A Novel Assay for Profiling GBM Cancer Model Heterogeneity and Drug Screening

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient-Derived Xenografts
2.2. Microtumor and Spheroid Culture
2.3. NanoString Custom Assay
2.4. Novel Molecular Subtype Classification: FastEMC

2.5. NanoString Normalization
2.6. Drug Screening
3. Results
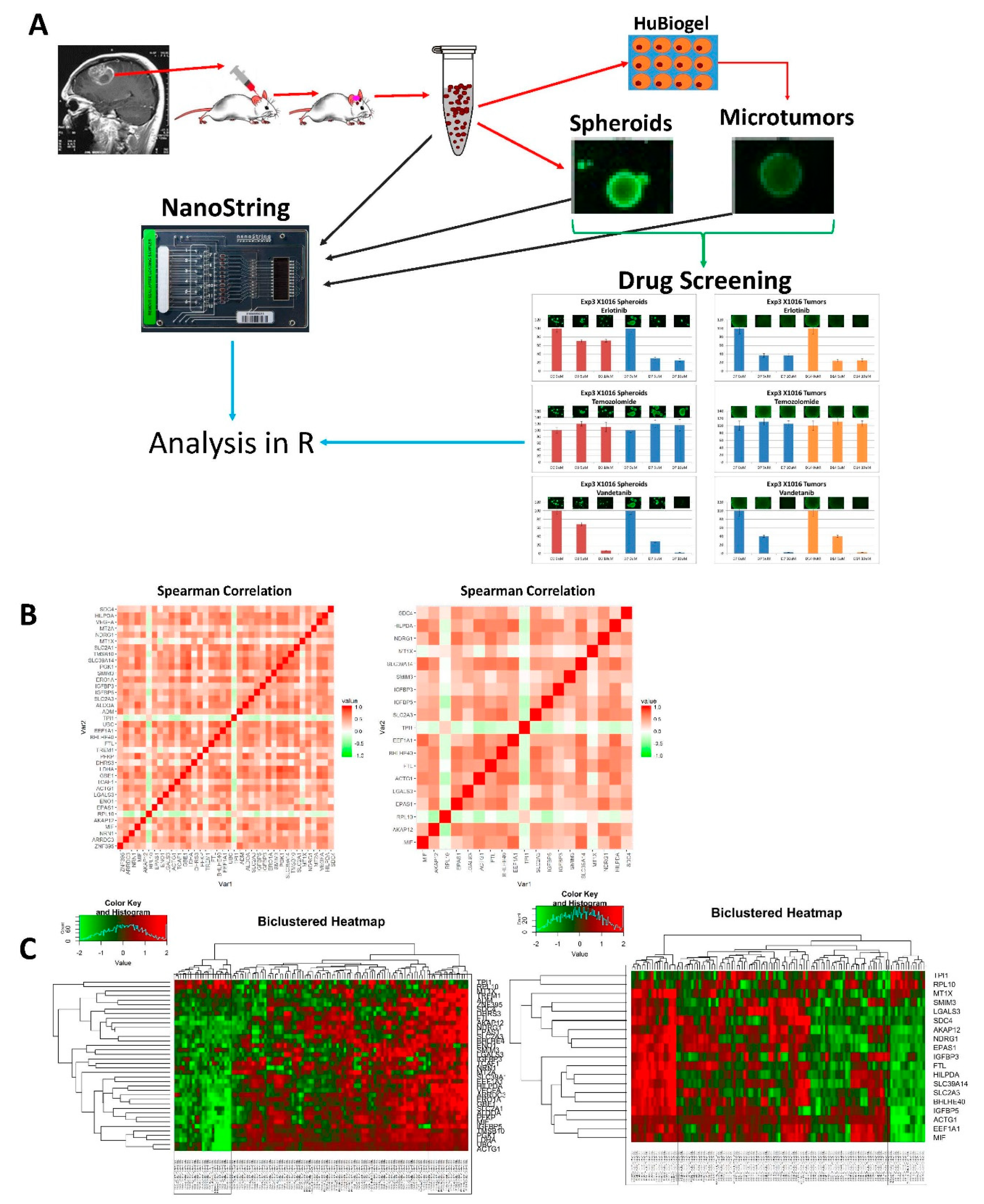
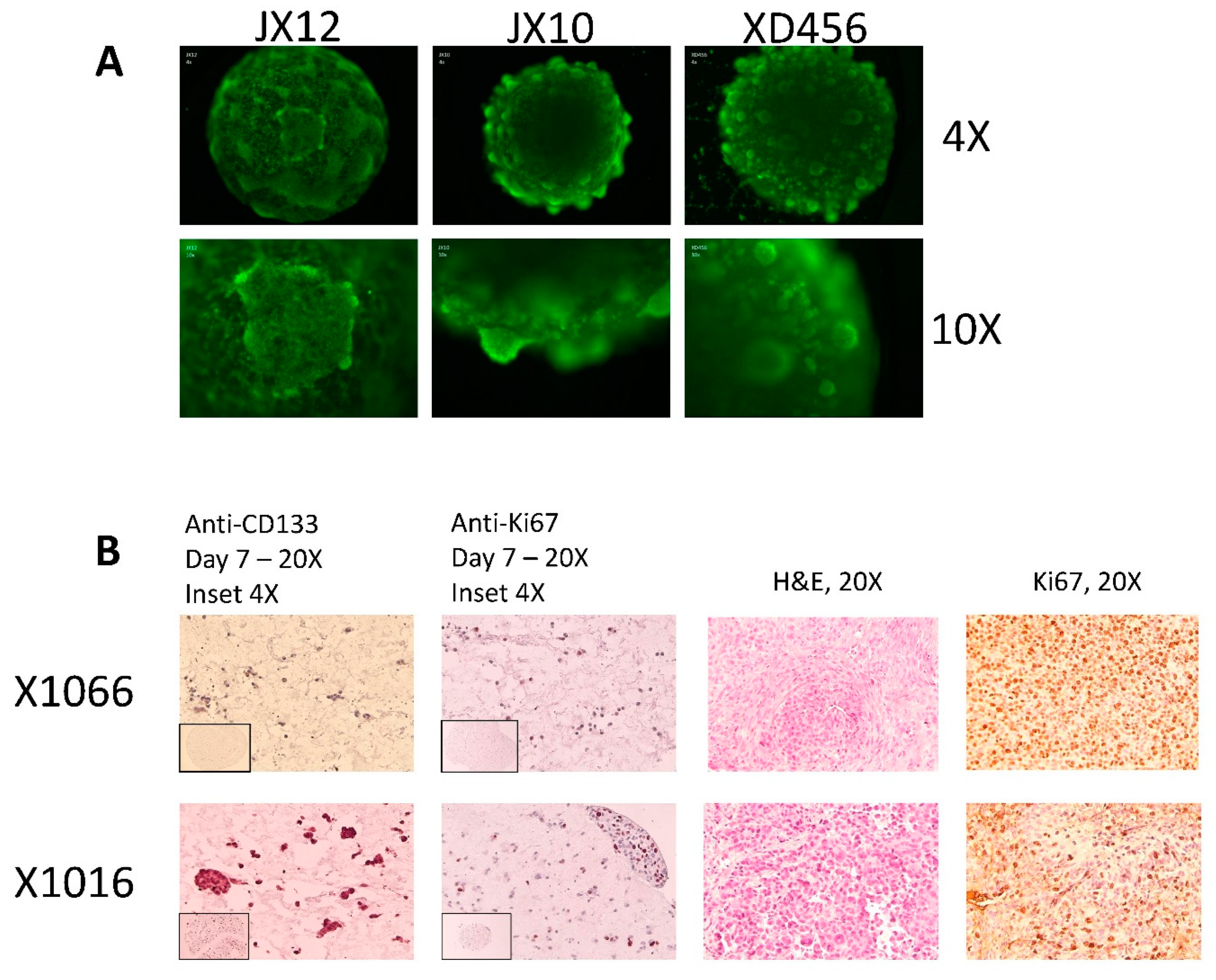
3.1. Microtumors and Spheroids
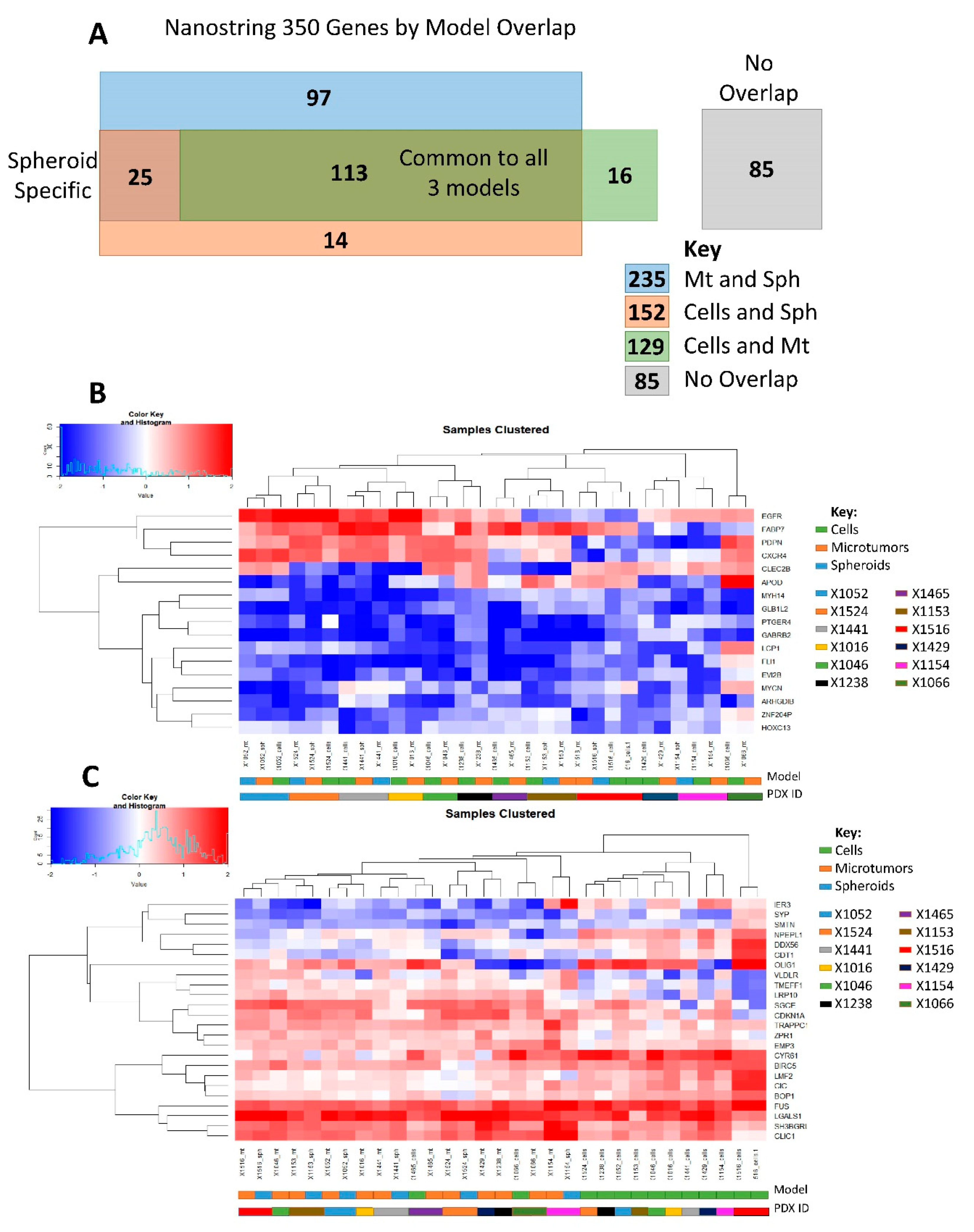
3.2. NanoString Model Specific and Independent Effects
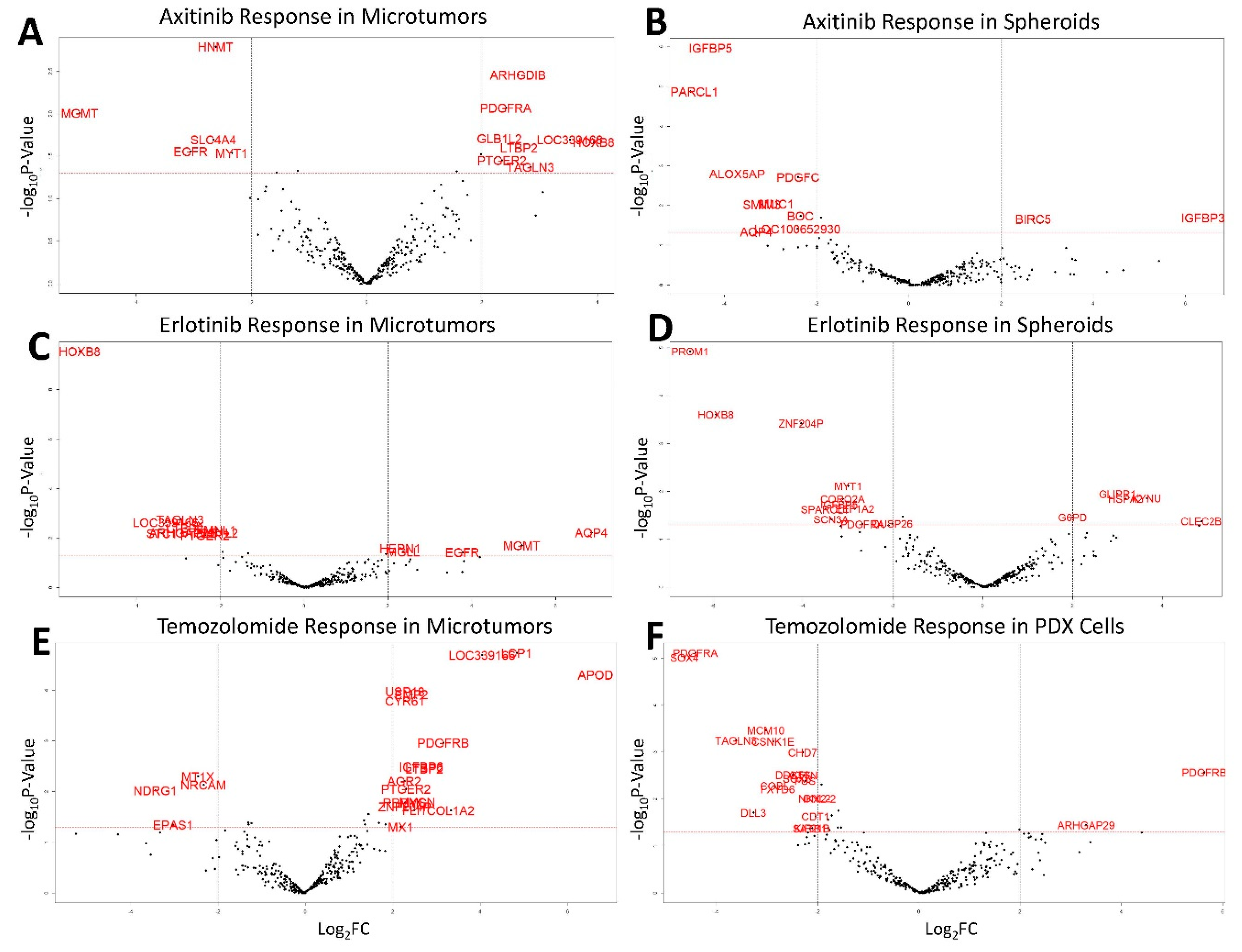
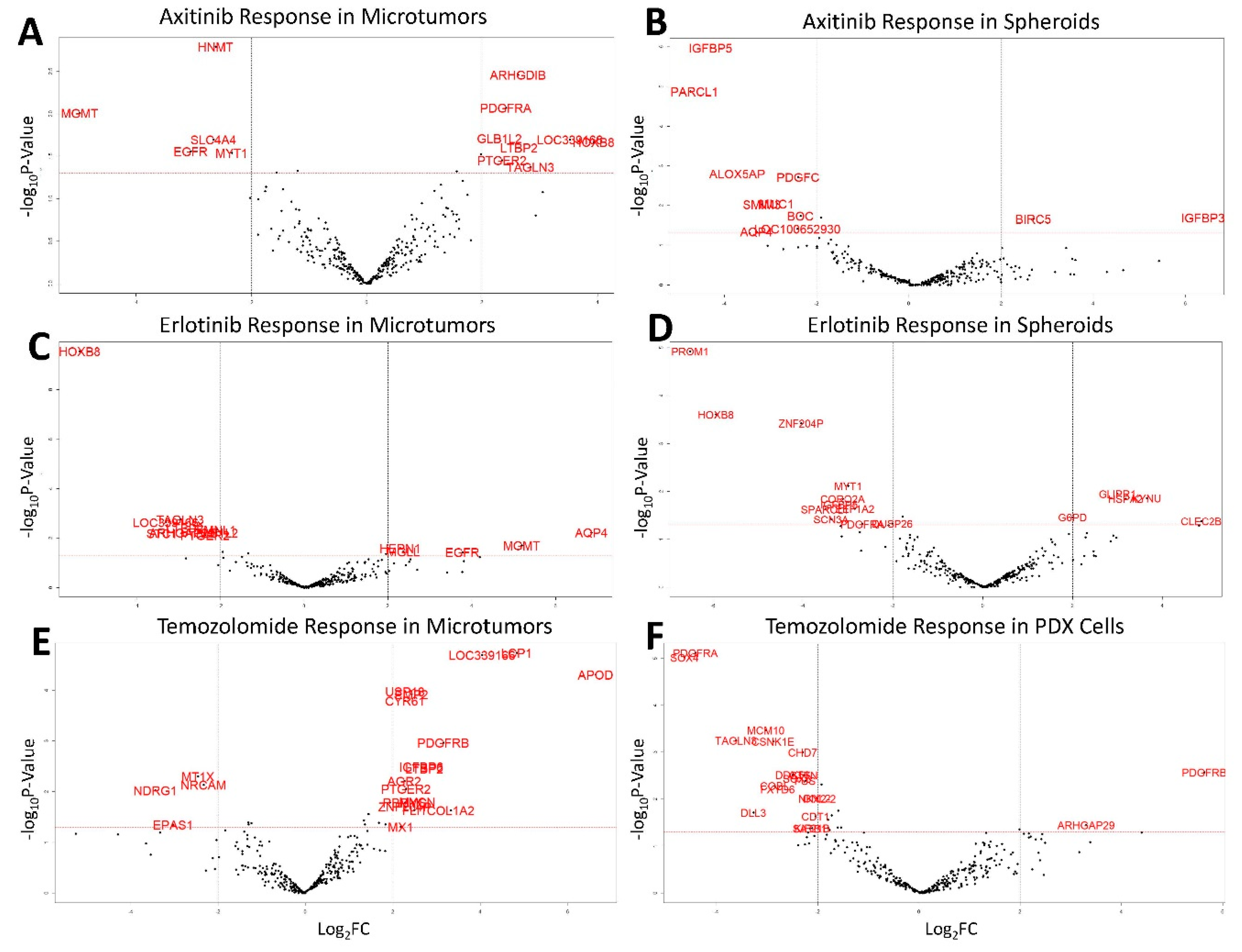
3.3. Drug Screening
3.4. Temozolomide Concordance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Carlo, D.T.; Cagnazzo, F.; Benedetto, N.; Morganti, R.; Perrini, P. Multiple high-grade gliomas: Epidemiology, management, and outcome. A systematic review and meta-analysis. Neurosurg. Rev. 2017, 42, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Stackhouse, C.T.; Gillespie, G.Y.; Willey, C.D. Cancer Explant Models. Curr. Top. Microbiol. Immunol. 2019, 1–30. [Google Scholar] [CrossRef]
- Cunha, P.P.; Costa, P.M.; Morais, C.M.; Lopes, I.R.; Cardoso, A.M.; Cardoso, A.L.; Mano, M.; Jurado, A.S.; Pedroso de Lima, M.C. High-throughput screening uncovers miRNAs enhancing glioblastoma cell susceptibility to tyrosine kinase inhibitors. Hum. Mol. Genet. 