841. [Google Scholar] [CrossRef]
- Masania, J.; Malczewska-Malec, M.; Razny, U.; Goralska, J.; Zdzienicka, A.; Kiec-Wilk, B.; Gruca, A.; Stancel-Mozwillo, J.; Dembinska-Kiec, A.; Rabbani, N.; et al. Dicarbonyl stress in clinical obesity. Glycoconj. J. 2016, 33, 581–589. [Google Scholar] [CrossRef]
- Nye, C.; Kim, J.; Kalhan, S.C.; Hanson, R.W. Reassessing triglyceride synthesis in adipose tissue. Trends Endocrinol. Metab. 2008, 19, 356–361. [Google Scholar] [CrossRef]
- Matafome, P.; Rodrigues, T.; Seica, R. Glycation and Hypoxia: Two Key Factors for Adipose Tissue Dysfunction. Curr. Med. Chem. 2015, 22, 2417–2437. [Google Scholar] [CrossRef]
- Bento, C.F.; Fernandes, R.; Matafome, P.; Sena, C.; Seiça, R.; Pereira, P. Methylglyoxal-induced imbalance in the ratio of vascular endothelial growth factor to angiopoietin 2 secreted by retinal pigment epithelial cells leads to endothelial dysfunction. Exp. Physiol. 2010, 95, 955–970. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef]
- Ye, J.; Gao, Z.; Yin, J.; He, Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am. J. Physiol. Endocrinol. Metab. 2007. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Woodward, M.; Tripp, E.; Goldberg, L.; Pyzik, R.; Yee, K.; Tansman, L.; Chen, X.; Mani, V.; et al. Elevated serum advanced glycation endproducts in obese indicate risk for the metabolic syndrome: A link between healthy and unhealthy obesity? J. Clin. Endocrinol. Metab. 2015, 100, 1957–1966. [Google Scholar] [CrossRef]
- Maessen, D.E.; Hanssen, N.M.; Lips, M.A.; Scheijen, J.L.; Willems van Dijk, K.; Pijl, H.; Stehouwer, C.D.; Schalkwijk, C.G. Energy restriction and Roux-en-Y gastric bypass reduce postprandial alpha-dicarbonyl stress in obese women with type 2 diabetes. Diabetologia 2016, 59, 2013–2017. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef]
- Matafome, P.; Santos-Silva, D.; Crisóstomo, J.; Rodrigues, T.; Rodrigues, L.; Sena, C.M.; Pereira, P.; Seiça, R. Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch. Physiol. Biochem. 2012, 118, 58–68. [Google Scholar] [CrossRef]
- Rodrigues, T.; Matafome, P.; Sereno, J.; Almeida, J.; Castelhano, J.; Gamas, L.; Neves, C.; Gonçalves, S.; Carvalho, C.; Arslanagic, A.; et al. Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Jia, X.; Wu, L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef]
- Nilsson, P.M. Early Vascular Ageing—A Concept in Development. Eur. Endocrinol. 2015, 11, 26–31. [Google Scholar] [CrossRef]
- van Sloten, T.T. Vascular dysfunction: At the heart of cardiovascular disease, cognitive impairment and depressive symptoms. Artery Res. 2017, 19, 18–23. [Google Scholar] [CrossRef]
- Jorgens, K.; Stoll, S.J.; Pohl, J.; Fleming, T.H.; Sticht, C.; Nawroth, P.P.; Hammes, H.P.; Kroll, J. High tissue glucose alters intersomitic blood vessels in zebrafish via methylglyoxal targeting the VEGF receptor signaling cascade. Diabetes 2015, 64, 213–225. [Google Scholar] [CrossRef]
- Samsonov, M.V.; Khapchaev, A.Y.; Vorotnikov, A.V.; Vlasik, T.N.; Yanushevskaya, E.V.; Sidorova, M.V.; Efremov, E.E.; Lankin, V.Z. Shirinsky VPImpact of Atherosclerosis- and Diabetes-Related Dicarbonyls on Vascular Endothelial Permeability: A Comparative Assessment. Oxid. Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Liu, H.; Yu, S.; Zhang, H.; Xu, J. Angiogenesis impairment in diabetes: Role of methylglyoxal-induced receptor for advanced glycation endproducts, autophagy and vascular endothelial growth factor receptor 2. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef]
- Ahmed, U.; Dobler, D.; Larkin, S.J.; Rabbani, N.; Thornalley, P.J. Reversal of hyperglycemia-induced angiogenesis deficit of human endothelial cells by overexpression of glyoxalase 1 in vitro. Ann. N. Y. Acad. Sci. 2008, 1126, 262–264. [Google Scholar] [CrossRef]
