Development of Personalized Therapeutic Strategies by Targeting Actionable Vulnerabilities in Metastatic and Chemotherapy-Resistant Breast Cancer PDXs

 , ,
, ,  , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Ethic Statements
2.2. PDX Tissue Bank Generation
2.3. Animals
2.4. PDX Culture
2.5. Exome-Sequencing
2.6. RNA-Sequencing
2.7. Drug Test
2.8. Western Blotting
2.9. Statistical Analysis
2.10. Data Access
3. Results
3.1. Breast Cancer PDXs Recapitulate the Biology of the Tumor of Origin
3.2. Each Metastatic PDX Represents a Unique Genomic Landscape Pattern
3.3. Transcriptional Analysis of PDX Cells Parallels Mutational Profile
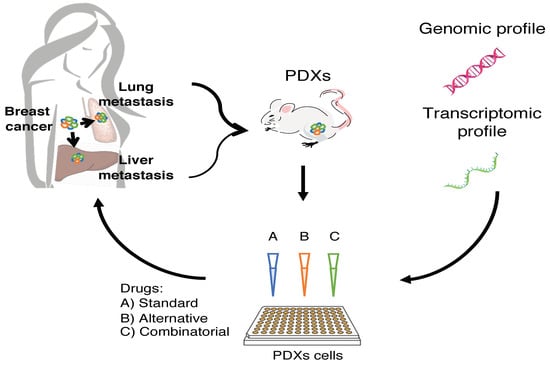
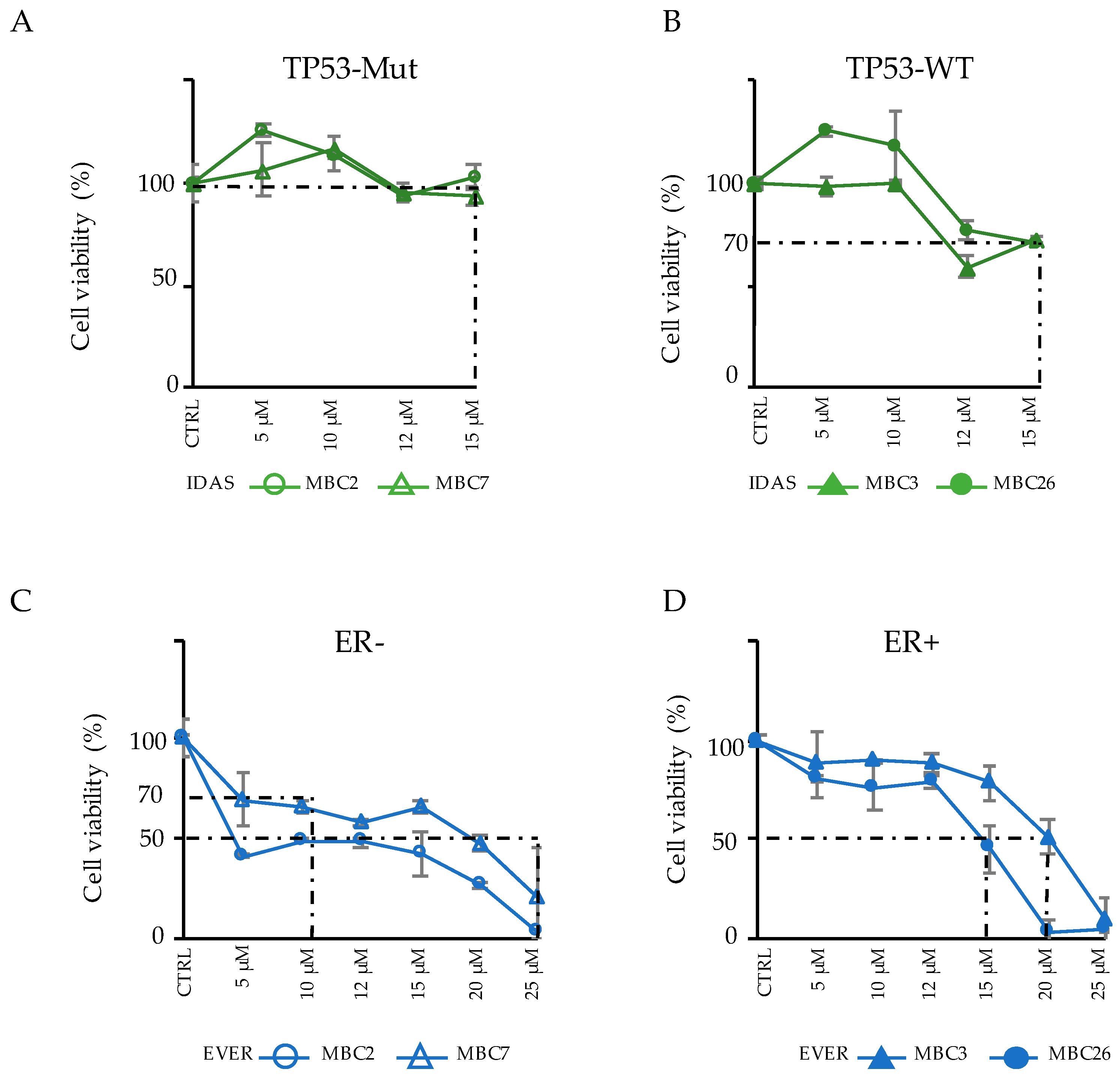
3.4. Integration of Genomic and Transcriptomic Profiles Predicts Resistance and Sensitivity to Treatments
3.5. Combinatorial Drug Administration Can Prioritize Therapeutic Regimen in Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ginsburg, O.; Bray, F.; Coleman, M.P.; Vanderpuye, V.; Eniu, A.; Kotha, S.R.; Sarker, M.; Huong, T.T.; Allemani, C.; Dvaladze, A.; et al. The global burden of women’s cancers: A grand challenge in global health. Lancet 2017, 389, 847–860. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Johnston, S.R. Enhancing Endocrine Therapy for Hormone Receptor-Positive Advanced Breast Cancer: Cotargeting Signaling Pathways. J. Natl. Cancer Inst. 2015. [Google Scholar] [CrossRef]
- Hosford, S.R.; Miller, T.W. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharmacogenom. Personal. Med. 2014, 7, 203–215. [Google Scholar]
- Palma, G.; Frasci, G.; Chirico, A.; Esposito, E.; Siani, C.; Saturnino, C.; Arra, C.; Ciliberto, G.; Giordano, A.; D’Aiuto, M. Triple negative breast cancer: Looking for the missing link between biology and treatments. Oncotarget 2015, 6, 26560–26574. [Google Scholar] [CrossRef]
- Sudhan, D.R.; Schwarz, L.J.; Guerrero-Zotano, A.; Formisano, L.; Nixon, M.J.; Croessmann, S.; Ericsson, P.I.G.; Sanders, M.; Balko, J.M.; Avogadri-Connors, F.; et al. Extended Adjuvant Therapy with Neratinib Plus Fulvestrant Blocks ER/HER2 Crosstalk and Maintains Complete Responses of ER(+)/HER2(+) Breast Cancers: Implications to the ExteNET Trial. Clin. Cancer Res. 2019, 25, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Nicosia, S.V.; Dave, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014, 344, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef] [PubMed]
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016, 6, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- D’Alesio, C.; Punzi, S.; Cicalese, A.; Fornasari, L.; Furia, L.; Riva, L.; Carugo, A.; Curigliano, G.; Criscitiello, C.; Pruneri, G.; et al. RNAi screens identify CHD4 as an essential gene in breast cancer growth. Oncotarget. 2016, 7, 80901–80915. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, N.; Somlo, G.; Wu, J.; Frankel, P.; Kai, M.; Liu, X.; Wu, S.V.; Nguyen, D.; Chan, N.; Hsieh, M.Y.; et al. Characterization of patient-derived tumor xenografts (PDXs) as models for estrogen receptor positive (ER+HER2- and ER+HER2+) breast cancers. J. Steroid Biochem. Mol. Biol. 2017, 170, 65–74. [Google Scholar] [CrossRef] [PubMed]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, N.; Lofgren, K.A.; Peterson, E.A.; Meier, D.R.; Jung, E.H.; Kenny, P.A. Mutant GATA3 Actively Promotes the Growth of Normal and Malignant Mammary Cells. Anticancer Res. 2018, 38, 4435–4441. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Wang, H.; Wang, Y.; He, Q.; Xia, X.; Hu, Z.Y.; Ouyang, Q. Clinical and genetic risk factors for Fulvestrant treatment in post-menopause ER-positive advanced breast cancer patients. J. Transl. Med. 2019. [Google Scholar] [CrossRef]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832. [Google Scholar] [CrossRef]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Li, L.; Wang, N.; Xiong, Y.; Li, Y.; Gu, Y. Relationship of Epidermal Growth Factor Receptor Expression with Clinical Symptoms and Metastasis of Invasive Breast Cancer. J. Interferon Cytokine Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Gendoo, D.M.A.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. A subgroup of microRNAs defines PTEN-deficient, triple-negative breast cancer patients with poorest prognosis and alterations in RB1, MYC, and Wnt signaling. Breast Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Aftab, A.; Shahzad, S.; Hussain, H.M.J.; Khan, R.; Irum, S.; Tabassum, S. CDKN2A/P16INK4A variants association with breast cancer and their in-silico analysis. Breast Cancer 2019, 26, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Y.; An, N.; Zhang, M. Feature genes in metastatic breast cancer identified by MetaDE and SVM classifier methods. Mol. Med. Rep. 2018, 17, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.W.; Zeng, Z.; Konopleva, M.; Verstovsek, S.; Ravandi, F.; Ferrajoli, A.; Ferrajoli, A.; Thomas, D.; Wierda, W.; Apostolidou, E.; et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res. 2006, 12, 5165–5173. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, L.; Gu, G.; Rechoum, Y.; Beyer, A.R.; Pejerrey, S.M.; Tsimelzon, A.; Wang, T.; Huffman, K.; Ludlow, A.; Ando, S.; et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res. Treat. 2016, 157, 253–265. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Cobb, P.; Malikayil, K.; Flaherty, D.; Andrew Johnson, C.; Raman, D.; Saleh, N.; Higgins, B.; Vara, B.A.; Johnston, J.N.; et al. MDM2 Antagonists Counteract Drug-Induced DNA Damage. EBioMedicine 2017, 24, 43–55. [Google Scholar] [CrossRef][Green Version]
