Autophagy in Neurotrauma: Good, Bad, or Dysregulated

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Traumatic Spinal Cord and Brain Injury and Their Injury Mechanisms
3. Autophagy, Autophagy Flux, and the Lysosomal Functions in Neurotrauma
3.1. Autophagy and Autophagy Flux
3.2. Impairment of Autophagy Flux in SCI and TBI
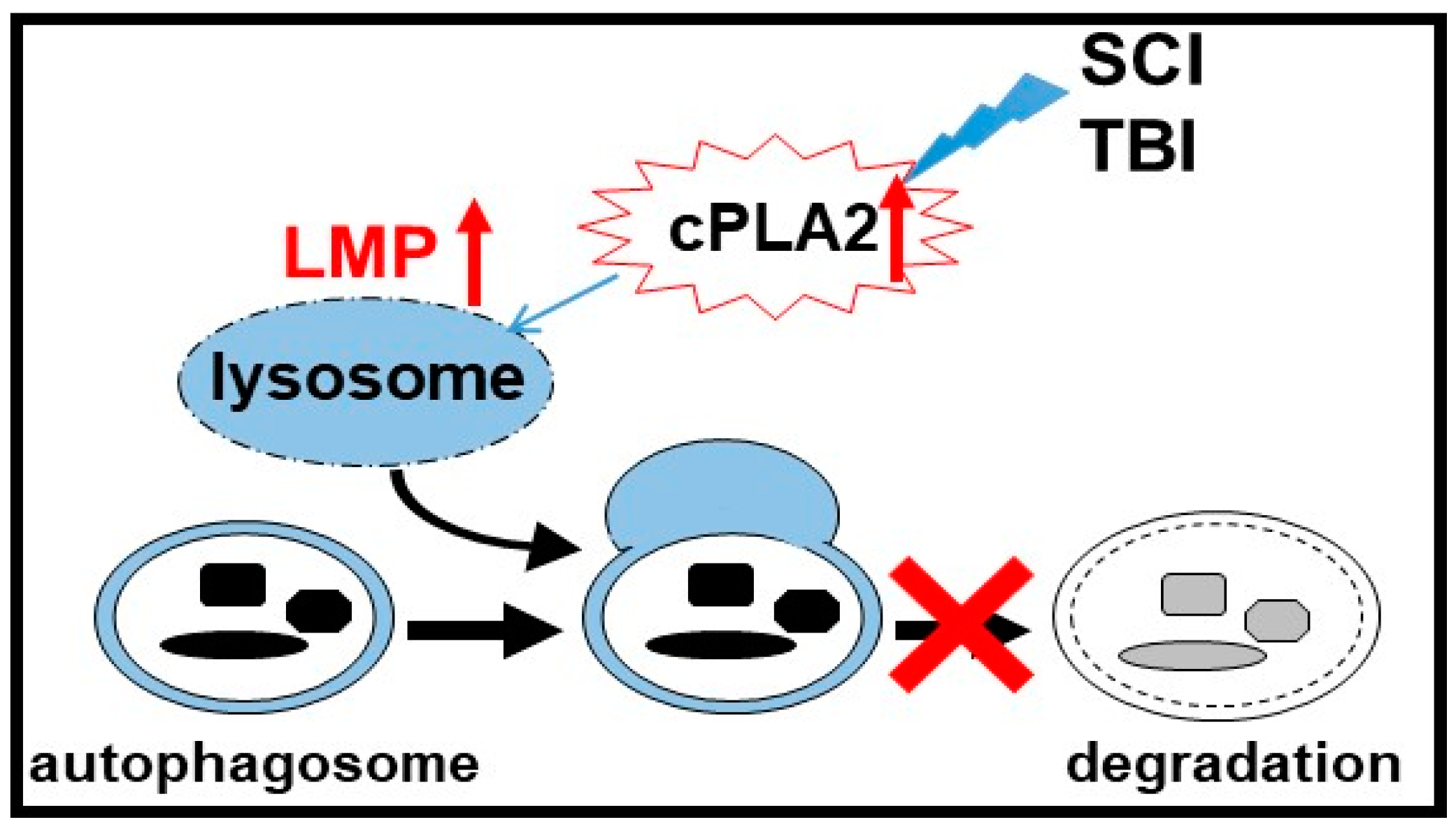
3.3. Lysosomal Functions In Neurotrauma
4. Beneficial or Detrimental Effects of Autophagy in CNS Cells After Trauma
4.1. Function of Autophagy in Neurons after Neurotrauma
4.2. Role of Autophagy in Oligodendrocyte Survival after Neurotrauma
4.3. Function of Autophagy in Microglia and Astrocytes after CNS Trauma
4.4. Beneficial or Detrimental Effects of Autophagy Activation in Neurotrauma
5. Therapeutic Potential of Autophagy–Lysosomal Pathway Modulation in Neurotrauma
5.1. Neuroprotection
5.2. Neuroinflammation
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid. Redox Signal. 2015, 23, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Wu, J. Modification of autophagy-lysosomal pathway as a neuroprotective treatment for spinal cord injury. Neural Regen. Res. 2015, 10, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Y.; Choi, H.M.C.; Sarkar, C.; Koh, E.Y.; Wu, J.; Lipinski, M.M. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sarkar, C.; Dinizo, M.; Faden, A.I.; Koh, E.Y.; Lipinski, M.M.; Wu, J. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015, 6, e1582. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 2014, 10, 2208–2222. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Blomgren, K.; Kroemer, G. Autophagy in acute brain injury. Nat. Rev. Neurosci. 2016, 17, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Smith, C.M.; Chen, Y.; Sullivan, M.L.; Kochanek, P.M.; Clark, R.S. Autophagy in acute brain injury: Feast, famine, or folly? Neurobiol. Dis. 2011, 43, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.S.; Bayir, H.; Kochanek, P.M.; Clark, R.S.B. The role of autophagy in acute brain injury: A state of flux? Neurobiol. Dis. 2019, 122, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ackery, A.; Tator, C.; Krassioukov, A. A global perspective on spinal cord injury epidemiology. J. Neurotrauma 2004, 21, 1355–1370. [Google Scholar] [CrossRef] [PubMed]
- Wood-Dauphinee, S.; Exner, G.; Bostanci, B.; Exner, G.; Glass, C.; Jochheim, K.A.; Kluger, P.; Koller, M.; Krishnan, K.R.; Post, M.W.; et al. Quality of life in patients with spinal cord injury--basic issues, assessment, and recommendations. Restor. Neurol. Neurosci. 2002, 20, 135–149. [Google Scholar] [PubMed]
- Tator, C.H. Experimental and clinical studies of the pathophysiology and management of acute spinal cord injury. J. Spinal Cord Med. 1996, 19, 206–214. [Google Scholar] [CrossRef]
- Tator, C.H.; Fehlings, M.G. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J. Neurosurg. 1991, 75, 15–26. [Google Scholar] [CrossRef]
- Beattie, M.S.; Farooqui, A.A.; Bresnahan, J.C. Review of current evidence for apoptosis after spinal cord injury. J. Neurotrauma 2000, 17, 915–925. [Google Scholar] [CrossRef]
- Osterholm, J.L. The pathophysiological response to spinal cord injury. The current status of related research. J. Neurosurg. 1974, 40, 5–33. [Google Scholar] [CrossRef]
- Mautes, A.E.; Weinzierl, M.R.; Donovan, F.; Noble, L.J. Vascular events after spinal cord injury: Contribution to secondary pathogenesis. Phys. Ther. 2000, 80, 673–687. [Google Scholar] [PubMed]
