Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis



Abstract
1. Introduction
2. Tissue Factor-Bearing EVs in Cancer-Associated Thrombosis
3. Platelets in Cancer-Associated Thrombosis
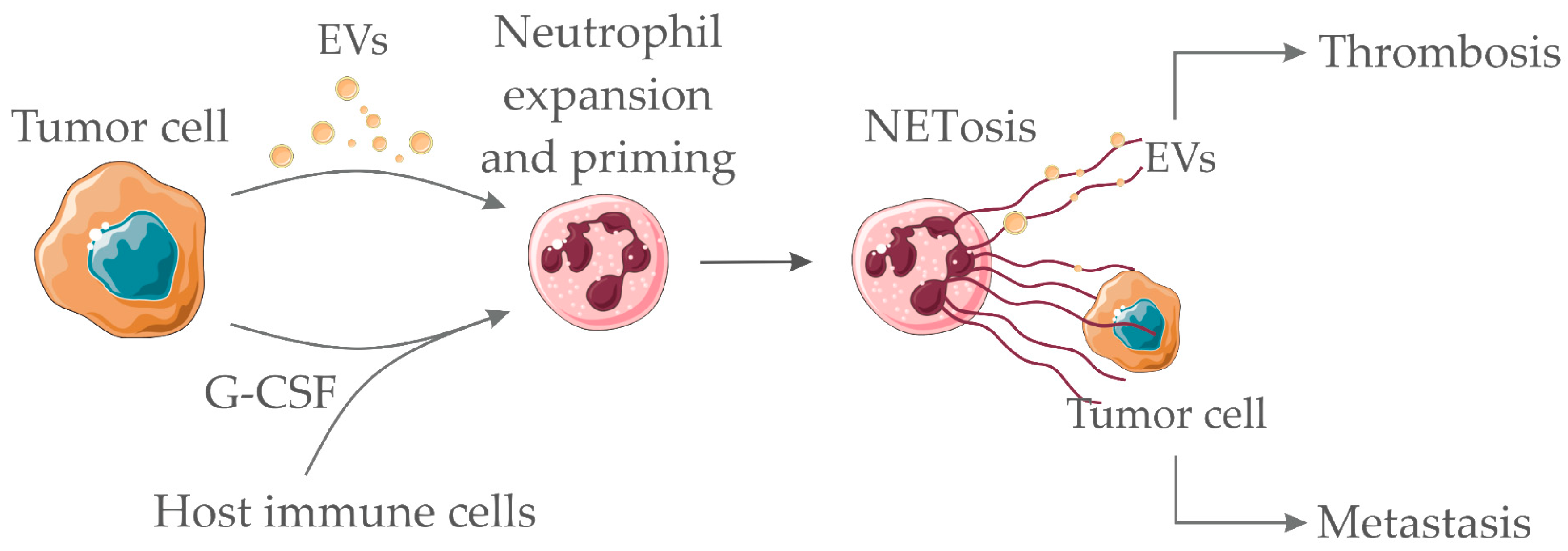
4. Neutrophils and NETs in Cancer-Associated Thrombosis
5. Polyphosphate-Bearing EVs
6. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Timp, J.F.; Braekkan, S.K.; Versteeg, H.H.; Cannegieter, S.C. Epidemiology of cancer-associated venous thrombosis. Blood 2013, 122, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.T.; Katholing, A.; Rietbrock, S.; Bamber, L.; Martinez, C. Epidemiology of first and recurrent venous thromboembolism in patients with active cancer. A population-based cohort study. Thromb. Haemost. 2017, 117, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Trousseau’s syndrome: Multiple definitions and multiple mechanisms. Blood 2007, 110, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Metharom, P.; Falasca, M.; Berndt, M.C. The History of Armand Trousseau and Cancer-Associated Thrombosis. Cancers 2019, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Kuderer, N.M.; Culakova, E.; Lyman, G.H.; Francis, C.W. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008, 111, 4902–4907. [Google Scholar] [CrossRef] [PubMed]
- Ay, C.; Pabinger, I.; Cohen, A.T. Cancer-associated venous thromboembolism: Burden, mechanisms, and management. Thromb. Haemost. 2017, 117, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.G.; Monteiro, R.Q. Activation of blood coagulation in cancer: Implications for tumour progression. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef] [PubMed]
- Abdol Razak, N.B.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 2018, 10, 380. [Google Scholar] [CrossRef]
- van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Nawaz, M.; Shah, N.; Zanetti, B.R.; Maugeri, M.; Silvestre, R.N.; Fatima, F.; Neder, L.; Valadi, H. Extracellular Vesicles and Matrix Remodeling Enzymes: The Emerging Roles in Extracellular Matrix Remodeling, Progression of Diseases and Tissue Repair. Cells 2018, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.G.; Oliveira, A.S.; Campos, L.C.; Bonamino, M.; Chammas, R.; Werneck, C.; Vicente, C.P.; Barcinski, M.A.; Petersen, L.C.; Monteiro, R.Q. Malignant transformation in melanocytes is associated with increased production of procoagulant microvesicles. Thromb. Haemost. 2011, 106, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost. 2016, 14, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Hron, G.; Kollars, M.; Weber, H.; Sagaster, V.; Quehenberger, P.; Eichinger, S.; Kyrle, P.A.; Weltermann, A. Tissue factor-positive microparticles: Cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb. Haemost. 2007, 97, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef] [PubMed]
- Menck, K.; Bleckmann, A.; Wachter, A.; Hennies, B.; Ries, L.; Schulz, M.; Balkenhol, M.; Pukrop, T.; Schatlo, B.; Rost, U.; et al. Characterisation of tumour-derived microvesicles in cancer patients’ blood and correlation with clinical outcome. J. Extracell. Vesicles 2017, 6, 1340745. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Rak, J. Microparticles in cancer. Semin. Thromb. Hemost. 2010, 36, 888–906. [Google Scholar] [CrossRef]
- Lima, L.G.; Leal, A.C.; Vargas, G.; Porto-Carreiro, I.; Monteiro, R.Q. Intercellular transfer of tissue factor via the uptake of tumor-derived microvesicles. Thromb. Res. 2013, 132, 450–456. [Google Scholar] [CrossRef]