- Li, H.; O’Meara, M.; Zhang, X.; Zhang, K.; Seyoum, B.; Yi, Z.; Kaufman, R.J. Monks TJ1,5, Wang JM7,2,8. Ameliorating Methylglyoxal-Induced Progenitor Cell Dysfunction for Tissue Repair in Diabetes. Diabetes 2019, 68, 1287–1302. [Google Scholar] [CrossRef]
- Brouwers, O.; Yu, L.; Niessen, P.; Slenter, J.; Jaspers, K.; Wagenaar, A.; Post, M.; Miyata, T.; Backes, W.; Stehouwer, C.; et al. Glyoxalase-1 overexpression partially prevents diabetes-induced impaired arteriogenesis in a rat hindlimb ligation model. Glycoconj. J. 2016, 33, 627–630. [Google Scholar] [CrossRef][Green Version]
- Nigro, C.; Leone, A.; Longo, M.; Prevenzano, I.; Fleming, T.H.; Nicolò, A.; Parrillo, L.; Spinelli, R.; Formisano, P.; Nawroth, P.P.; et al. Methylglyoxal accumulation de-regulates HoxA5 expression, thereby impairing angiogenesis in glyoxalase 1 knock-down mouse aortic endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 73–85. [Google Scholar] [CrossRef]
- Kim, J.; Kim, O.S.; Kim, C.S.; Kim, N.H.; Kim, J.S. Cytotoxic role of methylglyoxal in rat retinal pericytes: Involvement of a nuclear factor-kappaB and inducible nitric oxide synthase pathway. Chem. Biol. Interact. 2010, 188, 86–93. [Google Scholar] [CrossRef]
- Kim, J.; Kim, O.S.; Kim, C.S.; Sohn, E.; Jo, K.; Kim, J.S. Accumulation of argpyrimidine, a methylglyoxal-derived advanced glycation end product, increases apoptosis of lens epithelial cells both in vitro and in vivo. Exp. Mol. Med. 2012, 44, 167–175. [Google Scholar] [CrossRef]
- Chan, C.M.; Huang, D.Y.; Huang, Y.P.; Hsu, S.H.; Kang, L.Y.; Shen, C.M.; Lin, W.W. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. J. Cell. Mol. Med. 2016, 20, 1749–1760. [Google Scholar] [CrossRef]
- Kim, J.; Kim, C.S.; Lee, Y.M.; Jo, K.; Shin, S.D.; Kim, J.S. Methylglyoxal induces hyperpermeability of the blood-retinal barrier via the loss of tight junction proteins and the activation of matrix metalloproteinases. Graefe’s Arch. Clin. Exp. Ophthalmol. 2012, 250, 691–697. [Google Scholar] [CrossRef]
- Schlotterer, A.; Kolibabka, M.; Lin, J.; Acunman, K.; Dietrich, N.; Sticht, C.; Fleming, T.; Nawroth, P.; Hammes, H.P. Methylglyoxal induces retinopathy-type lesions in the absence of hyperglycemia: Studies in a rat model. FASEB J. 2019, 33, 4141–4153. [Google Scholar] [CrossRef]
- Hammes, H.P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef]
- Kolibabka, M.; Friedrichs, P.; Dietrich, N.; Fleming, T.; Schlotterer, A.; Hammes, H.P. Dicarbonyl Stress Mimics Diabetic Neurovascular Damage in the Retina. Exp. Clin. Endocrinol. Diabetes 2016, 124, 437–439. [Google Scholar] [CrossRef]
- Fosmark, D.S.; Berg, J.P.; Jensen, A.B.; Sandvik, L.; Agardh, E.; Agardh, C.D.; Hanssen, K.F. Increased retinopathy occurrence in type 1 diabetes patients with increased serum levels of the advanced glycation endproduct hydroimidazolone. Acta Ophthalmol. 2009, 87, 498–500. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef]
- Giacco, F.; Du, X.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M. Brownlee M.Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef]
- Rodrigues, L.; Matafome, P.; Crisóstomo, J.; Santos-Silva, D.; Sena, C.; Pereira, P.; Seiça, R. Advanced glycation end products and diabetic nephropathy: A comparative study using diabetic and normal rats with methylglyoxal-induced glycation. J. Physiol. Biochem. 2014, 70, 173–184. [Google Scholar] [CrossRef]
- Ikeda, Y.; Inagi, R.; Miyata, T.; Nagai, R.; Arai, M.; Miyashita, M.; Itokawa, M.; Fujita, T.; Nangaku, M. Glyoxalase I retards renal senescence. Am. J. Pathol. 2011, 179, 2810–2821. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Østergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; Russell, G.B.; Miller, M.E.; Rich, S.S.; Mauer, M. Early progression of diabetic nephropathy correlates with methylglyoxal-derived advanced glycation end products. Diabetes Care 2013, 36, 3234–3239. [Google Scholar] [CrossRef]
