
Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
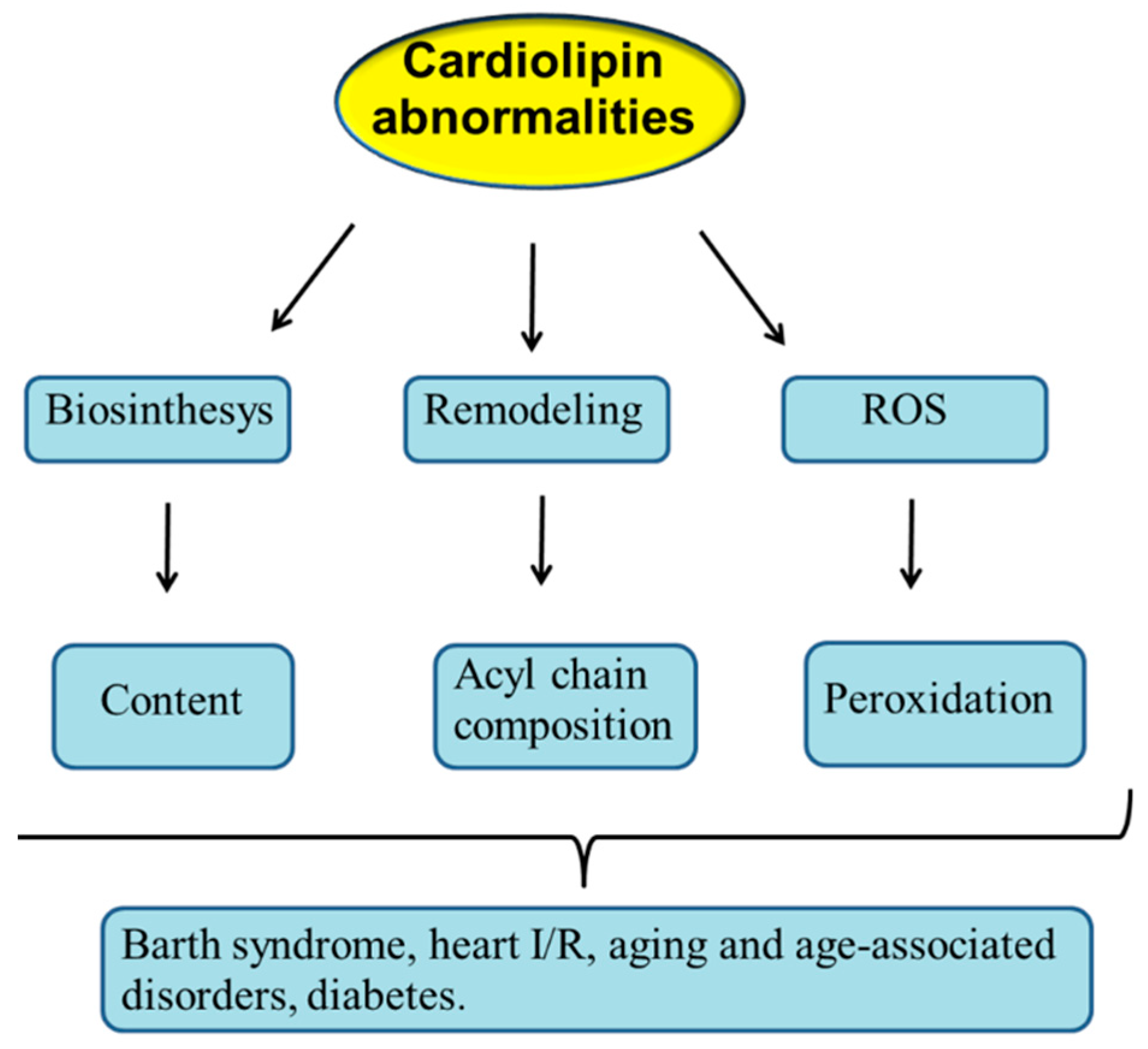
2. Cardiolipin Biosynthesis and Remodeling
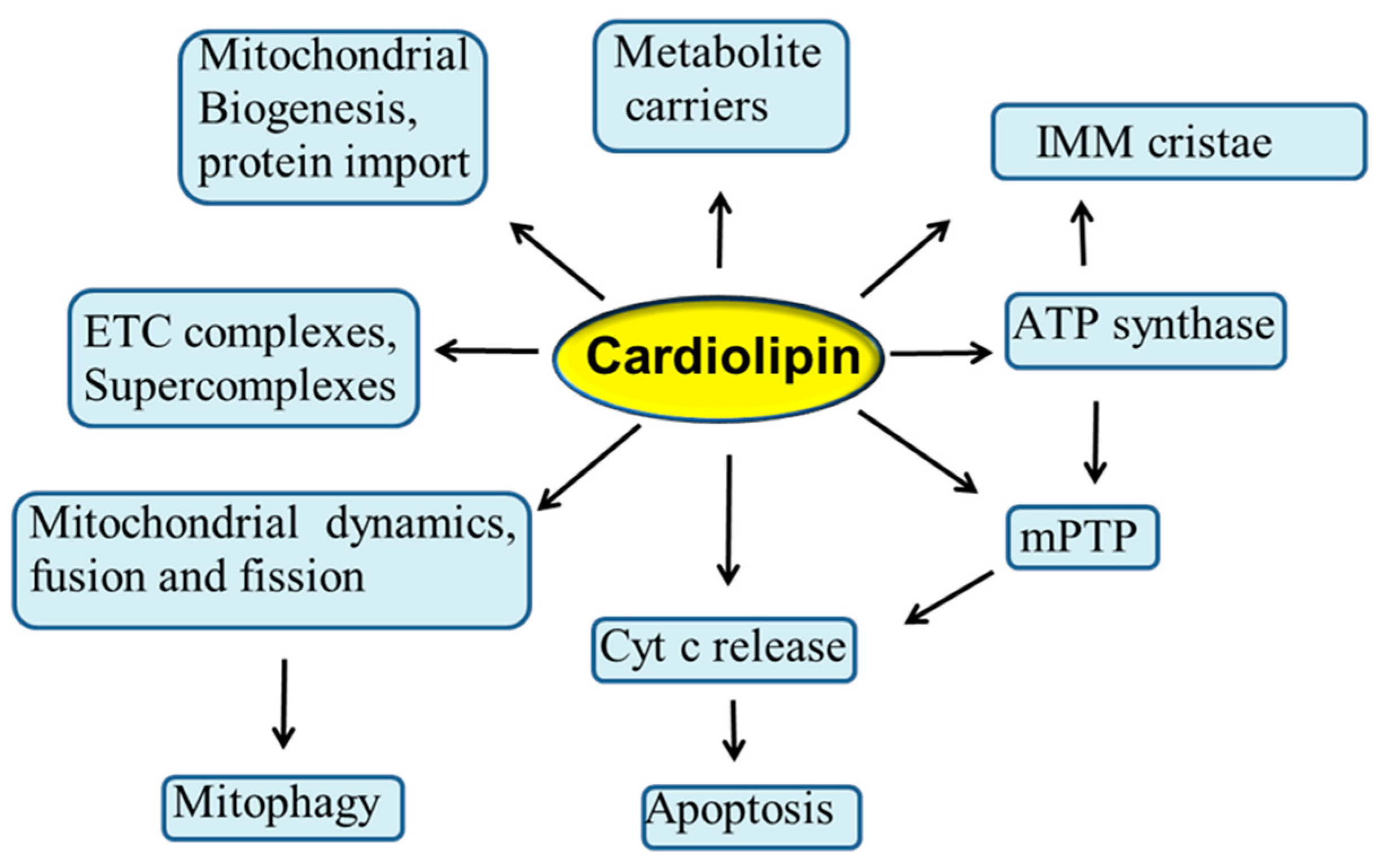
3. Cardiolipin and Mitochondrial Bioenergetics
3.1. Interaction of CL with Mitochondrial Substrate Carriers
3.2. Interaction of CL with ETC Complexes
3.3. Role of CL in Supercomplex Formation
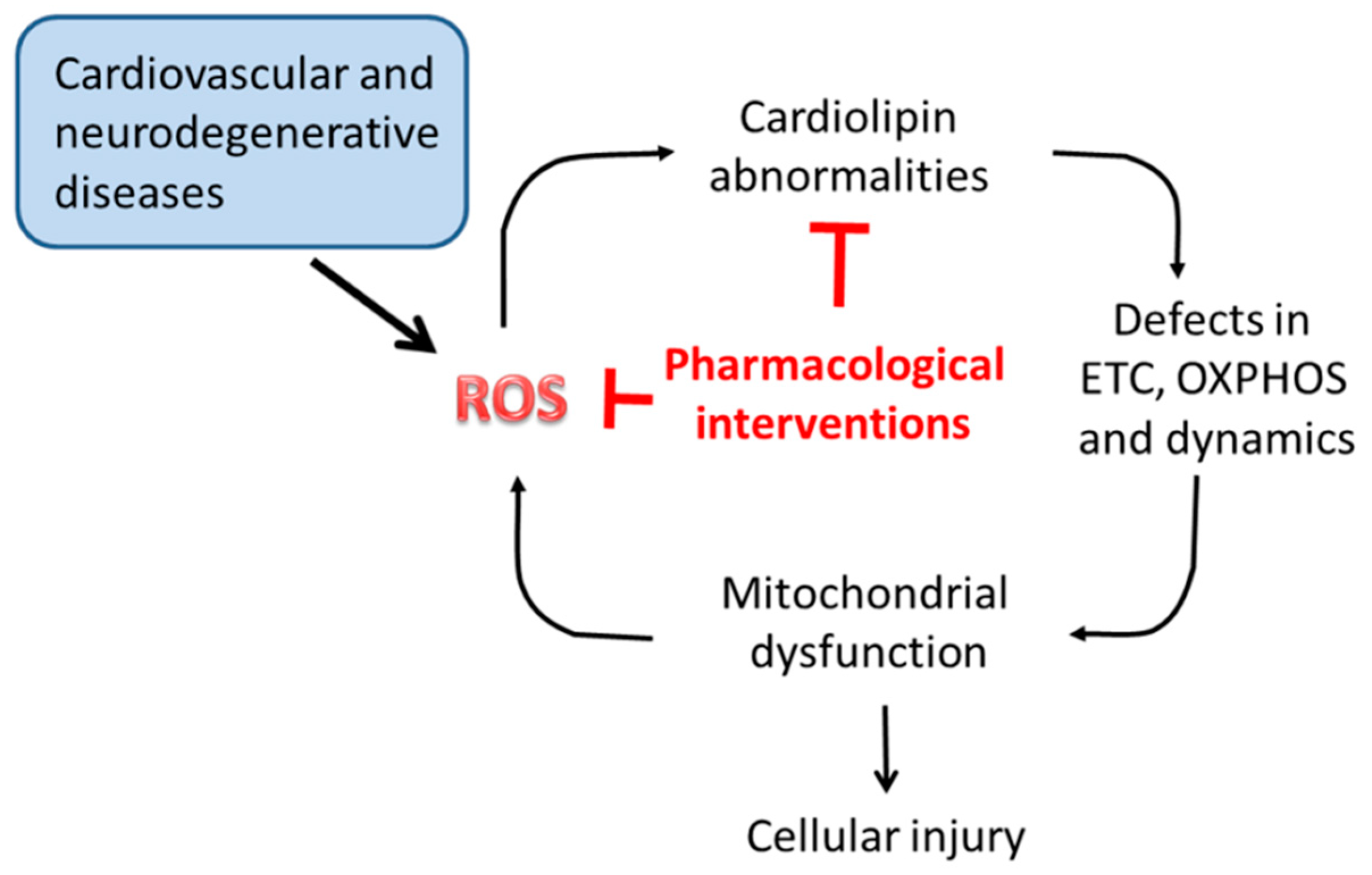
4. ROS and Cardiolipin Oxidation
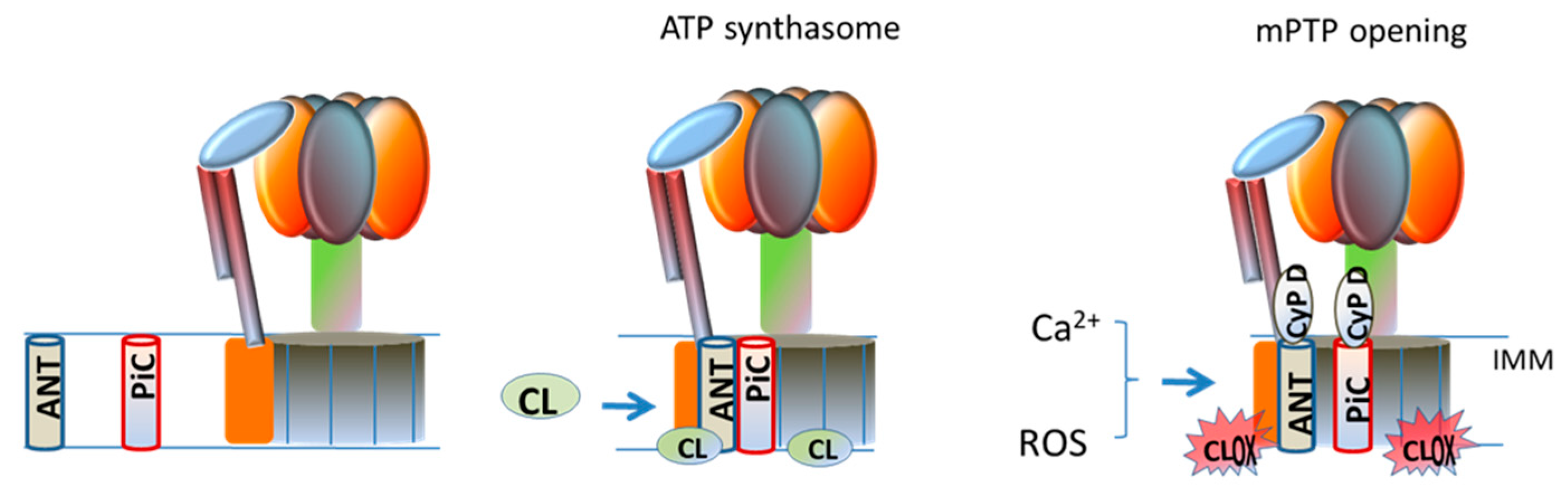
5. Interaction of Cardiolipin with ATP-Synthase and Implications for mPTP
6. Interaction of CL with Cytochrome C and Apoptosis
7. Role of Cardiolipin in Mitochondrial Dynamics
CL in Mitophagy
8. CL Alterations and Diseases
8.1. Barth Syndrome
8.2. Myocardial Ischemia/Reperfusion Injury
8.3. Diabetes
8.4. Parkinson’s Disease
9. Cardiolipin Targeting Therapeutic Agents in Disease
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pr. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Mejia, E.M.; Hatch, G.M. Mitocho, ndrial phospholipids: role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.W.; Claypool, S.M. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet. 2015, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ball, W.B.; Neff, J.K.; Gohil, V.M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2017, 592, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Elías-Wolff, F.; Lindén, M.; Lyubartsev, A.P.; Brandt, E.G. Curvature sensing by cardiolipin in simulated buckled membranes. Soft Matter 2019, 15, 792–802. [Google Scholar] [CrossRef]
- Claypool, S.M.; Koehler, C.M. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012, 37, 32–41. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Boil. 2017, 5. [Google Scholar] [CrossRef]
- Musatov, A.; Sedlák, E. Role of cardiolipin in stability of integral membrane proteins. Biochimie 2017, 142, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nature 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, K.; Modak, A.; Nangia, S.; Daman, T.H.; Gunsel, U.; Robinson, V.L.; Mokranjac, D.; May, E.R.; Alder, N.N. Cardiolipin mediates membrane and channel interactions of the mitochondrial TIM23 protein import complex receptor Tim50. Sci. Adv. 2017, 3, e1700532. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Greenberg, M.L. Mitochondrial membrane biogenesis: phospholipids and proteins go hand in hand: Figure 1. J. Cell Boil. 2009, 184, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U.; Tokarska-Schlattner, M.; Epand, R.M.; Boissan, M.; Lacombe, M.L.; Kagan, V.E. NME4/nucleoside diphosphate kinase D in cardiolipin signaling and mitophagy. Lab. Invest. 2018, 98, 228–232. [Google Scholar] [CrossRef]