- Muhsin-Sharafaldine, M.R.; Saunderson, S.C.; Dunn, A.C.; Faed, J.M.; Kleffmann, T.; McLellan, A.D. Procoagulant and immunogenic properties of melanoma exosomes, microvesicles and apoptotic vesicles. Oncotarget 2016, 7, 56279–56294. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Ettelaie, C.; Collier, M.E.; Featherby, S.; Benelhaj, N.E.; Greenman, J.; Maraveyas, A. Analysis of the potential of cancer cell lines to release tissue factor-containing microvesicles: Correlation with tissue factor and PAR2 expression. Thromb. J. 2016, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle 2009, 8, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Klement, G. Impact of oncogenes and tumor suppressor genes on deregulation of hemostasis and angiogenesis in cancer. Cancer Metastasis Rev. 2000, 19, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; May, L.; Lhotak, V.; Shahrzad, S.; Shirasawa, S.; Weitz, J.I.; Coomber, B.L.; Mackman, N.; Rak, J.W. Oncogenic events regulate tissue factor expression in colorectal cancer cells: Implications for tumor progression and angiogenesis. Blood 2005, 105, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Milsom, C.C.; Yu, J.L.; Mackman, N.; Micallef, J.; Anderson, G.M.; Guha, A.; Rak, J.W. Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: Effect on tumor initiation and angiogenesis. Cancer Res. 2008, 68, 10068–10076. [Google Scholar] [CrossRef]
- Hugo de Almeida, V.; Guimaraes, I.D.S.; Almendra, L.R.; Rondon, A.M.R.; Tilli, T.M.; de Melo, A.C.; Sternberg, C.; Monteiro, R.Q. Positive crosstalk between EGFR and the TF-PAR2 pathway mediates resistance to cisplatin and poor survival in cervical cancer. Oncotarget 2018, 9, 30594–30609. [Google Scholar] [CrossRef]
- Garnier, D.; Magnus, N.; Lee, T.H.; Bentley, V.; Meehan, B.; Milsom, C.; Montermini, L.; Kislinger, T.; Rak, J. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J. Biol. Chem. 2012, 287, 43565–43572. [Google Scholar] [CrossRef]
- White, T.A.; Pan, S.; Witt, T.A.; Simari, R.D. Murine strain differences in hemostasis and thrombosis and tissue factor pathway inhibitor. Thromb. Res. 2010, 125, 84–89. [Google Scholar] [CrossRef]
- Hisada, Y.; Mackman, N. Mouse models of cancer-associated thrombosis. Thromb. Res. 2018, 164, S48–S53. [Google Scholar] [CrossRef]
- Mizuno, T.; Tsukiya, T.; Takewa, Y.; Tatsumi, E. Differences in clotting parameters between species for preclinical large animal studies of cardiovascular devices. J. Artif. Organs 2018, 21, 138–141. [Google Scholar] [CrossRef]
- Mege, D.; Mezouar, S.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Microparticles and cancer thrombosis in animal models. Thromb. Res. 2016, 140, S21–S26. [Google Scholar] [CrossRef]
- Wang, J.G.; Geddings, J.E.; Aleman, M.M.; Cardenas, J.C.; Chantrathammachart, P.; Williams, J.C.; Kirchhofer, D.; Bogdanov, V.Y.; Bach, R.R.; Rak, J.; et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood 2012, 119, 5543–5552. [Google Scholar] [CrossRef]
- Thomas, G.M.; Panicot-Dubois, L.; Lacroix, R.; Dignat-George, F.; Lombardo, D.; Dubois, C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J. Exp. Med. 2009, 206, 1913–1927. [Google Scholar] [CrossRef]
- Thomas, G.M.; Brill, A.; Mezouar, S.; Crescence, L.; Gallant, M.; Dubois, C.; Wagner, D.D. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J. Thromb Haemost. 2015, 13, 1310–1319. [Google Scholar] [CrossRef]
- Hisada, Y.; Ay, C.; Auriemma, A.C.; Cooley, B.C.; Mackman, N. Human pancreatic tumors grown in mice release tissue factor-positive microvesicles that increase venous clot size. J. Thromb. Haemost. 2017, 15, 2208–2217. [Google Scholar] [CrossRef]
- Hisada, Y.; Grover, S.P.; Maqsood, A.; Houston, R.; Ay, C.; Noubouossie, D.F.; Cooley, B.C.; Wallen, H.; Key, N.S.; Thalin, C.; et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica 2019. [Google Scholar] [CrossRef]
- Stark, K.; Schubert, I.; Joshi, U.; Kilani, B.; Hoseinpour, P.; Thakur, M.; Grunauer, P.; Pfeiler, S.; Schmidergall, T.; Stockhausen, S.; et al. Distinct Pathogenesis of Pancreatic Cancer Microvesicle-Associated Venous Thrombosis Identifies New Antithrombotic Targets In Vivo. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 772–786. [Google Scholar] [CrossRef]
- Rautou, P.E.; Mackman, N. Microvesicles as risk markers for venous thrombosis. Expert Rev. Hematol. 2013, 6, 91–101. [Google Scholar] [CrossRef]
- Manly, D.A.; Wang, J.; Glover, S.L.; Kasthuri, R.; Liebman, H.A.; Key, N.S.; Mackman, N. Increased microparticle tissue factor activity in cancer patients with Venous Thromboembolism. Thromb. Res. 2010, 125, 511–512. [Google Scholar] [CrossRef]