2017, 26, 4375–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartararo, C.E.; Reznik, E.; de Carvalho, A.C.; Mikkelsen, T.; Stockwell, B.R. High-Throughput Screening of Patient-Derived Cultures Reveals Potential for Precision Medicine in Glioblastoma. ACS Med. Chem. Lett. 2015, 6, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Akay, M.; Hite, J.; Avci, N.G.; Fan, Y.; Akay, Y.; Lu, G.; Zhu, J.J. Drug Screening of Human GBM Spheroids in Brain Cancer Chip. Sci. Rep. 2018, 8, 15423. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.; Chalmers, A.J.; Gomez-Roman, N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers 2019, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.N.; Anderson, J.C.; Duarte, C.W.; Shevin, R.S.; Langford, C.P.; Singh, R.; Gillespie, G.Y.; Willey, C.D. Combinatorial Drug Testing in 3D Microtumors Derived from GBM Patient-Derived Xenografts Reveals Cytotoxic Synergy in Pharmacokinomics-informed Pathway Interactions. Sci. Rep. 2018, 8, 8412. [Google Scholar] [CrossRef]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Gilbert, A.N.; Shevin, R.S.; Anderson, J.C.; Langford, C.P.; Eustace, N.; Gillespie, G.Y.; Singh, R.; Willey, C.D. Generation of Microtumors Using 3D Human Biogel Culture System and Patient-derived Glioblastoma Cells for Kinomic Profiling and Drug Response Testing. J. Vis. Exp. 2016, e54026. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Huse, J.T.; Berman, S.H.; Pedraza, A.; Zhang, J.; Suehara, Y.; Viale, A.; Cavatore, M.; Heguy, A.; Szerlip, N.; et al. Quantitative assessment of intragenic receptor tyrosine kinase deletions in primary glioblastomas: Their prevalence and molecular correlates. Acta Neuropathol. 2014, 127, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Bergström, T.; Jiang, Y.; Johansson, P.; Marinescu, V.D.; Lindberg, N.; Segerman, A.; Wicher, G.; Niklasson, M.; Baskaran, S.; et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EbioMedicine 2015, 2, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2019, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Azzalini, A.; Menardi, G. Clustering via Nonparametric Density Estimation: The R Package pdfCluster. J. Stat. Softw. 2014, 57, 1–26. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, S.C.; Kim, S.J.; Park, C.H.; Jeung, H.C.; Kim, Y.B.; Ahn, J.B.; Chung, H.C.; Rha, S.Y. Identification of a radiosensitivity signature using integrative metaanalysis of published microarray data for NCI-60 cancer cells. BMC Genom. 2012, 13, 348. [Google Scholar] [CrossRef]
- Speers, C.; Zhao, S.; Liu, M.; Bartelink, H.; Pierce, L.J.; Feng, F.Y. Development and Validation of a Novel Radiosensitivity Signature in Human Breast Cancer. Clin. Cancer Res. 2015, 21, 3667–3677. [Google Scholar] [CrossRef]
- Meng, J.; Li, P.; Zhang, Q.; Yang, Z.; Fu, S. A radiosensitivity gene signature in predicting glioma prognostic via EMT pathway. Oncotarget 2014, 5, 4683–4693. [Google Scholar] [CrossRef] [Green Version]
- Duarte, C.W.; Willey, C.D.; Zhi, D.; Cui, X.; Harris, J.J.; Vaughan, L.K.; Mehta, T.; McCubrey, R.O.; Khodarev, N.N.; Weichselbaum, R.R.; et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS ONE 2012, 7, e29653. [Google Scholar] [CrossRef] [PubMed]
- Alcoser, S.Y.; Kimmel, D.J.; Borgel, S.D.; Carter, J.P.; Dougherty, K.M.; Hollingshead, M.G. Real-time PCR-based assay to quantify the relative amount of human and mouse tissue present in tumor xenografts. BMC Biotechnol. 2011, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Gronych, J.; Warnatz, H.J.; Hutter, B.; Gröbner, S.; Ryzhova, M.; Pfaff, E.; Hovestadt, V.; Weinberg, F.; Halbach, S.; et al. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar]
- Olar, A.; Sulman, E.P. Molecular Markers in Low Grade Glioma-Toward Tumor Reclassification. Semin. Radiat. Oncol. 