- Zhang, B.; Gensel, J.C. Is neuroinflammation in the injured spinal cord different than in the brain? Examining intrinsic differences between the brain and spinal cord. Exp. Neurol. 2014, 258, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pajoohesh-Ganji, A.; Stoica, B.A.; Dinizo, M.; Guanciale, K.; Faden, A.I. Delayed expression of cell cycle proteins contributes to astroglial scar formation and chronic inflammation after rat spinal cord contusion. J. Neuroinflammation 2012, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.X.L.; Wald, M.M.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths; Centers for Disease Control and Prevention NcfIPaC: Atlanta, GA, USA, 2010. [Google Scholar]
- Dutton, R.P.; Stansbury, L.G.; Leone, S.; Kramer, E.; Hess, J.R.; Scalea, T.M. Trauma mortality in mature trauma systems: Are we doing better? An analysis of trauma mortality patterns, 1997-2008. J. Trauma 2010, 69, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Panter, S.S.; Faden, A.I. Biochemical changes and secondary injury from stroke and trauma, Chapter 4. In Principles and Practice of Restorative Neurology; Butterworth’s: New York, NY, USA, 1992; pp. 32–52. [Google Scholar]
- Keane, R.W.; Davis, A.R.; Dietrich, W.D. Inflammatory and apoptotic signaling after spinal cord injury. J. Neurotrauma 2006, 23, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Springer, J.E. Neuroprotection and acute spinal cord injury: A reappraisal. NeuroRx 2004, 1, 80–100. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Progressive damage after brain and spinal cord injury: Pathomechanisms and treatment strategies. Prog. Brain Res. 2007, 161, 125–141. [Google Scholar]
- Sidaros, A.; Engberg, A.W.; Sidaros, K.; Liptrot, M.G.; Herning, M.; Petersen, P.; Paulson, O.B.; Jernigan, T.L.; Rostrup, E. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: A longitudinal study. Brain 2008, 131, 559–572. [Google Scholar] [CrossRef]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Faden, A.I. Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Faden, A.I. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010, 7, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; Madathil, S.K.; Saatman, K.E. Genetic manipulation of cell death and neuroplasticity pathways in traumatic brain injury. Neurotherapeutics 2012, 9, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E. Key note lecture: Toward a mechanistic taxonomy for cell death programs. Stroke 2007, 38, 652–660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bredesen, D.E. Programmed cell death mechanisms in neurological disease. Curr. Mol. Med. 2008, 8, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Frankel, L.B.; Lund, A.H. MicroRNA regulation of autophagy. Carcinogenesis 2012, 33, 2018–2025. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Tan, X.; Jing, H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012, 8, 873–882. [Google Scholar] [CrossRef]
- Alqurashi, N.; Hashimi, S.M.; Wei, M.Q. Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics. Int. J. Mol. Sci. 2013, 14, 3874–3900. [Google Scholar] [CrossRef]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kagedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef]