- Campello, E.; Spiezia, L.; Radu, C.M.; Bulato, C.; Castelli, M.; Gavasso, S.; Simioni, P. Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb. Res. 2011, 127, 473–477. [Google Scholar] [CrossRef]
- Tesselaar, M.E.; Romijn, F.P.; Van Der Linden, I.K.; Prins, F.A.; Bertina, R.M.; Osanto, S. Microparticle-associated tissue factor activity: A link between cancer and thrombosis? J. Thromb. Haemost. 2007, 5, 520–527. [Google Scholar] [CrossRef]
- Khorana, A.A.; Francis, C.W.; Menzies, K.E.; Wang, J.G.; Hyrien, O.; Hathcock, J.; Mackman, N.; Taubman, M.B. Plasma tissue factor may be predictive of venous thromboembolism in pancreatic cancer. J. Thromb. Haemost. 2008, 6, 1983–1985. [Google Scholar] [CrossRef]
- Faille, D.; Bourrienne, M.C.; de Raucourt, E.; de Chaisemartin, L.; Granger, V.; Lacroix, R.; Panicot-Dubois, L.; Hammel, P.; Levy, P.; Ruszniewski, P.; et al. Biomarkers for the risk of thrombosis in pancreatic adenocarcinoma are related to cancer process. Oncotarget 2018, 9, 26453–26465. [Google Scholar] [CrossRef]
- Toth, B.; Liebhardt, S.; Steinig, K.; Ditsch, N.; Rank, A.; Bauerfeind, I.; Spannagl, M.; Friese, K.; Reininger, A.J. Platelet-derived microparticles and coagulation activation in breast cancer patients. Thromb. Haemost. 2008, 100, 663–669. [Google Scholar] [CrossRef]
- Fricke, A.; Ullrich, P.V.; Cimniak, A.F.V.; Becherer, C.; Follo, M.; Heinz, J.; Scholber, J.; Herget, G.W.; Hauschild, O.; Wittel, U.A.; et al. Levels of activated platelet-derived microvesicles in patients with soft tissue sarcoma correlate with an increased risk of venous thromboembolism. BMC Cancer 2017, 17, 527. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Orbe, J.; Roncal, C.; Alvarez-Hernandez, M.; Martinez de Lizarrondo, S.; Alves, M.T.; Garcia Mata, J.; Paramo, J.A. Tissue factor expressed by microparticles is associated with mortality but not with thrombosis in cancer patients. Thromb. Haemost. 2013, 110, 598–608. [Google Scholar] [CrossRef]
- Auwerda, J.J.; Yuana, Y.; Osanto, S.; de Maat, M.P.; Sonneveld, P.; Bertina, R.M.; Leebeek, F.W. Microparticle-associated tissue factor activity and venous thrombosis in multiple myeloma. Thromb. Haemost. 2011, 105, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Claussen, C.; Rausch, A.V.; Lezius, S.; Amirkhosravi, A.; Davila, M.; Francis, J.L.; Hisada, Y.M.; Mackman, N.; Bokemeyer, C.; Schmalfeldt, B.; et al. Microvesicle-associated tissue factor procoagulant activity for the preoperative diagnosis of ovarian cancer. Thromb. Res. 2016, 141, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.G.; Prendergast, E.; Geddings, J.E.; Walts, A.E.; Agadjanian, H.; Hisada, Y.; Karlan, B.Y.; Mackman, N.; Walsh, C.S. Evaluation of venous thrombosis and tissue factor in epithelial ovarian cancer. Gynecol Oncol 2017, 146, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Gezelius, E.; Flou Kristensen, A.; Bendahl, P.O.; Hisada, Y.; Risom Kristensen, S.; Ek, L.; Bergman, B.; Wallberg, M.; Falkmer, U.; Mackman, N.; et al. Coagulation biomarkers and prediction of venous thromboembolism and survival in small cell lung cancer: A sub-study of RASTEN—A randomized trial with low molecular weight heparin. PLoS ONE 2018, 13, e0207387. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.; Ay, C.; Mackman, N.; Bertina, R.M.; Kaider, A.; Marosi, C.; Key, N.S.; Barcel, D.A.; Scheithauer, W.; Kornek, G.; et al. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J. Thromb. Haemost. 2012, 10, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Measurement of tissue factor activity in extracellular vesicles from human plasma samples. Res. Pract. Thromb. Haemost. 2019, 3, 44–48. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Khorana, A.A. Risk assessment for cancer-associated thrombosis: What is the best approach? Thromb. Res. 2012, 129, S10–S15. [Google Scholar] [CrossRef]
- Starling, N.; Rao, S.; Cunningham, D.; Iveson, T.; Nicolson, M.; Coxon, F.; Middleton, G.; Daniel, F.; Oates, J.; Norman, A.R. Thromboembolism in patients with advanced gastroesophageal cancer treated with anthracycline, platinum, and fluoropyrimidine combination chemotherapy: A report from the UK National Cancer Research Institute Upper Gastrointestinal Clinical Studies Group. J. Clin. Oncol. 2009, 27, 3786–3793. [Google Scholar] [CrossRef]