- Wang, X.J.; Ma, S.B.; Liu, Z.F.; Li, H.; Gao, W.Y. Elevated levels of alpha-dicarbonyl compounds in the plasma of type II diabetics and their relevance with diabetic nephropathy. J. Chromatogr. B 2019, 1106, 19–25. [Google Scholar] [CrossRef]
- Perco, P.; Ju, W.; Kerschbaum, J.; Leierer, J.; Menon, R.; Zhu, C.; Kretzler, M.; Mayer, G.; Rudnicki, M. Identification of dicarbonyl and L-xylulose reductase as a therapeutic target in human chronic kidney disease. JCI Insight 2019. [Google Scholar] [CrossRef]
- Makinen, V.P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segrè, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014. [Google Scholar] [CrossRef]
- van Sloten, T.T.; Henry, R.M.; Dekker, J.M.; Nijpels, G.; Unger, T.; Schram, M.T.; Stehouwer, C.D. Endothelial dysfunction plays a key role in increasing cardiovascular risk in type 2 diabetes: The Hoorn study. Hypertension 2014, 64, 1299–1305. [Google Scholar] [CrossRef]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef]
- Mirra, P.; Nigro, C.; Prevenzano, I.; Procopio, T.; Leone, A.; Raciti, G.A.; Andreozzi, F.; Longo, M.; Fiory, F.; Beguinot, F.; et al. The role of miR-190a in methylglyoxal-induced insulin resistance in endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 440–449. [Google Scholar] [CrossRef]
- Nigro, C.; Mirra, P.; Prevenzano, I.; Leone, A.; Fiory, F.; Longo, M.; Cabaro, S.; Oriente, F.; Beguinot, F.; Miele, C. miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal. Int. J. Mol. Sci. 2018, 19, 522. [Google Scholar] [CrossRef]
- Mukohda, M.; Morita, T.; Okada, M.; Hara, Y.; Yamawaki, H. Long-term methylglyoxal treatment causes endothelial dysfunction of rat isolated mesenteric artery. J. Vet. Med. Sci. 2013, 75, 151–157. [Google Scholar] [CrossRef]
- Turkseven, S.; Ertuna, E.; Yetik-Anacak, G.; Yasa, M. Methylglyoxal causes endothelial dysfunction: The role of endothelial nitric oxide synthase and AMP-activated protein kinase alpha. J. Basic Clin. Physiol. Pharmacol. 2014, 25, 109–115. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Desai, K.M.; Wu, L. Methylglyoxal scavengers attenuate endothelial dysfunction induced by methylglyoxal and high concentrations of glucose. Br. J. Pharmacol. 2010, 161, 1843–1856. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Haenen, G.; Miyata, T.; Brownlee, M.; Stehouwer, C.D.; De Mey, J.G.; Schalkwijk, C.G. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia 2010, 53, 989–1000. [Google Scholar] [CrossRef]
- Sena, C.M.; Matafome, P.; Crisóstomo, J.; Rodrigues, L.; Fernandes, R.; Pereira, P.; Seiça, R.M. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol. Res. 2012, 65, 497–506. [Google Scholar] [CrossRef]
- Dhar, I.; Dhar, A.; Wu, L.; Desai, K.M. Methylglyoxal, a reactive glucose metabolite, increases renin angiotensin aldosterone and blood pressure in male Sprague-Dawley rats. Am. J. Hypertens. 2014, 27, 308–316. [Google Scholar] [CrossRef]
- Yang, Y.; Konduru, A.S.; Cui, N.; Yu, L.; Trower, T.C.; Shi, W.; Shi, Y.; Jiang, C. Acute exposure of methylglyoxal leads to activation of KATP channels expressed in HEK293 cells. Acta Pharmacol. Sin. 2014, 35, 58–64. [Google Scholar] [CrossRef]
- Li, S.S.; Wu, Y.; Jin, X.; Jiang, C. The SUR2B subunit of rat vascular KATP channel is targeted by miR-9a-3p induced by prolonged exposure to methylglyoxal. Am. J. Physiol. Cell Physiol. 2015. [Google Scholar] [CrossRef]