- You, Z.; Savitz, S.I.; Yang, J.; Degterev, A.; Yuan, J.; Cuny, G.D.; Moskowitz, M.A.; Whalen, M.J. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2008, 28, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Tao, Y.; Zhang, S.; Wang, J.; Feng, X. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 2014, 266, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I. Neuroprotection and traumatic brain injury: Theoretical option or realistic proposition. Curr. Opin. Neurol. 2002, 15, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Leal, W.; Corkill, D.J.; Freire, M.A.; Picanco-Diniz, C.W.; Perry, V.H. Astrocytosis, microglia activation, oligodendrocyte degeneration, and pyknosis following acute spinal cord injury. Exp. Neurol. 2004, 190, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E.; van der Maesen, K.; Somera, F.P. Macrophage and microglial responses to cytokines in vitro: Phagocytic activity, proteolytic enzyme release, and free radical production. J. Neurosci. Res. 1998, 54, 68–78. [Google Scholar] [CrossRef]
- Suh, H.S.; Kim, M.O.; Lee, S.C. Inhibition of Granulocyte-Macrophage Colony-Stimulating Factor Signaling and Microglial Proliferation by Anti-CD45RO: Role of Hck Tyrosine Kinase and Phosphatidylinositol 3-Kinase/Akt. J. Immunol. 2005, 174, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Popovich, P.G.; Wei, P.; Stokes, B.T. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J. Comp. Neurol. 1997, 377, 443–464. [Google Scholar] [CrossRef]
- Watanabe, T.; Yamamoto, T.; Abe, Y.; Saito, N.; Kumagai, T.; Kayama, H. Differential activation of microglia after experimental spinal cord injury. J. Neurotrauma 1999, 16, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.L.; Parrish, M.E.; Springer, J.E.; Doty, K.; Dossett, L. Acute inflammatory response in spinal cord following impact injury. Exp. Neurol. 1998, 151, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, F.; Conrad, A.; Kohl, A.; Korf, H.W.; Hailer, N.P. Clodronate inhibits the secretion of proinflammatory cytokines and NO by isolated microglial cells and reduces the number of proliferating glial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp. Neurol. 2004, 189, 241–251. [Google Scholar] [CrossRef]
- Delves, P.J.; Roitt, I.M. The immune system. First of two parts. N. Engl. J. Med. 2000, 343, 37–49. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Tani, M.; Krivacic, K.; Chernosky, A.; Kelner, G.S.; Maciejewski, D.; Maki, R.; Ransohoff, R.M.; Stokes, B.T. Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J. Neurosci. Res. 1998, 53, 368–376. [Google Scholar] [CrossRef]
- Pan, J.Z.; Ni, L.; Sodhi, A.; Aguanno, A.; Young, W.; Hart, R.P. Cytokine activity contributes to induction of inflammatory cytokine mRNAs in spinal cord following contusion. J. Neurosci. Res. 2002, 68, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Olschowka, J.A.; Wrathall, J.R. Increase of interleukin-1beta mRNA and protein in the spinal cord following experimental traumatic injury in the rat. Brain Res. 1997, 759, 190–196. [Google Scholar] [CrossRef]
- Byrnes, K.R.; Garay, J.; Di Giovanni, S.; De Biase, A.; Knoblach, S.M.; Hoffman, E.P.; Movsesyan, V.; Faden, A.I. Expression of two temporally distinct microglia-related gene clusters after spinal cord injury. Glia 2006, 53, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Papandreou, M.E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A. Neuronal autophagy: A housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. P62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, A.; Kanno, H.; Ozawa, H.; Yamaya, S.; Itoi, E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J. Neurotrauma 2012, 29, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Yamaya, S.; Itoi, E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine 2011, 36, E1427–E1434. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, F.; Yone, K.; Kawabata, N.; Sakakima, H.; Matsuda, F.; Ishidou, Y.; Maeda, S.; Abematsu, M.; Komiya, S.; Setoguchi, T. Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: Functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy 2011, 7, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.H.; Wang, L.; Guo, Z.J.; Bai, L.; Zhang, R.P.; Shuang, W.B.; Jia, Y.J.; Wang, J.; Li, X.Y.; Liu, Q. Valproic acid reduces autophagy and promotes functional recovery after spinal cord injury in rats. Neurosci. Bull. 2013, 29, 484–492. [Google Scholar] [CrossRef]
- Munoz-Galdeano, T.; Reigada, D.; Del Aguila, A.; Velez, I.; Caballero-Lopez, M.J.; Maza, R.M.; Nieto-Diaz, M. Cell Specific Changes of Autophagy in a Mouse Model of Contusive Spinal Cord Injury. Front. Cell. Neurosci. 2018, 12, 164. [Google Scholar] [CrossRef]
- Chen, H.C.; Fong, T.H.; Lee, A.W.; Chiu, W.T. Autophagy is activated in injured neurons and inhibited by methylprednisolone after experimental spinal cord injury. Spine 2012, 37, 470–475. [Google Scholar] [CrossRef]
- Hou, H.; Zhang, L.; Zhang, L.; Tang, P. Acute spinal cord injury in rats should target activated autophagy. J. Neurosurg. Spine 2014, 20, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Wang, Z.G.; Wu, F.Z.; Kong, X.X.; Yang, J.; Lin, B.B.; Zhu, S.P.; Lin, L.; Gan, C.S.; Fu, X.B. Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol. Neurobiol. 2013, 48, 452–464. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, C.; Meng, B.; Tang, T.S.; Yang, H.L. Changes in autophagy proteins in a rat model of spinal cord injury. Chin. J. Traumatol. 2014, 17, 193–197. [Google Scholar]
- Berliocchi, L.; Maiaru, M.; Varano, G.P.; Russo, R.; Corasaniti, M.T.; Bagetta, G.; Tassorelli, C. Spinal autophagy is differently modulated in distinct mouse models of neuropathic pain. Mol. Pain 2015, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Lin, J.H.; Muharram, A.; Liu, W.G. Beclin-1-mediated autophagy protects spinal cord neurons against mechanical injury-induced apoptosis. Apoptosis 2014, 19, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Itoi, E. Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol. Dis. 2009, 33, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fu, Q.; Shen, B.; Huang, X.; Wang, K.; He, P.; Li, F.; Zhang, F.; Shen, H. Enhanced p62 expression triggers concomitant autophagy and apoptosis in a rat chronic spinal cord compression model. Mol. Med. Rep. 2014, 9, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fukuda, T.; Iwadate, K. Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: Association with time between the injury and death. Am. J. Forensic. Med. Pathol. 2014, 35, 38–44. [Google Scholar] [CrossRef]
- Clark, R.S.; Bayir, H.; Chu, C.T.; Alber, S.M.; Kochanek, P.M.; Watkins, S.C. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef]
- Au, A.K.; Aneja, R.K.; Bayir, H.; Bell, M.J.; Janesko-Feldman, K.; Kochanek, P.M.; Clark, R.S.B. Autophagy Biomarkers Beclin 1 and p62 are Increased in Cerebrospinal Fluid after Traumatic Brain Injury. Neurocrit. Care 2017, 26, 348–355. [Google Scholar] [CrossRef]
- Lai, Y.; Hickey, R.W.; Chen, Y.; Bayir, H.; Sullivan, M.L.; Chu, C.T.; Kochanek, P.M.; Dixon, C.E.; Jenkins, L.W.; Graham, S.H.; et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J. Cereb. Blood Flow Metab. 2008, 28, 540–550. [Google Scholar] [CrossRef]
- Diskin, T.; Tal-Or, P.; Erlich, S.; Mizrachy, L.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J. Neurotrauma 2005, 22, 750–762. [Google Scholar] [CrossRef]