- Simanek, R.; Vormittag, R.; Ay, C.; Alguel, G.; Dunkler, D.; Schwarzinger, I.; Steger, G.; Jaeger, U.; Zielinski, C.; Pabinger, I. High platelet count associated with venous thromboembolism in cancer patients: Results from the Vienna Cancer and Thrombosis Study (CATS). J. Thromb. Haemost. 2010, 8, 114–120. [Google Scholar] [CrossRef]
- Gucer, F.; Moser, F.; Tamussino, K.; Reich, O.; Haas, J.; Arikan, G.; Petru, E.; Winter, R. Thrombocytosis as a prognostic factor in endometrial carcinoma. Gynecol. Oncol. 1998, 70, 210–214. [Google Scholar] [CrossRef]
- Monreal, M.; Fernandez-Llamazares, J.; Pinol, M.; Julian, J.F.; Broggi, M.; Escola, D.; Abad, A. Platelet count and survival in patients with colorectal cancer--a preliminary study. Thromb. Haemost. 1998, 79, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Chadha, A.S.; Kocak-Uzel, E.; Das, P.; Minsky, B.D.; Delclos, M.E.; Mahmood, U.; Guha, S.; Ahmad, M.; Varadhachary, G.R.; Javle, M.; et al. Paraneoplastic thrombocytosis independently predicts poor prognosis in patients with locally advanced pancreatic cancer. Acta. Oncol. 2015, 54, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, Z.; Josa, V.; Toth, A.; Szilasi, Z.; Tihanyi, B.; Zarand, A.; Harsanyi, L.; Szallasi, Z. Paraneoplastic thrombocytosis in gastrointestinal cancer. Platelets 2016, 27, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Takahashi, T.; Nakamura, K.; Tsuyuoka, R.; Okuno, Y.; Enomoto, T.; Fukumoto, M.; Imura, H. Thrombocytosis in patients with tumors producing colony-stimulating factor. Blood 1992, 80, 2052–2059. [Google Scholar] [PubMed]
- Avraham, H.; Banu, N.; Scadden, D.T.; Abraham, J.; Groopman, J.E. Modulation of megakaryocytopoiesis by human basic fibroblast growth factor. Blood 1994, 83, 2126–2132. [Google Scholar] [PubMed]
- Sasaki, Y.; Takahashi, T.; Miyazaki, H.; Matsumoto, A.; Kato, T.; Nakamura, K.; Iho, S.; Okuno, Y.; Nakao, K. Production of thrombopoietin by human carcinomas and its novel isoforms. Blood 1999, 94, 1952–1960. [Google Scholar] [PubMed]
- Casella, I.; Feccia, T.; Chelucci, C.; Samoggia, P.; Castelli, G.; Guerriero, R.; Parolini, I.; Petrucci, E.; Pelosi, E.; Morsilli, O.; et al. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood 2003, 101, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.L.; Nick, A.M.; McNeish, I.A.; Balkwill, F.; Han, H.D.; Bottsford-Miller, J.; Rupairmoole, R.; Armaiz-Pena, G.N.; Pecot, C.V.; Coward, J.; et al. Paraneoplastic thrombocytosis in ovarian cancer. N. Engl. J. Med. 2012, 366, 610–618. [Google Scholar] [CrossRef]
- Ikeda, M.; Furukawa, H.; Imamura, H.; Shimizu, J.; Ishida, H.; Masutani, S.; Tatsuta, M.; Satomi, T. Poor prognosis associated with thrombocytosis in patients with gastric cancer. Ann. Surg. Oncol. 2002, 9, 287–291. [Google Scholar] [CrossRef]
- Ji, Y.; Sheng, L.; Du, X.; Qiu, G.; Su, D. Elevated platelet count is a strong predictor of poor prognosis in stage I non-small cell lung cancer patients. Platelets 2015, 26, 138–142. [Google Scholar] [CrossRef]
- Plantureux, L.; Mege, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers 2018, 10, 441. [Google Scholar] [CrossRef]
- Mantur, M.; Kemona, H.; Kozlowski, R.; Kemona-Chetnik, I. Effect of tumor stage and nephrectomy on CD62P expression and sP-selectin concentration in renal cancer. Neoplasma 2003, 50, 262–265. [Google Scholar] [PubMed]
- Xu, X.R.; Carrim, N.; Neves, M.A.; McKeown, T.; Stratton, T.W.; Coelho, R.M.; Lei, X.; Chen, P.; Xu, J.; Dai, X.; et al. Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb. J. 2016, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Felding-Habermann, B.; Habermann, R.; Saldivar, E.; Ruggeri, Z.M. Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J. Biol. Chem. 1996, 271, 5892–5900. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, P.; Alonso-Escolano, D.; Radomski, M.W. Platelet--cancer interactions: Mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br. J. Pharmacol. 2004, 143, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Zigrino, P.; Brucker, C.; Bourdon, C.; Freund, M.; De Arcangelis, A.; Abrams, S.I.; Orend, G.; Gachet, C.; Mangin, P.H. Platelet integrin alpha6beta1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 2016, 1, e88245. [Google Scholar] [CrossRef] [PubMed]