- Jo-Watanabe, A.; Ohse, T.; Nishimatsu, H.; Takahashi, M.; Ikeda, Y.; Wada, T.; Shirakawa, J.; Nagai, R.; Miyata, T.; Nagano, T.; et al. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell 2014, 13, 519–528. [Google Scholar] [CrossRef]
- Tikellis, C.; Pickering, R.J.; Tsorotes, D.; Huet, O.; Cooper, M.E.; Jandeleit-Dahm, K.; Thomas, M.C. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes 2014, 63, 3915–3925. [Google Scholar] [CrossRef]
- Muniyappa, R.; Srinivas, P.R. Dicarbonyl stress and atherosclerosis: Is it all RAGE? Diabetes 2014, 63, 3587–3589. [Google Scholar] [CrossRef][Green Version]
- Soro-Paavonen, A.; Watson, A.M.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef]
- Berlanga, J.; Cibrian, D.; Guillén, I.; Freyre, F.; Alba, J.S.; Lopez-Saura, P.; Merino, N.; Aldama, A.; Quintela, A.M.; Triana, M.E.; et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 2005, 109, 83–95. [Google Scholar] [CrossRef]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shaheen, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Glycation of LDL by methylglyoxal increases arterial atherogenicity: A possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef]
- Rabbani, N.; Chittari, M.V.; Bodmer, C.W.; Zehnder, D.; Ceriello, A.; Thornalley, P.J. Increased glycation and oxidative damage to apolipoprotein B100 of LDL cholesterol in patients with type 2 diabetes and effect of metformin. Diabetes 2010, 59, 1038–1045. [Google Scholar] [CrossRef][Green Version]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.; Thornalley, P.J.; Rabbani, N. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr. Diabetes 2014. [Google Scholar] [CrossRef]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef]
- Heier, M.; Margeirsdottir, H.D.; Torjesen, P.A.; Seljeflot, I.; Stensæth, K.H.; Gaarder, M.; Brunborg, C.; Hanssen, K.F.; Dahl-Jørgensen, K. The advanced glycation end product methylglyoxal-derived hydroimidazolone-1 and early signs of atherosclerosis in childhood diabetes. Diabetes Vasc. Dis. Res. 2015, 12, 139–145. [Google Scholar] [CrossRef]
- Sethi, S.; Rivera, O.; Oliveros, R.; Chilton, R. Aortic stiffness: Pathophysiology, clinical implications, and approach to treatment. Integr. Blood Press. Control 2014, 7, 29–34. [Google Scholar] [CrossRef]
- Hanssen, N.M.J.; Scheijen, J.L.J.M.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D.A.; Schalkwijk, C.G. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease in Individuals With Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes 2017, 66, 2278–2283. [Google Scholar] [CrossRef]
- Hanssen, N.M.J.; Westerink, J.; Scheijen, J.L.J.M.; van der Graaf, Y.; Stehouwer, C.D.A.; Schalkwijk, C.G. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease and Mortality in Individuals With Type 2 Diabetes. Diabetes Care 2018, 41, 1689–1695. [Google Scholar] [CrossRef]
- Peters, A.S.; Lercher, M.; Fleming, T.H.; Nawroth, P.P.; Bischoff, M.S.; Dihlmann, S.; Böckler, D.; Hakimi, M. Reduced glyoxalase 1 activity in carotid artery plaques of nondiabetic patients with increased hemoglobin A1c level. J. Vasc. Surg. 2016, 64, 990–994. [Google Scholar] [CrossRef]
- Schulman, C.I.; Uribarri, J.; Cai, W.; Manning, R.; Landy, D.C.; Gallardo, M.; Castillo, A.; Namias, N.; Striker, G.E.; Livingstone, A.; et al. Increased circulating advanced glycation end products (AGEs) in acute trauma patients. Clin. Chem. Lab. Med. 2014, 52, 103–108. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Ghosh, A.; Kar, M. Methylglyoxal increase in uremia with special reference to snakebite-mediated acute renal failure. Clin. Chim. Acta 2008, 391, 13–17. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Mishra, R.; Mukherjee, D.; Mishra, R.; Kar, M. Snakebite mediated acute kidney injury, prognostic predictors, oxidative and carbonyl stress: A prospective study. Indian J. Nephrol. 2016, 26, 427–433. [Google Scholar] [CrossRef]