- Dudek, J.; Hartmann, M.; Rehling, P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2019, 1865, 810–821. [Google Scholar] [CrossRef]
- Ott, M.; Zhivotovsky, B.; Orrenius, S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007, 14, 1243–1247. [Google Scholar] [CrossRef]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signaling platform to launch apoptosis. Biochim. Biophys. Acta. (BBA)-Biomembr. 2009, 1788, 2022–2031. [Google Scholar] [CrossRef]
- Lucken-Ardjomande, S.; Montessuit, S.; Martinou, J.C. Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death Differ. 2008, 15, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell calcium 2009, 45, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox Signal. 2014, 20, 1925–1953. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Chu, C.T.; Tyurina, Y.Y.; Cheikhi, A.; Bayir, H. Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 2014, 179, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Jalmar, O.; François-Moutal, L.; García-Sáez, A.J.; Perry, M.; Granjon, T.; Gonzalvez, F.; Gottlieb, E.; Ayala-Sanmartin, J.; Klösgen, B.; Schwille, P.; et al. Caspase-8 binding to cardiolipin in giant unilamellar vesicles provides a functional docking platform for bid. PLoS ONE 2013, 8, e55250. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef] [PubMed]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, J.; Yang, K.; Zhongdan, Z.; Abendschein, D.R.; Gross, R.W. Alterations in Myocardial Cardiolipin Content and Composition Occur at the Very Earliest Stages of Diabetes: A Shotgun Lipidomics Study. Biochemistry 2007, 46, 6417–6428. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, G.J.; McMaster, C.R. Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem. Phys. Lipids 2015, 193, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef] [PubMed]
- Pointer, C.B.; Klegeris, A. Cardiolipin in Central Nervous System Physiology and Pathology. Cell Mol. Neurobiol. 2017, 37, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. Am. J. Physiol. Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [PubMed]