2015, 25, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ayob, M.; Kendall, G. A Monte Carlo Hyper-Heuristic To Optimise Component Placement Sequencing For Multi Head Placement Machine. In Proceedings of the International Conference on Intelligent Technologies, InTech’03, ChiangMai, Thailand, 17 December 2003; pp. 132–141. [Google Scholar]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef]
- Kritikou, I.; Giannopoulou, E.; Koutras, A.K.; Labropoulou, V.T.; Kalofonos, H.P. The combination of antitumor drugs, exemestane and erlotinib, induced resistance mechanism in H358 and A549 non-small cell lung cancer (NSCLC) cell lines. Pharm. Biol. 2013, 52, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Neri, A.; Marmiroli, S.; Tassone, P.; Lombardi, L.; Nobili, L.; Verdelli, D.; Civallero, M.; Cosenza, M.; Bertacchini, J.; Federico, M.; et al. The oral protein-kinase C beta inhibitor enzastaurin (LY317615) suppresses signalling through the AKT pathway, inhibits proliferation and induces apoptosis in multiple myeloma cell lines. Leuk. Lymphoma 2008, 49, 1374–1383. [Google Scholar] [CrossRef]
- Perazzoli, G.; Prados, J.; Ortiz, R.; Caba, O.; Cabeza, L.; Berdasco, M.; Gónzalez, B.; Melguizo, C. Temozolomide Resistance in Glioblastoma Cell Lines: Implication of MGMT, MMR, P-Glycoprotein and CD133 Expression. PLoS ONE 2015, 10, e0140131. [Google Scholar] [CrossRef]