- Liu, C.L.; Chen, S.; Dietrich, D.; Hu, B.R. Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 2008, 28, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Erlich, S.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 2007, 26, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.J.; Li, P.; Ning, Y.L.; Zhao, Y.; Peng, Y.; Yang, N.; Zhao, Z.A.; Chen, J.F.; Zhou, Y.G. Impaired autophagic flux is associated with the severity of trauma and the role of A2AR in brain cells after traumatic brain injury. Cell Death Dis. 2018, 9, 252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Li, S.X.; Chen, X.P.; Yang, L.; Zhang, Y.G.; Liu, R.; Tao, L.Y. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci. Bull. 2008, 24, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, S.; Dunn, W.A.; Jr Hayes, R.L.; Wang, K.K. Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem. Biophys. Res. Commun. 2008, 373, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Tyurin, V.A.; Tyurina, Y.Y.; Viner, R.; Ritov, V.; Amoscato, A.A.; Zhao, Q.; Zhang, X.J.; Janesko-Feldman, K.L.; Alexander, H.; et al. Selective early cardiolipin peroxidation after traumatic brain injury: An oxidative lipidomics analysis. Ann. Neurol. 2007, 62, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Hickey, R.W.; Bayir, H.; Watkins, S.C.; Tyurin, V.A.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Ren, J.; Gibson, G.; et al. Starving neurons show sex difference in autophagy. J. Biol. Chem. 2009, 284, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Futter, C.E.; Patel, S. Methods for monitoring lysosomal morphology. Methods. Cell Biol. 2015, 126, 1–19. [Google Scholar]
- de Duve, D. The peroxisome: A new cytoplasmic organelle. Proc. R. Soc. Lond.Ser. B Biol. Sci. 1969, 173, 71–83. [Google Scholar]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Ledesma, M.D.; Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 2016, 32, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Muela, N.; Hernandez-Pinto, A.M.; Serrano-Puebla, A.; Garcia-Ledo, L.; Latorre, S.H.; de la Rosa, E.J.; Boya, P. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015, 22, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T.; Oikawa, S. The role of lysosomal rupture in neuronal death. Prog. Neurobiol. 2009, 89, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. When lysosomes get old. Exp. Gerontol. 2000, 35, 119–131. [Google Scholar] [CrossRef]
- Yin, Y.; Li, E.; Sun, G.; Yan, H.Q.; Foley, L.M.; Andrzejczuk, L.A.; Attarwala, I.Y.; Hitchens, T.K.; Kiselyov, K.; Dixon, C.E.; et al. Effects of DHA on Hippocampal Autophagy and Lysosome Function After Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 2454–2470. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Simonyi, A.; Sun, A.Y.; Sun, G.Y. Phospholipases A2 and neural membrane dynamics: Implications for Alzheimer’s disease. J. Neurochem. 2011, 116, 813–819. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Yang, H.C.; Rosenberger, T.A.; Horrocks, L.A. Phospholipase A2 and its role in brain tissue. J. Neurochem. 1997, 69, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.D.; Schievella, A.R.; Nalefski, E.A.; Lin, L.L. Cytosolic phospholipase A2. J. Lipid Mediat. Cell Signal. 