- Pennington, E.R.; Funai, K.; Brown, D.A.; Shaikh, S.R. The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. et Biophys. Acta (BBA) - Mol. Cell Boil. Lipids 2019, 1864, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Mejia, E.M.; Nguyen, H.; Hatch, G.M. Mammalian cardiolipin biosynthesis. Chem. Phys. Lipids 2014, 179, 11–16. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006, 580, 5450–5455. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 2017, 52, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Schlattner, U.; Tokarska-Schlattner, M.; Rousseau, D.; Boissan, M.; Mannella, C.; Epand, R.; Lacombe, M.-L. Mitochondrial cardiolipin/phospholipid trafficking: The role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 2014, 179, 32–41. [Google Scholar] [CrossRef]
- Liu, J.; Dai, Q.; Chen, J.; Durrant, D.; Freeman, A.; Liu, T.; Grossman, D.; Lee, R.M. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 2003, 1, 892–902. [Google Scholar]
- Epand, R.F.; Tokarska-Schlattner, M.; Schlattner, U.; Wallimann, T.; Epand, R.M. Cardiolipin clusters and membrane domain formation induced by mitochondrial proteins. J. Mol. Biol. 2007, 365, 968–980. [Google Scholar] [CrossRef]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2016, 1863, 2362–2378. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M. Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta. 2009, 1788, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Nury, H.; Dahout-Gonzalez, C.; Trezeguet, V.; Lauquin, G.; Brandolin, G.; Pebay-Peyroula, E. Relations Between Structure and Function of the Mitochondrial ADP/ATP Carrier. Annu. Rev. Biochem. 2006, 75, 713–741. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Nett, J.H.; Trumpower, B.L.; Hunte, C. Specific roles of protein–phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001, 20, 6591–6600. [Google Scholar] [CrossRef]
- Musatov, A.; Robinson, N.C. Bound Cardiolipin Is Essential for Cytochrome c Oxidase Proton Translocation. Biochemistry 2014, 105, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Pöyry, S.; Cramariuc, O.; Postila, P.A.; Kaszuba, K.; Sarewicz, M.; Osyczka, A.; Vattulainen, I.; Rog, T. Atomistic simulations indicate cardiolipin to have an integral role in the structure of the cytochrome bc1 complex. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2013, 1827, 769–778. [Google Scholar] [CrossRef]
- Enríquez, J.A. Supramolecular Organization of Respiratory Complexes. Annu. Rev. Physiol. 2016, 78, 533–561. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 427–443. [Google Scholar] [CrossRef]