- Rantanen, V.; Grénman, S.; Kulmala, J.; Grénman, R. Comparative evaluation of cisplatin and carboplatin sensitivity in endometrial adenocarcinoma cell lines. Br. J. Cancer 1994, 69, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
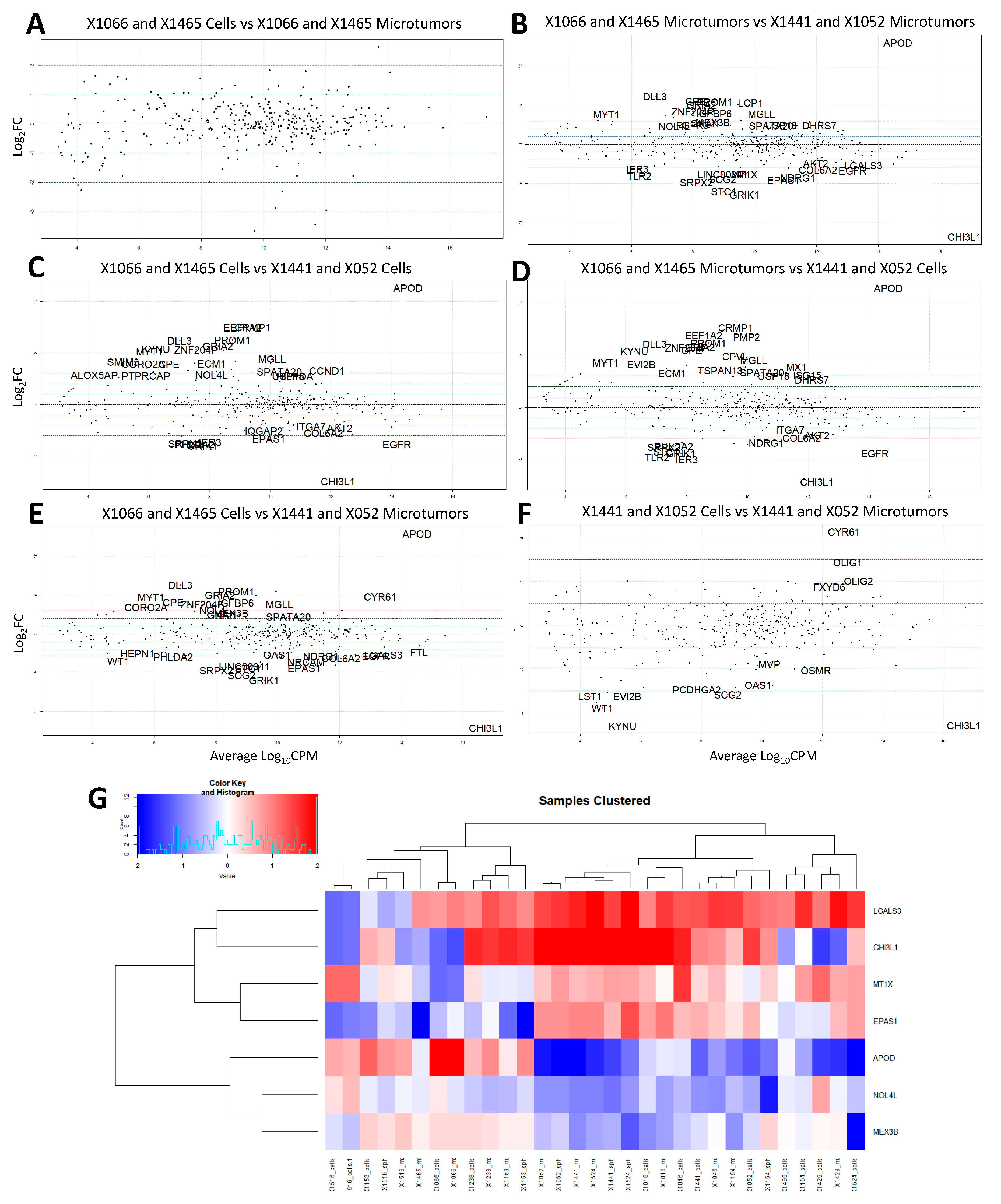

{kind=link}
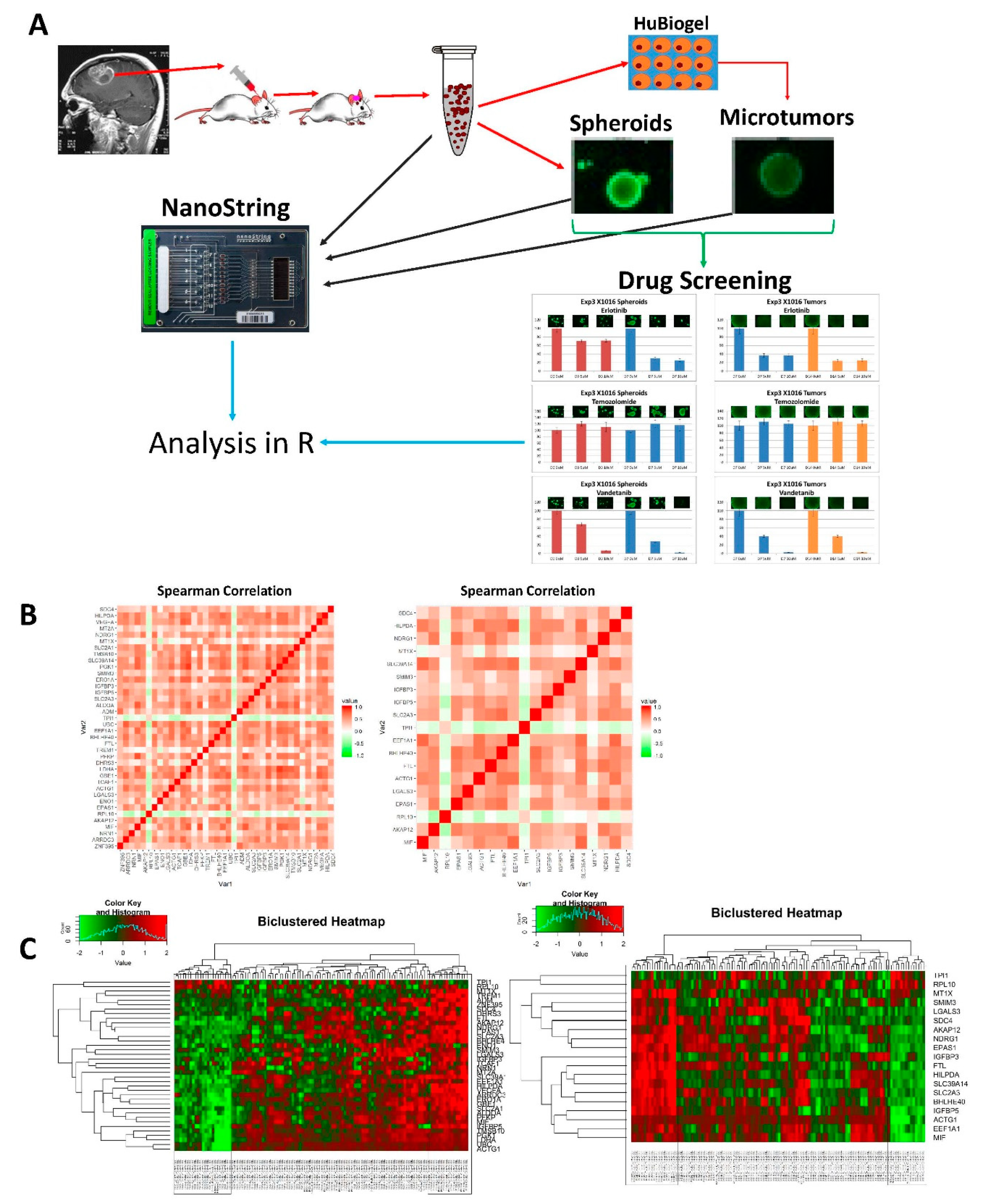
{kind=link}
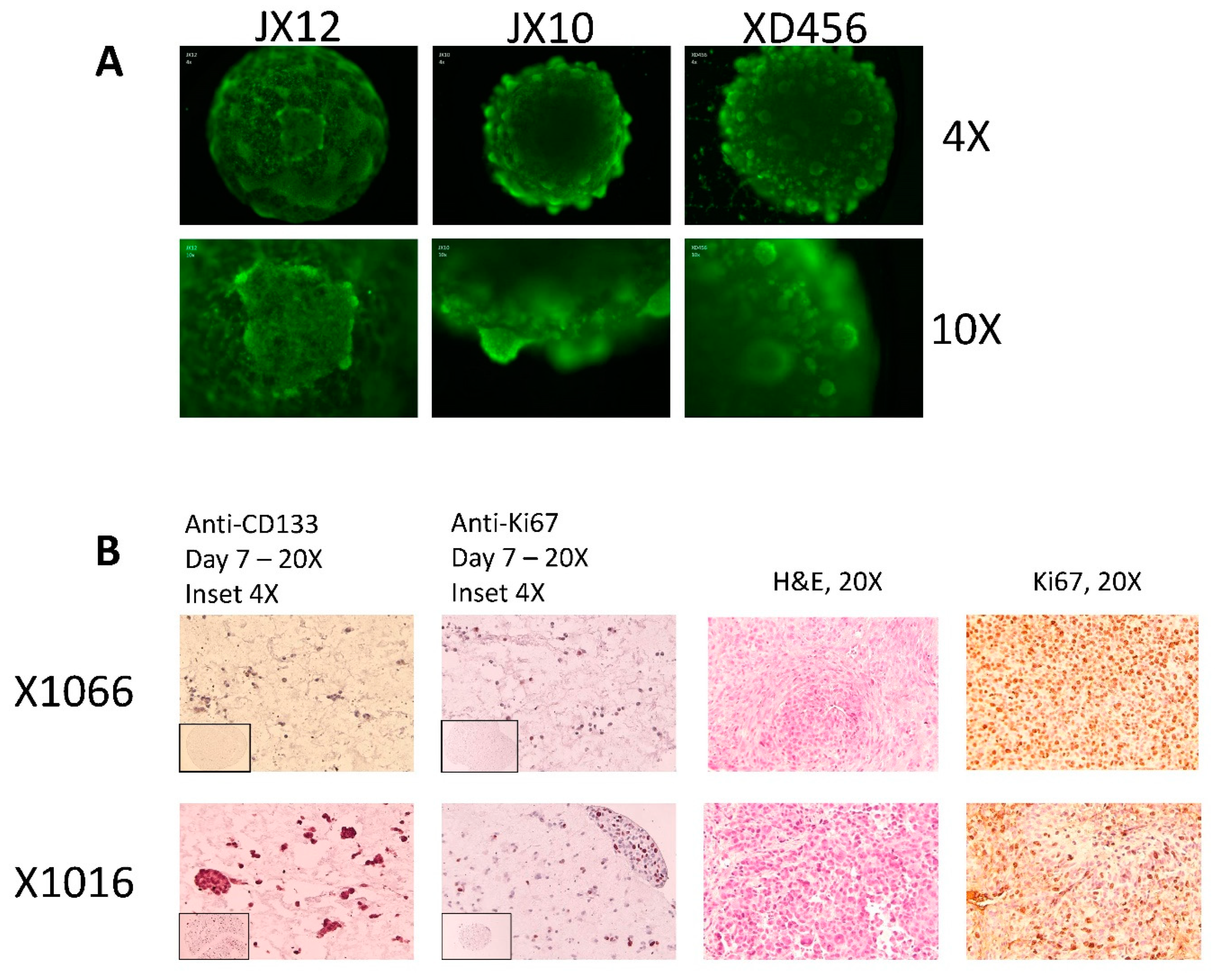
{kind=link}
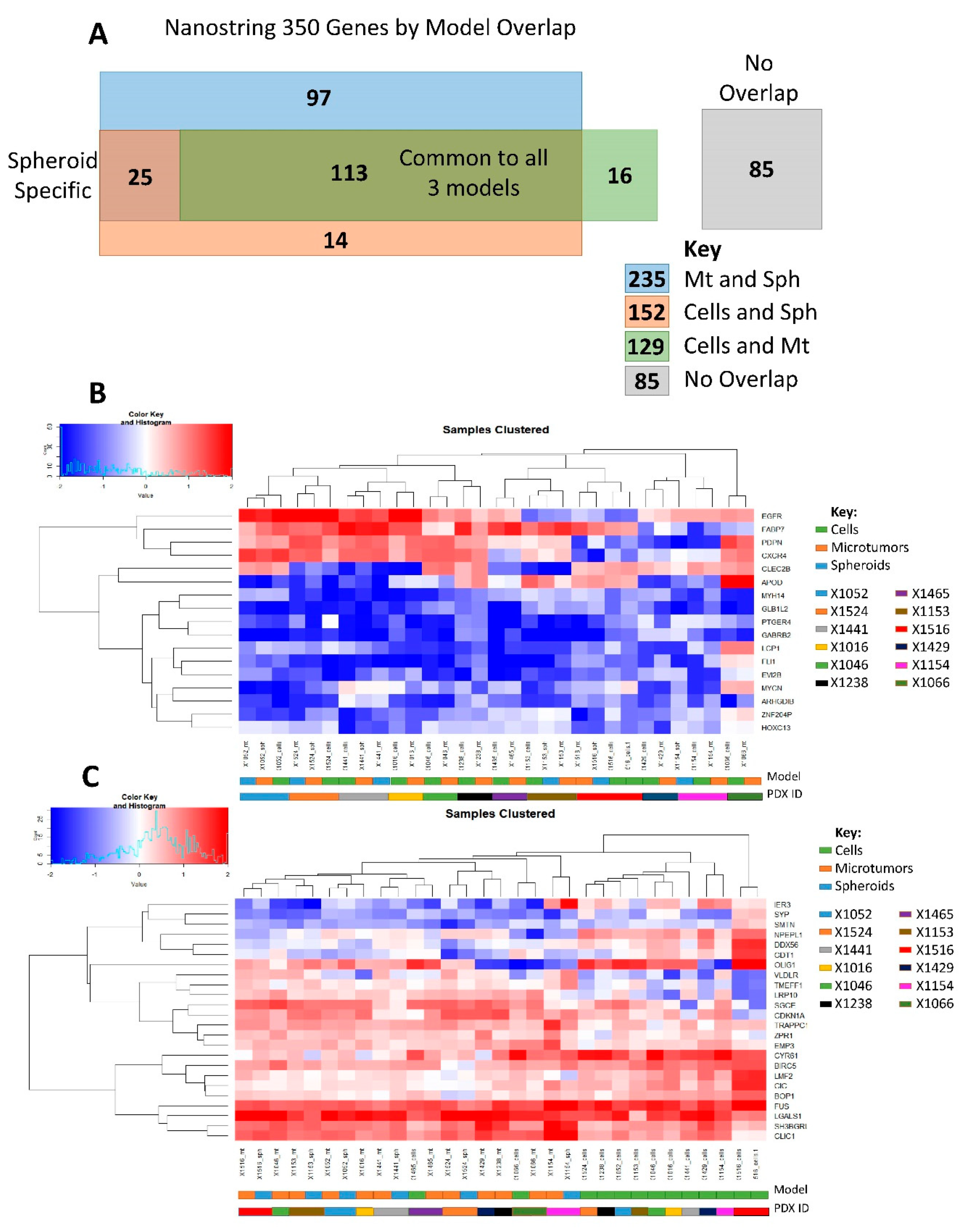
{kind=link}
{kind=link}
| Signature | Genes in Signature | Source(s) |
|---|---|---|
| 1. Novel Gene Expression Molecular Subtype | 100 | FastEMC |
| 2. Gene Expression Molecular Subtype | 23 | Drs. Cameron Brennan and Jason Huse (Kastenhuber et al., 2014) [12,14] |
| 3. Gene Expression Molecular Subtype | 28 | Patel et al., 2014 [13] |
| 4. Cell Cycle Progression | 4 | Patel et al., 2014 [13] |
| 5. Curated Genes of Interest | 32 | In-house |
| 6. Genes Down-Regulated in Radiation Sensitive vs. Radiation Resistant | 26 | Kim, Rha et al., 2012 [18] |
| 7. Genes Up-Regulated in Radiation Sensitive vs. Radiation Resistant | 39 | Kim, Rha et al., 2012 [18] |
| 8. Positive Correlation in Radiation Resistance | 37 | Speers, Zhao et al., 2015 [19] |
| 9. Negative Correlation in Radiation Resistance | 37 | Speers, Zhao et al., 2015 [19] |
| 10. Radiation Sensitivity EMT Pathway | 15 | Meng, Fu et al., 2014 [20] |
| 11. Hypoxia | 19 | Patel et al., 2014 [13] |