- Boukerche, H.; Berthier-Vergnes, O.; Penin, F.; Tabone, E.; Lizard, G.; Bailly, M.; McGregor, J.L. Human melanoma cell lines differ in their capacity to release ADP and aggregate platelets. Br. J. Haematol. 1994, 87, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef]
- Nie, D.; Che, M.; Zacharek, A.; Qiao, Y.; Li, L.; Li, X.; Lamberti, M.; Tang, K.; Cai, Y.; Guo, Y.; et al. Differential expression of thromboxane synthase in prostate carcinoma: Role in tumor cell motility. Am. J. Pathol. 2004, 164, 429–439. [Google Scholar] [CrossRef]
- Kajita, S.; Ruebel, K.H.; Casey, M.B.; Nakamura, N.; Lloyd, R.V. Role of COX-2, thromboxane A2 synthase, and prostaglandin I2 synthase in papillary thyroid carcinoma growth. Mod. Pathol. 2005, 18, 221–227. [Google Scholar] [CrossRef]
- Moussa, O.; Yordy, J.S.; Abol-Enein, H.; Sinha, D.; Bissada, N.K.; Halushka, P.V.; Ghoneim, M.A.; Watson, D.K. Prognostic and functional significance of thromboxane synthase gene overexpression in invasive bladder cancer. Cancer Res. 2005, 65, 11581–11587. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Suzuki, T.; Takahashi, Y.; Ukai, M.; Tauchi, K.; Fujii, T.; Horikawa, N.; Minamimura, T.; Tabuchi, Y.; Morii, M.; et al. Upregulation of thromboxane synthase in human colorectal carcinoma and the cancer cell proliferation by thromboxane A2. FEBS Lett 2006, 580, 3368–3374. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.C.; Gately, K.; Cummins, R.; Kay, E.; O’Byrne, K.J.; Pidgeon, G.P. Examination of thromboxane synthase as a prognostic factor and therapeutic target in non-small cell lung cancer. Mol. Cancer 2011, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- de Leval, X.; Benoit, V.; Delarge, J.; Julemont, F.; Masereel, B.; Pirotte, B.; Merville, M.P.; David, J.L.; Dogne, J.M. Pharmacological evaluation of the novel thromboxane modulator BM-567 (II/II). Effects of BM-567 on osteogenic sarcoma-cell-induced platelet aggregation. Prostaglandins Leukot. Essent. Fatty Acids 2003, 68, 55–59. [Google Scholar] [CrossRef]
- Nieman, M.T. Protease-activated receptors in hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Tassi Yunga, S.; Sylman, J.L.; Zilberman-Rudenko, J.; Shirai, T.; Hebert, J.F.; Kayton, R.; Zhang, Y.; Nan, X.; Shatzel, J.J.; et al. The role of coagulation and platelets in colon cancer-associated thrombosis. Am. J. Physiol. Cell Physiol. 2019, 316, C264–C273. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.S.; Gouvea, I.E.; Silva, M.C.; Castro, E.D.; de Paula, C.A.; Okamoto, D.; Oliveira, L.; Peres, G.B.; Ottaiano, T.; Facina, G.; et al. Cathepsin K induces platelet dysfunction and affects cell signaling in breast cancer–molecularly distinct behavior of cathepsin K in breast cancer. BMC Cancer 2016, 16, 173. [Google Scholar] [CrossRef]
- Verbovsek, U.; Van Noorden, C.J.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015, 35, 71–84. [Google Scholar] [CrossRef]
- Honn, K.V.; Cavanaugh, P.; Evens, C.; Taylor, J.D.; Sloane, B.F. Tumor cell-platelet aggregation: Induced by cathepsin B-like proteinase and inhibited by prostacyclin. Science 1982, 217, 540–542. [Google Scholar] [CrossRef]
- Sebastiano, M.; Momi, S.; Falcinelli, E.; Bury, L.; Hoylaerts, M.F.; Gresele, P. A novel mechanism regulating human platelet activation by MMP-2-mediated PAR1 biased signaling. Blood 2017, 129, 883–895. [Google Scholar] [CrossRef]