- A Letts, J.; A Sazanov, L. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Boil. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Claypool, S.M. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. et Biophys. Acta (BBA) - Biomembr. 2009, 1788, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.R.; Eid, R.; Boucher, E.; Miller, K.A.; Mandato, C.A.; Greenwood, M.T. Stress is an agonist for the induction of programmed cell death: A review. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2019, 1866, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000, 466, 323–326. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species generated by the mitochondrial respiratory chain affect the complex III activity via cardiolipin peroxidation in beef-heart submitochondrial particles. Mitochondrion 2001, 1, 151–159. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Uchida, K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Mehrotra, A.; Rigoni, G.; Soriano, M. Who and how in the regulation of mitochondrial cristae shape and function. Biochem. Biophys. Res. Commun. 2018, 500, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Rampelt, H.; Zerbes, R.M.; van der Laan, M.; Pfanner, N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Haines, T.H.; A Dencher, N. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 2002, 528, 35–39. [Google Scholar] [CrossRef]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Azzolini, M.; Szabò, I.; Zoratti, M. The mitochondrial permeabilità transition pore in AD 2016: An update. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2515–2530. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Checchetto, V.; Szabò, I.; Bernardi, P. F-ATP Synthase and the Permeability Transition Pore: Fewer doubts, more certainties. FEBS Lett. 2019, 593, 1542–1553. [Google Scholar] [CrossRef]
- Eble, K.S.; Coleman, W.B.; Hantgan, R.R.; Cunningham, C.C. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J. Boil. Chem. 1990, 265, 19434–19440. [Google Scholar]
- Duncan, A.L.; Robinson, A.J.; Walker, J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. 2016, 113, 8687–8692. [Google Scholar] [CrossRef]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. 2014, 111, 10396–10397. [Google Scholar] [CrossRef] [PubMed]
- Claypool, S.M.; Oktay, Y.; Boontheung, P.; Loo, J.A.; Koehler, C.M. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 2008, 182, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The C Ring of the F1Fo ATP Synthase Forms the Mitochondrial Permeability Transition Pore: A Critical Appraisal. Front. Oncol. 2014, 4, 234. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Pereverzev, M.O.; Vygodina, T.V.; Konstantinov, A.A.; Skulachev, V.P. Cytochrome c, an ideal antioxidant. Biochem. Soc. Trans. 2003, 31, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, E.K.J.; Wallace, C.J.A.; Kinnunen, P.K.J. Phospholipid-Cytochrome c Interaction: EVIDENCE FOR THE EXTENDED LIPID ANCHORAGE. J. Boil. Chem. 2002, 277, 8822–8826. [Google Scholar] [CrossRef]