- Kumagai, T.; Nangaku, M.; Kojima, I.; Nagai, R.; Ingelfinger, J.R.; Miyata, T.; Fujita, T.; Inagi, R. Glyoxalase I overexpression ameliorates renal ischemia-reperfusion injury in rats. Am. J. Physiol. Ren. Physiol. 2009. [Google Scholar] [CrossRef]
- Pieroh, P.; Koch, M.; Wagner, D.C.; Boltze, J.; Ehrlich, A.; Ghadban, C.; Hobusch, C.; Birkenmeier, G.; Dehghani, F. Temporal Dynamics of Glyoxalase 1 in Secondary Neuronal Injury. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Pieroh, P.; Wagner, D.C.; Alessandri, B.; Dabbagh Nazari, M.; Ehrlich, A.; Ghadban, C.; Hobusch, C.; Birkenmeier, G.; Dehghani, F. Comparative Examination of Temporal Glyoxalase 1 Variations Following Perforant Pathway Transection, Excitotoxicity, and Controlled Cortical Impact Injury. Neurotox. Res. 2018, 33, 412–421. [Google Scholar] [CrossRef]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef]
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Outeiro, T.F. The sour side of neurodegenerative disorders: The effects of protein glycation. J. Pathol. 2010, 221, 13–25. [Google Scholar] [CrossRef]
- Kikuchi, S.; Shinpo, K.; Moriwaka, F.; Makita, Z.; Miyata, T.; Tashiro, K. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: Synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J. Neurosci. Res. 1999, 57, 280–289. [Google Scholar] [CrossRef]
- Pamplona, R.; Dalfó, E.; Ayala, V.; Bellmunt, M.J.; Prat, J.; Ferrer, I.; Portero-Otín, M. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 2005, 280, 21522–21530. [Google Scholar] [CrossRef]
- Dalfo, E.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.Z.; Zhou, X.W. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J. Neuropathol. Exp. Neurol. 2005, 64, 816–830. [Google Scholar] [CrossRef]
- Castellani, R.; Smith, M.A.; Richey, P.L.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Munch, G.; Lüth, H.J.; Wong, A.; Arendt, T.; Hirsch, E.; Ravid, R.; Riederer, P. Crosslinking of alpha-synuclein by advanced glycation endproducts—An early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 2000, 20, 253–257. [Google Scholar] [CrossRef]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef]
- Woltjer, R.L.; Maezawa, I.; Ou, J.J.; Montine, K.S.; Montine, T.J. Advanced glycation endproduct precursor alters intracellular amyloid-beta/A beta PP carboxy-terminal fragment aggregation and cytotoxicity. J. Alzheimer’s Dis. 2003, 5, 467–476. [Google Scholar] [CrossRef]
- Ko, S.Y.; Ko, H.A.; Chu, K.H.; Shieh, T.M.; Chi, T.C.; Chen, H.I.; Chang, W.C.; Chang, S.S. The Possible Mechanism of Advanced Glycation End Products (AGEs) for Alzheimer’s Disease. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Ko, S.Y.; Lin, Y.P.; Lin, Y.S.; Chang, S.S. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic. Biol. Med. 2010, 49, 474–480. [Google Scholar] [CrossRef]
- Yan, S.S.; Chen, D.; Yan, S.; Guo, L.; Du, H.; Chen, J.X. RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front. Biosci. 2012, 4, 240–250. [Google Scholar] [CrossRef]
- Li, X.H.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.Z.; Zhou, X.W. Glycation exacerbates the neuronal toxicity of beta-amyloid. Cell Death Dis. 2013. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Szego, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Y.; Li, Y.; Xiao, S.; Song, D.; Qing, H.; Li, Q.; Rajput, A.H. Occurrence and distribution of salsolinol-like compound, 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (ADTIQ) in parkinsonian brains. J. Neural Transm. 2012, 119, 435–441. [Google Scholar] [CrossRef]