- Santos-Martinez, M.J.; Medina, C.; Gilmer, J.F.; Radomski, M.W. Matrix metalloproteinases in platelet function: Coming of age. J. Thromb. Haemost. 2008, 6, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Inoue, K.; Fuller, G.L.; Garcia, A.; Eble, J.A.; Pohlmann, S.; Inoue, O.; Gartner, T.K.; Hughan, S.C.; Pearce, A.C.; Laing, G.D.; et al. A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 2006, 107, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Mir Seyed Nazari, P.; Riedl, J.; Pabinger, I.; Ay, C. The role of podoplanin in cancer-associated thrombosis. Thromb. Res. 2018, 164, S34–S39. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Inoue, K.; Kato, Y.; Inoue, O.; Kaneko, M.K.; Mishima, K.; Yatomi, Y.; Yamazaki, Y.; Narimatsu, H.; Ozaki, Y. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 2007, 282, 25993–26001. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Preusser, M.; Nazari, P.M.; Posch, F.; Panzer, S.; Marosi, C.; Birner, P.; Thaler, J.; Brostjan, C.; Lotsch, D.; et al. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood 2017, 129, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Inoue, O.; Tamura, S.; Tsukiji, N.; Sasaki, T.; Endo, H.; Satoh, K.; Osada, M.; Sato-Uchida, H.; Fujii, H.; et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J. Thromb. Haemost. 2017, 15, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Mege, D.; Panicot-Dubois, L.; Ouaissi, M.; Robert, S.; Sielezneff, I.; Sastre, B.; Dignat-George, F.; Dubois, C. The origin and concentration of circulating microparticles differ according to cancer type and evolution: A prospective single-center study. Int. J. Cancer 2016, 138, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.G.; Sandim, V.; Almeida, V.H.; Rondon, A.M.R.; Succar, B.B.; Hottz, E.D.; Leal, A.C.; Vercoza, B.R.F.; Rodrigues, J.C.F.; Bozza, P.T.; et al. Breast-cancer extracellular vesicles induce platelet activation and aggregation by tissue factor-independent and -dependent mechanisms. Thromb. Res. 2017, 159, 24–32. [Google Scholar] [CrossRef]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J. Cancer 2015, 136, 462–475. [Google Scholar] [CrossRef]
- Budnik, I.; Brill, A. Immune Factors in Deep Vein Thrombosis Initiation. Trends Immunol. 2018, 39, 610–623. [Google Scholar] [CrossRef]
- Connolly, G.C.; Khorana, A.A.; Kuderer, N.M.; Culakova, E.; Francis, C.W.; Lyman, G.H. Leukocytosis, thrombosis and early mortality in cancer patients initiating chemotherapy. Thromb. Res. 2010, 126, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Blix, K.; Jensvoll, H.; Braekkan, S.K.; Hansen, J.B. White blood cell count measured prior to cancer development is associated with future risk of venous thromboembolism-the Tromso study. PLoS ONE 2013, 8, e73447. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, M.; Cohen, H.W.; Billett, H.H. Persistent neutrophilia is a marker for an increased risk of venous thrombosis. J. Thromb. Thrombolysis 2016, 42, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, P.; Riondino, S.; Formica, V.; Cereda, V.; Tosetto, L.; La Farina, F.; Valente, M.G.; Vergati, M.; Guadagni, F.; Roselli, M. Venous thromboembolism risk prediction in ambulatory cancer patients: Clinical significance of neutrophil/lymphocyte ratio and platelet/lymphocyte ratio. Int J. Cancer 2015, 136, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Artoni, A.; Abbattista, M.; Bucciarelli, P.; Gianniello, F.; Scalambrino, E.; Pappalardo, E.; Peyvandi, F.; Martinelli, I. Platelet to Lymphocyte Ratio and Neutrophil to Lymphocyte Ratio as Risk Factors for Venous Thrombosis. Clin. Appl. Thromb. Hemost. 2018, 24, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, H.E.; Paz, L.H.; Wang, Y.; Oramas, D.M.; Simons, C.R.; Tafur, A.J. Performance of Current Thromboembolism Risk Assessment Tools in Patients With Gastric Cancer and Validity After First Treatment. Clin. Appl. Thromb. Hemost. 2018, 24, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.J.; Ritchie, W.G.; Lynch, P.R. Venous endothelial damage produced by massive sticking and emigration of leukocytes. Am. J. Pathol. 1974, 74, 507–532. [Google Scholar]
- Stewart, G.J. Neutrophils and deep venous thrombosis. Haemostasis 1993, 23, 127–140. [Google Scholar] [CrossRef]
- Gross, P.L.; Furie, B.C.; Merrill-Skoloff, G.; Chou, J.; Furie, B. Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J. Leukoc. Biol. 2005, 78, 1318–1326. [Google Scholar] [CrossRef]