- Sinibaldi, F.; Howes, B.D.; Piro, M.C.; Polticelli, F.; Bombelli, C.; Ferri, T.; Coletta, M.; Smulevich, G.; Santucci, R. Extended cardiolipin anchorage to cytochrome c: a model for protein–mitochondrial membrane binding. JBIC J. Boil. Inorg. Chem. 2010, 15, 689–700. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E.; et al. Peroxidase Activity and Structural Transitions of Cytochrome c Bound to Cardiolipin-Containing Membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Kakimoto, Y.; Furuta, S.; Itoh, K.; Sesaki, H.; Endo, T.; Tamura, Y. Maintenance of Cardiolipin and Crista Structure Requires Cooperative Functions of Mitochondrial Dynamics and Phospholipid Transport. Cell Rep. 2019, 26, 518–528.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.J.; Tyurina, Y.Y.; Mohammadyani, D.; Kapralov, A.A.; Anthonymuthu, T.S.; Qu, F.; Amoscato, A.A.; Sparvero, L.J.; Tyurin, V.A.; Planas-Iglesias, J.; et al. Known unknowns of cardiolipin signaling: The best is yet to come. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Stepanyants, N.; Macdonald, P.J.; Francy, C.A.; Mears, J.A.; Qi, X.; Ramachandran, R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol. Biol. Cell 2015, 26, 3104–3116. [Google Scholar] [CrossRef] [PubMed]
- Bustillo-Zabalbeitia, I.; Montessuit, S.; Raemy, E.; Basáñez, G.; Terrones, O.; Martinou, J.-C. Specific Interaction with Cardiolipin Triggers Functional Activation of Dynamin-Related Protein 1. PLOS ONE 2014, 9, e102738. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.-L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Ades, L.C.; Gedeon, A.K.; Wilson, M.J.; Latham, M.; Partington, M.W.; Mulley, J.C.; Nelson, J.; Lui, K.; Sillence, D.O. Barth syndrome: Clinical features and confirmation of gene localisation to distal Xq28. Am. J. Med Genet. 1993, 45, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial Respiratory Chain Supercomplexes Are Destabilized in Barth Syndrome Patients. J. Mol. Boil. 2006, 361, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Mejia, E.M.; Cole, L.K.; Hatch, G.M. Cardiolipin metabolism and the role it plays in heart failure and mitochondrial supercomplex formation. Cardiovasc. Hematol. Disord.-Drug Targets 2014, 14, 98–106. [Google Scholar] [CrossRef]