| 12. Stemness | 21 | Patel et al., 2014 [13] |
| 13. IFN/STAT1 Signaling | 7 | Willey, Gillespie et al. 2012 [21] |
| 14. PanCancer Internal Reference Genes | 7 | NanoString |
| 15. PTGER2/ptger2 Human/Mouse Reference | 2 | Alcoser et al., 2011 [22] |
| 16. Other—Curated List | 32 | Trabelsi et al., 2016; Patel et al., 2014; Olar, Sulman et al., 2015 [13,23,24] |
| Drug | Target/Mechanism | Concentrations |
|---|---|---|
| Axitinib | VEGFR tyrosine kinase inhibitor | 0, 5, 10 μM |
| Erlotinib | EGFR tyrosine kinase inhibitor | 0, 5, 10 μM |
| Temozolomide | Alkylating agent | 0, 5, 10 μM |
| Carboplatin | Platinum-based antineoplastic | 0, 5, 10 μM |
| Enzastaurin | Protein kinase C beta | 0, 5, 10 μM |
| Vandetanib | VEGFR2 tyrosine kinase inhibitor | 0, 5, 10 μM |
| 350 Gene Panel | 113 Core Genes | |||||||
|---|---|---|---|---|---|---|---|---|
| PDX_ID | Cells a to Mts b | Cells to Sph c | Mt to Sph | PDX_ID | Cells to Mts | Cells to Sph | Mt to Sph | |
| X1516 | 0.652 | 0.649 | 0.642 | X1516 | 0.884 | 0.884 | 0.985 | |
| X1154 | 0.772 | 0.658 | 0.847 | X1154 | 0.89 | 0.816 | 0.924 | |
| X1238 | 0.796 | X1238 | 0.926 | |||||
| X1046 | 0.840 | X1429 | 0.934 | |||||
| X1429 | 0.845 | X1524 | 0.941 | 0.942 | 0.962 | |||
| X1524 | 0.857 | 0.874 | 0.943 | X1046 | 0.954 | |||