1995, 12, 83–117. [Google Scholar] [CrossRef]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.Q.; Zhou, Y.; Halabisky, B.; Cisse, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.C. Cytosolic phospholipase A(2): Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W.; O’Regan, M.H. A potentially critical role of phospholipases in central nervous system ischemic, traumatic, and neurodegenerative disorders. Brain Res. Rev. 2004, 44, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, Y.C.; Kim, S.J.; Choi, M.S.; Tsai, P.C.; Saha, A.; Wei, H.; Xu, Y.; Xiao, Y.J.; Zhang, P.; et al. Production of lysophosphatidylcholine by cPLA2 in the brain of mice lacking PPT1 is a signal for phagocyte infiltration. Hum. Mol. Genet. 2007, 16, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.K.; Deng, L.X.; Zhang, Y.P.; Lu, Q.B.; Wang, X.F.; Hu, J.G.; Oakes, E.; Bonventre, J.V.; Shields, C.B.; Xu, X.M. Cytosolic phospholipase A2 protein as a novel therapeutic target for spinal cord injury. Ann. Neurol. 2014, 75, 644–658. [Google Scholar] [CrossRef]
- Li, Y.; Jones, J.W.; Choi, H.M.C.; Sarkar, C.; Kane, M.A.; Koh, E.Y.; Lipinski, M.M.; Wu, J. cPLA2 activation contributes to lysosomal defects leading to impairment of autophagy after spinal cord injury. Cell Death Dis. 2019, in press. [Google Scholar]
- Sarkar, C.; Jones, J.W.; Hegdekar, N.; Thayer, J.A.; Kumar, A.; Faden, A.I.; Kane, M.A.; Lipinski, M.M. PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy 2019, 1–20. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A. Brain phospholipases A2: A perspective on the history. Prostaglandin. Leukot. Essent. Fatty Acids 2004, 71, 161–169. [Google Scholar] [CrossRef]
- Gijon, M.A.; Leslie, C.C. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J. Leukoc. Biol. 1999, 65, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A(2) research: From cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Ong, W.Y.; Horrocks, L.A. Inhibitors of brain phospholipase A2 activity: Their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol. Rev. 2006, 58, 591–620. [Google Scholar] [CrossRef]
- Berliocchi, L.; Russo, R.; Maiaru, M.; Levato, A.; Bagetta, G.; Corasaniti, M.T. Autophagy impairment in a mouse model of neuropathic pain. Mol. Pain 2011, 7, 83. [Google Scholar] [CrossRef]
- Zhang, E.; Yi, M.H.; Ko, Y.; Kim, H.W.; Seo, J.H.; Lee, Y.H.; Lee, W.; Kim, D.W. Expression of LC3 and Beclin 1 in the spinal dorsal horn following spinal nerve ligation-induced neuropathic pain. Brain Res. 2013, 1519, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Larner, S.F.; Hayes, R.L.; McKinsey, D.M.; Pike, B.R.; Wang, K.K. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J. Neurochem. 2004, 88, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Ohri, S.S.; Hetman, M.; Whittemore, S.R. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol. Dis. 2013, 58, 29–37. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Lipinski, M.M.; Py, B.F.; Yuan, J. Endoplasmic reticulum stress response in cell death and cell survival. In Apoptosis: Physiology and Pathology, 1st ed.; Reed, J.C., Green, D.R., Eds.; Cambridge University Press: Cambridge, UK, 2011; pp. 51–62. [Google Scholar]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Ribas, V.T.; Schnepf, B.; Challagundla, M.; Koch, J.C.; Bahr, M.; Lingor, P. Early and sustained activation of autophagy in degenerating axons after spinal cord injury. Brain Pathol. 2015, 25, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef]
- Beattie, M.S.; Hermann, G.E.; Rogers, R.C.; Bresnahan, J.C. Cell death in models of spinal cord injury. Prog. Brain Res. 2002, 137, 37–47. [Google Scholar] [PubMed]