- Huang, Y.; Powers, C.; Madala, S.K.; Greis, K.D.; Haffey, W.D.; Towbin, J.A.; Purevjav, E.; Javadov, S.; Strauss, A.W.; Khuchua, Z. Cardiac metabolic pathways affected in the mouse model of barth syndrome. PLoS One 2015, 10, e0128561. [Google Scholar] [CrossRef]
- Acehan, D.; Xu, Y.; Stokes, D.L.; Schlame, M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 2007, 87, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. New Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Consolini, A.E.; Ragone, M.I.; Bonazzola, P.; Colareda, G.A. Mitochondrial Bioenergetics During Ischemia and Reperfusion. Mitochondrial Dyn. Cardiovasc. Med. 2017, 982, 141–167. [Google Scholar]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Boil. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed Res. Int. 2015, 2015, 864946–864995. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Serena, D.; Ruggiero, F.M. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic. Biol. Med. 1999, 27, 42–50. [Google Scholar] [CrossRef]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell Mol. Med. 2017, 21, 2643–2653. [Google Scholar] [CrossRef]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp. Diabetes. Res. 2012, 2012, 703538. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Morales, N.; Rovira-Llopis, S.; Escribano-Lopez, I.; Bañuls, C.; Lopez-Domenech, S.; Falcón, R.; De Maranon, A.M.; Sola, E.; Jover, A.; Roldan, I.; et al. Role of Oxidative Stress and Mitochondrial Dysfunction in Skeletal Muscle in Type 2 Diabetic Patients. Curr. Pharm. Des. 2016, 22, 2650–2656. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 2010, 24, 6–15. [Google Scholar] [CrossRef]
- Chang, W.; Hatch, G.M.; Wang, Y.; Yu, F.; Wang, M. The relationship between phospholipids and insulin resistance: From clinical to experimental studies. J. Cell Mol. Med. 2019, 23, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Filippi, B.M.; Abraham, M.A.; Silva, P.N.; Rasti, M.; Lapierre, M.P.; Bauer, P.V.; Rocheleau, J.V.; Lam, T.K. Dynamin-Related Protein 1-Dependent Mitochondrial Fission Changes in the Dorsal Vagal Complex Regulate Insulin Action. Cell Rep. 2017, 18, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: from pathology to molecular disease mechanisms. Free. Radic. Boil. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef]
- Ghio, S.; Kamp, F.; Cauchi, R.; Giese, A.; Vassallo, N. Interaction of α-synuclein with biomembranes in Parkinson’s disease —role of cardiolipin. Prog. Lipid Res. 2016, 61, 73–82. [Google Scholar] [CrossRef]