| X1016 | 0.892 | X1016 | 0.955 | |||||
| X1153 | 0.908 | 0.910 | 0.974 | X1153 | 0.959 | 0.965 | 0.987 | |
| X1441 | 0.922 | 0.938 | 0.961 | X1441 | 0.965 | 0.984 | 0.976 | |
| X1052 | 0.925 | 0.933 | 0.965 | X1052 | 0.966 | 0.962 | 0.98 | |
| X1465 | 0.949 | X1066 | 0.978 | |||||
| X1066 | 0.958 | X1465 | 0.984 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stackhouse, C.T.; Rowland, J.R.; Shevin, R.S.; Singh, R.; Gillespie, G.Y.; Willey, C.D. A Novel Assay for Profiling GBM Cancer Model Heterogeneity and Drug Screening. Cells 2019, 8, 702. https://doi.org/10.3390/cells8070702
Stackhouse CT, Rowland JR, Shevin RS, Singh R, Gillespie GY, Willey CD. A Novel Assay for Profiling GBM Cancer Model Heterogeneity and Drug Screening. Cells. 2019; 8(7):702. https://doi.org/10.3390/cells8070702
Chicago/Turabian StyleStackhouse, Christian T., James R. Rowland, Rachael S. Shevin, Raj Singh, G. Yancey Gillespie, and Christopher D. Willey. 2019. "A Novel Assay for Profiling GBM Cancer Model Heterogeneity and Drug Screening" Cells 8, no. 7: 702. https://doi.org/10.3390/cells8070702
APA StyleStackhouse, C. T., Rowland, J. R., Shevin, R. S., Singh, R., Gillespie, G. Y., & Willey, C. D. (2019). A Novel Assay for Profiling GBM Cancer Model Heterogeneity and Drug Screening. Cells, 8(7), 702. https://doi.org/10.3390/cells8070702