- Beeri, M.S.; Moshier, E.; Schmeidler, J.; Godbold, J.; Uribarri, J.; Reddy, S.; Sano, M.; Grossman, H.T.; Cai, W.; Vlassara, H. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech. Ageing Dev. 2011, 132, 583–587. [Google Scholar] [CrossRef]
- Srikanth, V.; Westcott, B.; Forbes, J.; Phan, T.G.; Beare, R.; Venn, A.; Pearson, S.; Greenaway, T.; Parameswaran, V.; Münch, G. Methylglyoxal, cognitive function and cerebral atrophy in older people. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 68–73. [Google Scholar] [CrossRef]
- Yaffe, K.; Lindquist, K.; Schwartz, A.V.; Vitartas, C.; Vittinghoff, E.; Satterfield, S.; Simonsick, E.M.; Launer, L.; Rosano, C.; Cauley, J.A.; et al. Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology 2011, 77, 1351–1356. [Google Scholar] [CrossRef]
- Southern, L.; Williams, J.; Esiri, M.M. Immunohistochemical study of N-epsilon-carboxymethyl lysine (CML) in human brain: Relation to vascular dementia. BMC Neurol. 2007. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.H.; Tu, Y.; Sun, H.T.; Liang, H.Q.; Cheng, S.X.; Zhang, S. Abeta-AGE aggravates cognitive deficit in rats via RAGE pathway. Neuroscience 2014, 257, 1–10. [Google Scholar] [CrossRef]
- More, S.S.; Vartak, A.P.; Vince, R. Restoration of glyoxalase enzyme activity precludes cognitive dysfunction in a mouse model of Alzheimer’s disease. ACS Chem. Neurosci. 2013, 4, 330–338. [Google Scholar] [CrossRef]
- Toyoda, Y.; Erkut, C.; Pan-Montojo, F.; Boland, S.; Stewart, M.P.; Müller, D.J.; Wurst, W.; Hyman, A.A.; Kurzchalia, T.V. Products of the Parkinson’s disease-related glyoxalase DJ-1, D-lactate and glycolate, support mitochondrial membrane potential and neuronal survival. Biol. Open 2014, 3, 777–784. [Google Scholar] [CrossRef]
- Sharma, N.; Rao, S.P.; Kalivendi, S.V. The deglycase activity of DJ-1 mitigates alpha-synuclein glycation and aggregation in dopaminergic cells: Role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic. Biol. Med. 2019, 135, 28–37. [Google Scholar] [CrossRef]
- Kuhla, B.; Lüth, H.J.; Haferburg, D.; Weick, M.; Reichenbach, A.; Arendt, T.; Münch, G. Pathological effects of glyoxalase I inhibition in SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 2006, 83, 1591–1600. [Google Scholar] [CrossRef]
- Bélanger, M.; Yang, J.; Petit, J.M.; Laroche, T.; Magistretti, P.J.; Allaman, I. Role of the glyoxalase system in astrocyte-mediated neuroprotection. J. Neurosci. 2011, 31, 18338–18352. [Google Scholar] [CrossRef]
- Ishige, K.; Schubert, D.; Sagara, Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001, 30, 433–446. [Google Scholar] [CrossRef]
- Myhrstad, M.C.; Carlsen, H.; Nordström, O.; Blomhoff, R.; Moskaug, J.Ø. Flavonoids increase the intracellular glutathione level by transactivation of the gamma-glutamylcysteine synthetase catalytical subunit promoter. Free Radic. Biol. Med. 2002, 32, 386–393. [Google Scholar] [CrossRef]
- Frandsen, J.; Narayanasamy, P. Flavonoid Enhances the Glyoxalase Pathway in Cerebellar Neurons to Retain Cellular Functions. Sci. Rep. 2017. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. https://doi.org/10.3390/cells8070749
Nigro C, Leone A, Fiory F, Prevenzano I, Nicolò A, Mirra P, Beguinot F, Miele C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells. 2019; 8(7):749. https://doi.org/10.3390/cells8070749
Chicago/Turabian StyleNigro, Cecilia, Alessia Leone, Francesca Fiory, Immacolata Prevenzano, Antonella Nicolò, Paola Mirra, Francesco Beguinot, and Claudia Miele. 2019. "Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging" Cells 8, no. 7: 749. https://doi.org/10.3390/cells8070749
APA StyleNigro, C., Leone, A., Fiory, F., Prevenzano, I., Nicolò, A., Mirra, P., Beguinot, F., & Miele, C. (2019). Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells, 8(7), 749. https://doi.org/10.3390/cells8070749