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barceló-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell Biol. 2005, 25, 10190–10201. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fabuel, I.; Martin-Martin, L.; Resch-Beusher, M.; Azkona, G.; Sanchez-Pernaute, R.; Bolaños, J.P. Mitochondrial respiratory chain disorganization in Parkinson’s disease-relevant PINK1 and DJ1 mutants. Neurochem. Int. 2017, 109, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Kapralov, A.A.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Belikova, N.A.; Vlasova, I.I.; Maeda, A.; et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C: protection against apoptosis versus delayed oxidative stress in Parkinson disease. J. Biol. Chem. 2009, 284, 15951–15969. [Google Scholar] [PubMed]
- Perier, C.; Tieu, K.; Guegan, C.; Caspersen, C.; Jackson-Lewis, V.; Carelli, V.; Vila, M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc. Natl. Acad. Sci. 2005, 102, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.; Paradies, G. Mitochondrial dysfunction in rat brain with agingInvolvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Chu, C.T.; Bayir, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J.; Qi, S.; Liu, Z.; Zhang, X.; Zheng, Y.; Andersen, J.-P.; Zhang, W.; Strong, R.; Martinez, P.A.; et al. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell 2019, 18, e12941. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Szeto, H.H.; Birk, A.V. Serendipity and the Discovery of Novel Compounds That Restore Mitochondrial Plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef]
- Petcherski, A.; Trudeau, K.M.; Wolf, D.M.; Segawa, M.; Lee, J.; Taddeo, E.P.; Deeney, J.T.; Liesa, M. Elamipretide Promotes Mitophagosome Formation and Prevents Its Reduction Induced by Nutrient Excess in INS1 β-cells. J. Mol. Boil. 2018, 430, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Shi, J.; Gupta, R.C.; Sabbah, H.N.; Hale, S.L.; Kloner, R.A. Bendavia, a Mitochondria-targeting Peptide, Improves Postinfarction Cardiac Function, Prevents Adverse Left Ventricular Remodeling, and Restores Mitochondria-related Gene Expression in Rats. J. Cardiovasc. Pharmacol. 2014, 64, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. et Biophys. Acta (BBA) - Mol. Cell Boil. Lipids 2016, 1861, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC: Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Moro, N.; Ruggiero, F.M.; Paradies, G. Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome c release. Free Radic. Biol. Med. 2009, 47, 969–974. [Google Scholar] [CrossRef]