- Saraswat Ohri, S.; Bankston, A.N.; Mullins, S.A.; Liu, Y.; Andres, K.R.; Beare, J.E.; Howard, R.M.; Burke, D.A.; Riegler, A.S.; Smith, A.E.; et al. Blocking Autophagy in Oligodendrocytes Limits Functional Recovery after Spinal Cord Injury. J. Neurosci. 2018, 38, 5900–5912. [Google Scholar] [CrossRef]
- Smith, C.M.; Mayer, J.A.; Duncan, I.D. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J. Neurosci. 2013, 33, 8088–8100. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Byrnes, K.R. Role of microglia in neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Messer, J.S. The cellular autophagy/apoptosis checkpoint during inflammation. Cell. Mol. Life Sci. 2017, 74, 1281–1296. [Google Scholar] [CrossRef]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef]
- Sil, P.; Muse, G.; Martinez, J. A ravenous defense: Canonical and non-canonical autophagy in immunity. Curr. Opin. Immunol. 2018, 50, 21–31. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Shin, D.M.; Yuk, J.M.; Shi, G.; Choi, D.K.; Lee, S.H.; Huang, S.M.; Kim, J.M.; Kim, C.D.; Lee, J.H.; et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J. Immunol. 2011, 186, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Su, Y.C.; Hu, C.W.; Lei, H.Y. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013, 20, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Terro, F.; Janet, T.; Rioux Bilan, A.; Paccalin, M.; Page, G. Involvement of interleukin-1beta in the autophagic process of microglia: Relevance to Alzheimer’s disease. J. Neuroinflammation 2013, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xian, W.; Zhou, H.; Chen, L.; Pei, Z. Potential protective effects of autophagy activated in MPP+ treated astrocytes. Exp. Ther. Med. 2016, 12, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Pla, A.; Pascual, M.; Guerri, C. Autophagy Constitutes a Protective Mechanism against Ethanol Toxicity in Mouse Astrocytes and Neurons. PLoS ONE 2016, 11, e0153097. [Google Scholar] [CrossRef]
- Cao, L.; Fu, M.; Kumar, S.; Kumar, A. Methamphetamine potentiates HIV-1 gp120-mediated autophagy via Beclin-1 and Atg5/7 as a pro-survival response in astrocytes. Cell Death Dis. 2016, 7, e2425. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Girelli, S.; Scopa, C.; Buonocore, G.; Longini, M.; Balduini, W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010, 6, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Fong, T.H.; Hsu, P.W.; Chiu, W.T. Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. J. Surg. Res. 2013, 179, e203–e210. [Google Scholar] [CrossRef]
- Hu, L.Y.; Sun, Z.G.; Wen, Y.M.; Cheng, G.Z.; Wang, S.L.; Zhao, H.B.; Zhang, X.R. ATP-mediated protein kinase B Akt/mammalian target of rapamycin mTOR/p70 ribosomal S6 protein p70S6 kinase signaling pathway activation promotes improvement of locomotor function after spinal cord injury in rats. Neuroscience 2010, 169, 1046–1062. [Google Scholar] [CrossRef]
- Walker, C.L.; Walker, M.J.; Liu, N.K.; Risberg, E.C.; Gao, X.; Chen, J.; Xu, X.M. Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS ONE 2012, 7, e30012. [Google Scholar] [CrossRef]
- Wahl, S.E.; McLane, L.E.; Bercury, K.K.; Macklin, W.B.; Wood, T.L. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J.Neurosci. 2014, 34, 4453–4465. [Google Scholar] [CrossRef]
- Lee, D.H.; Luo, X.; Yungher, B.J.; Bray, E.; Lee, J.K.; Park, K.K. Mammalian target of rapamycin’s distinct roles and effectiveness in promoting compensatory axonal sprouting in the injured CNS. J. Neurosci. 2014, 34, 15347–15355. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Isaka, M.; Hamaishi, M.; Imai, K.; Orihashi, K.; Sueda, T. Trehalose protects against spinal cord ischemia in rabbits. J. Vasc. Surg. 2014, 60, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef]