- Darbousset, R.; Thomas, G.M.; Mezouar, S.; Frere, C.; Bonier, R.; Mackman, N.; Renne, T.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 2012, 120, 2133–2143. [Google Scholar] [CrossRef]
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Opneja, A.; Nayak, L. The role of neutrophils in thrombosis. Thromb. Res. 2018, 170, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.A.; Lyon, M.; Simpson, D.; Mason, D.; Beynon, R.J.; Moots, R.J.; Wright, H.L. Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 423. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef]
- Waisberg, M.; Molina-Cruz, A.; Mizurini, D.M.; Gera, N.; Sousa, B.C.; Ma, D.; Leal, A.C.; Gomes, T.; Kotsyfakis, M.; Ribeiro, J.M.; et al. Plasmodium falciparum infection induces expression of a mosquito salivary protein (Agaphelin) that targets neutrophil function and inhibits thrombosis without impairing hemostasis. PLoS Pathog 2014, 10, e1004338. [Google Scholar] [CrossRef]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [PubMed]
- Elaskalani, O.; Abdol Razak, N.B.; Metharom, P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun. Signal. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Mizurini, D.M.; Aslan, J.S.; Gomes, T.; Ma, D.; Francischetti, I.M.; Monteiro, R.Q. Salivary Thromboxane A2-Binding Proteins from Triatomine Vectors of Chagas Disease Inhibit Platelet-Mediated Neutrophil Extracellular Traps (NETs) Formation and Arterial Thrombosis. PLoS Negl. Trop. Dis. 2015, 9, e0003869. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Dyer, M.R.; Chen, Q.; Haldeman, S.; Yazdani, H.; Hoffman, R.; Loughran, P.; Tsung, A.; Zuckerbraun, B.S.; Simmons, R.L.; Neal, M.D. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 2018, 8, 2068. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.; Doerfler, W.R.; Ellis, J.T.; Liang, X.; Ross, M.A.; Wallace, C.T.; Sperry, J.L.; Lotze, M.T.; et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 678. [Google Scholar] [CrossRef]
- Thalin, C.; Demers, M.; Blomgren, B.; Wong, S.L.; von Arbin, M.; von Heijne, A.; Laska, A.C.; Wallen, H.; Wagner, D.D.; Aspberg, S. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb. Res. 2016, 139, 56–64. [Google Scholar] [CrossRef]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Daullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Thalin, C.; Lundstrom, S.; Seignez, C.; Daleskog, M.; Lundstrom, A.; Henriksson, P.; Helleday, T.; Phillipson, M.; Wallen, H.; Demers, M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 2018, 13, e0191231. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.D.; Gu, J.Y.; Jung, H.S.; Kim, Y.J.; Kim, H.K. Contact System Activation and Neutrophil Extracellular Trap Markers: Risk Factors for Portal Vein Thrombosis in Patients With Hepatocellular Carcinoma. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618825310. [Google Scholar] [CrossRef] [PubMed]
- Grilz, E.; Mauracher, L.M.; Posch, F.; Konigsbrugge, O.; Zochbauer-Muller, S.; Marosi, C.; Lang, I.; Pabinger, I.; Ay, C. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br. J. Haematol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Hasler, P.; Holzgreve, W.; Gebhardt, S.; Hahn, S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 2005, 66, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R., Jr. Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Gu, J.; Kim, J.E.; Nam, Y.; Song, J.W.; Kim, H.K. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE 2019, 14, e0216055. [Google Scholar] [CrossRef]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef]
- Schoergenhofer, C.; Schwameis, M.; Wohlfarth, P.; Brostjan, C.; Abrams, S.T.; Toh, C.H.; Jilma, B. Granulocyte colony-stimulating factor (G-CSF) increases histone-complexed DNA plasma levels in healthy volunteers. Clin. Exp. Med. 2017, 17, 243–249. [Google Scholar] [CrossRef]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [PubMed]