- Mekhloufi, J.; Bonnefont-Rousselot, D.; Yous, S.; Lesieur, D.; Couturier, M.; Thérond, P.; Legrand, A.; Jore, D.; Gardès-Albert, M. Antioxidant activity of melatonin and a pinoline derivative on linoleate model system. J. Pineal Res. 2005, 39, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; de la Torre-Hernandez, J.M.; Consuegra-Sanchez, L.; Piccolo, R.; Gonzalez-Gonzalez, J.; Garcia-Camarero, T.; Del Mar Garcia-Saiz, M.; Aldea-Perona, A.; Reiter, R.J. Usefulness of Early Treatment With Melatonin to Reduce Infarct Size in Patients With ST-Segment Elevation Myocardial Infarction Receiving Percutaneous Coronary Intervention (From the Melatonin Adjunct in the Acute Myocardial Infarction Treated With Angioplasty Trial). Am. J. Cardiol. 2017, 120, 522–526. [Google Scholar]
- Wongprayoon, P.; Govitrapong, P. Melatonin as a mitochondrial protector in neurodegenerative diseases. Cell. Mol. Life Sci. 2017, 74, 3999–4014. [Google Scholar] [CrossRef] [PubMed]
- Feniouk, B.A.; Skulachev, V.P. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017, 10, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Antonenko, Y.N.; Cherepanov, D.A.; Chernyak, B.V.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; Korshunova, G.A.; Lyamzaev, K.G.; Pletjushkina, O.Y.; et al. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs). Biochim. et Biophys. Acta (BBA) - Bioenerg. 2010, 1797, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Cationic antioxidants as a powerful tool against mitochondrial oxidative stress. Biochem. Biophys. Res. Commun. 2013, 441, 275–279. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, R.; Lemaire, S.D.; Danon, A. Cardiolipin at the heart of stress response across kingdoms. Plant Signal. Behav. 2014, 9, e29228. [Google Scholar] [CrossRef] [PubMed][Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. https://doi.org/10.3390/cells8070728
Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells. 2019; 8(7):728. https://doi.org/10.3390/cells8070728
Chicago/Turabian StyleParadies, Giuseppe, Valeria Paradies, Francesca M. Ruggiero, and Giuseppe Petrosillo. 2019. "Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects" Cells 8, no. 7: 728. https://doi.org/10.3390/cells8070728
APA StyleParadies, G., Paradies, V., Ruggiero, F. M., & Petrosillo, G. (2019). Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells, 8(7), 728. https://doi.org/10.3390/cells8070728