- Pi, H.; Li, M.; Tian, L.; Yang, Z.; Yu, Z.; Zhou, Z. Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci. Rep. 2017, 7, 43466. [Google Scholar] [CrossRef]
- Fang, J.; Zhu, Y.; Wang, H.; Cao, B.; Fei, M.; Niu, W.; Zhou, Y.; Wang, X.; Li, X.; Zhou, M. Baicalin Protects Mice Brain From Apoptosis in Traumatic Brain Injury Model Through Activation of Autophagy. Front. Neurosci. 2018, 12, 1006. [Google Scholar] [CrossRef]
- Wang, Z.F.; Gao, C.; Chen, W.; Gao, Y.; Wang, H.C.; Meng, Y.; Luo, C.L.; Zhang, M.Y.; Chen, G.; Chen, X.P.; et al. Salubrinal offers neuroprotection through suppressing endoplasmic reticulum stress, autophagy and apoptosis in a mouse traumatic brain injury model. Neurobiol. Learn. Mem. 2019, 161, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, Z.; Fang, Z.; Lin, W.; Wu, S.; Yang, F.; Li, Y.; Fu, H.; Gao, H.; Li, S. Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J. Neuroinflammation 2018, 15, 310. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Zhou, M.; Li, X.; Fang, Z.; Gao, H.; Li, Y.; Hu, W. Valproic Acid Attenuates Traumatic Brain Injury-Induced Inflammation in Vivo: Involvement of Autophagy and the Nrf2/ARE Signaling Pathway. Front. Mol. Neurosci. 2018, 11, 117. [Google Scholar] [CrossRef]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef]
- Randall-Demllo, S.; Chieppa, M.; Eri, R. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef] [PubMed]
- Marselli, L.; Bugliani, M.; Suleiman, M.; Olimpico, F.; Masini, M.; Petrini, M.; Boggi, U.; Filipponi, F.; Syed, F.; Marchetti, P. Beta-Cell inflammation in human type 2 diabetes and the role of autophagy. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 130–136. [Google Scholar] [CrossRef]
- Pan, L.; Li, Y.; Jia, L.; Qin, Y.; Qi, G.; Cheng, J.; Qi, Y.; Li, H.; Du, J. Cathepsin S deficiency results in abnormal accumulation of autophagosomes in macrophages and enhances Ang II-induced cardiac inflammation. PLoS ONE 2012, 7, e35315. [Google Scholar] [CrossRef] [PubMed]
- Junkins, R.D.; McCormick, C.; Lin, T.J. The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy 2014, 10, 538–547. [Google Scholar] [CrossRef]
- Francois, A.; Rioux Bilan, A.; Quellard, N.; Fernandez, B.; Janet, T.; Chassaing, D.; Paccalin, M.; Terro, F.; Page, G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J. Neuroinflammation 2014, 11, 139. [Google Scholar] [CrossRef]
- von Leden, R.E.; Yauger, Y.J.; Khayrullina, G.; Byrnes, K.R. Central Nervous System Injury and Nicotinamide Adenine Dinucleotide Phosphate Oxidase: Oxidative Stress and Therapeutic Targets. J. Neurotrauma 2016, 34, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2014, 22, 377. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. A genome-wide analysis reveals differential regulation of autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. https://doi.org/10.3390/cells8070693
Wu J, Lipinski MM. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells. 2019; 8(7):693. https://doi.org/10.3390/cells8070693
Chicago/Turabian StyleWu, Junfang, and Marta M. Lipinski. 2019. "Autophagy in Neurotrauma: Good, Bad, or Dysregulated" Cells 8, no. 7: 693. https://doi.org/10.3390/cells8070693
APA StyleWu, J., & Lipinski, M. M. (2019). Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells, 8(7), 693. https://doi.org/10.3390/cells8070693