- Yeo, B.; Redfern, A.D.; Mouchemore, K.A.; Hamilton, J.A.; Anderson, R.L. The dark side of granulocyte-colony stimulating factor: A supportive therapy with potential to promote tumour progression. Clin. Exp. Metastasis 2018, 35, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.J.; Smith, S.A.; Morrissey, J.H. Polyphosphate in thrombosis, hemostasis, and inflammation. Res. Pract. Thromb. Haemost. 2019, 3, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Nickel, K.F.; Ronquist, G.; Langer, F.; Labberton, L.; Fuchs, T.A.; Bokemeyer, C.; Sauter, G.; Graefen, M.; Mackman, N.; Stavrou, E.X.; et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood 2015, 126, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Rayzman, V.; Nolte, M.W.; Nickel, K.F.; Bjorkqvist, J.; Jamsa, A.; Hardy, M.P.; Fries, M.; Schmidbauer, S.; Hedenqvist, P.; et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci. Transl. Med. 2014, 6, 222ra217. [Google Scholar] [CrossRef] [PubMed]
- Nickel, K.F.; Labberton, L.; Long, A.T.; Langer, F.; Fuchs, T.A.; Stavrou, E.X.; Butler, L.M.; Renne, T. The polyphosphate/factor XII pathway in cancer-associated thrombosis: Novel perspectives for safe anticoagulation in patients with malignancies. Thromb. Res. 2016, 141, S4–S7. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Schattner, M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 271. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Hamidi, A.; Olsson, A.K. Platelets, NETs and cancer. Thromb. Res. 2018, 164, S148–S152. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | TF Measurement | VTE |
|---|---|---|
| Pancreatic, non–small cell lung, ovarian, colorectal and breast [15,41,42] | TF antigen (IFC) | Yes |
| Colon, lung, bladder, pancreatic, prostate, rectal, bile duct, brain, cholangio, liver, lymphoma, renal cell, testis and other types of cancer [15,41,42] | TF activity (FXa generation assay) | Yes |
| Gastrointestinal, lung, pancreatic, prostatic, breast, liver, uterine and brain [15,41,42] | TF antigen (FACS) | Yes |
| Pancreatic [43,44,45] | TF activity (FXa generation assay), TF antigen (FACS or ELISA) | Yes |
| Breast [43,46] | TF activity (FXa generation assay), TF antigen (FACS) | Yes/ No |
| Soft tissue sarcoma [47] | TF antigen (FACS) | No |
| Non-Hodgkin lymphoma, colorectal, breast, stomach, lung and pancreatic [48] | TF antigen (ELISA), TF activity (FXa generation assay) | No |
| Multiple myelomas [49] | TF activity (FXa generation assay) | No |
| Ovarian [50,51] | TF antigen (ELISA), TF activity (FXa generation assay or FGT) | No |
| Small cell lung cancer [52] | TF activity (FXa generation assay) | No |
| Gastric, colorectal and brain [53] | TF activity (FXa generation assay) | No |
| EVs-Linked Molecules | Suggested Effect |
|---|---|
| TF | Activation of the extrinsic pathway, fibrin generation, and platelet activation/aggregation in a thrombin-dependent manner |
| Podoplanin | Platelet aggregation |
| PSGL-1 | Accumulation of the EVs at the site of thrombosis through binding to platelets via P-selectin |
| Integrins | Accumulation of the EVs at the site of thrombosis through binding to platelets |
| Unknown | Neutrophil activation and NETs release |
| Unknown | Binding to NETs |
| PolyP | Activation of the contact pathway, fibrin generation, and platelet activation/aggregation in a thrombin-dependent manner |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, V.H.; Rondon, A.M.R.; Gomes, T.; Monteiro, R.Q. Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis. Cells 2019, 8, 716. https://doi.org/10.3390/cells8070716
Almeida VH, Rondon AMR, Gomes T, Monteiro RQ. Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis. Cells. 2019; 8(7):716. https://doi.org/10.3390/cells8070716
Chicago/Turabian StyleAlmeida, Vitor H., Araci M. R. Rondon, Tainá Gomes, and Robson Q. Monteiro. 2019. "Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis" Cells 8, no. 7: 716. https://doi.org/10.3390/cells8070716
APA StyleAlmeida, V. H., Rondon, A. M. R., Gomes, T., & Monteiro, R. Q. (2019). Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis. Cells, 8(7), 716. https://doi.org/10.3390/cells8070716