- Chen, X.; Paudyal, S.C.; Chin, R.I.; You, Z. PCNA promotes processive DNA end resection by Exo1. Nucl. Acids Res. 2013, 41, 9325–9338. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, T.J.; Meier, J.B.; Jansen, V.M. Current Landscape of Targeted Therapies for Hormone-Receptor Positive, HER2 Negative Metastatic Breast Cancer. Front. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mikula-Pietrasik, J.; Witucka, A.; Pakula, M.; Uruski, P.; Begier-Krasinska, B.; Niklas, A.; Tykarski, A.; Ksiazek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine. Oncol. Lett. 2019, 17, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Stossel, C.; Atias, D.; Buzhor, E.; Halperin, S.; Cohen, K.; Raitses-Gurevich, M.; Glick, Y.; Raskin, S.; Yehuda, D.; et al. Recapitulating the clinical scenario of BRCA-associated pancreatic cancer in pre-clinical models. Int. J. Cancer 2018, 143, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Zhang, G.; Kim, H.S.; Stinson, R.M.; Bechara, R.; Zhang, C.; Chen, Z.; Saba, N.F.; Pakkala, S.; Pillai, R.; et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Erriquez, J.; Olivero, M.; Mittica, G.; Scalzo, M.S.; Vaira, M.; De Simone, M.; Ponzone, R.; Katsaros, D.; Aglietta, M.; Calogero, R.; et al. Xenopatients show the need for precision medicine approach to chemotherapy in ovarian cancer. Oncotarget 2016, 7, 26181–26191. [Google Scholar] [CrossRef] [PubMed]
- Francis, O.L.; Milford, T.A.; Beldiman, C.; Payne, K.J. Fine-tuning patient-derived xenograft models for precision medicine approaches in leukemia. J. Investig. Med. 2016, 64, 740–744. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef]
- Marangoni, E.; Vincent-Salomon, A.; Auger, N.; Degeorges, A.; Assayag, F.; de Cremoux, P.; de Plater, L.; Guyader, C.; De Pinieux, G.; Judde, J.G.; et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin. Cancer Res. 2007, 13, 3989–3998. [Google Scholar] [CrossRef]
- Wheler, J.J.; Parker, B.A.; Lee, J.J.; Atkins, J.T.; Janku, F.; Tsimberidou, A.M.; Zinner, R.; Subbiah, V.; Fu, S.; Schwab, R.; et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget 2014, 5, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Andersen, J.N.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med. 2011, 17, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Glas, A.M.; Wessels, L.F.; Witteveen, A.T.; Peterse, J.L.; van’t Veer, L.J. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 15901–15905. [Google Scholar] [CrossRef] [PubMed]
- Cerk, S.; Schwarzenbacher, D.; Adiprasito, J.B.; Stotz, M.; Hutterer, G.C.; Gerger, A.; Ling, H.; Calin, G.A.; Pichler, M. Current Status of Long Non-Coding RNAs in Human Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1485. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Malouf, G.G.; Chen, Y.; Zhang, J.; Yao, H.; Valero, V.; Weinstein, J.N.; Spano, J.P.; Meric-Bernstam, F.; Khayat, D.; et al. Comprehensive analysis of long non-coding RNAs in human breast cancer clinical subtypes. Oncotarget 2014, 5, 9864–9876. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007, 608, 1–22. [Google Scholar]
- Yin, S.; Bhattacharya, R.; Cabral, F. Human mutations that confer paclitaxel resistance. Mol. Cancer Ther. 2010, 9, 327–335. [Google Scholar] [CrossRef]
- Xu, J.H.; Hu, S.L.; Shen, G.D.; Shen, G. Tumor suppressor genes and their underlying interactions in paclitaxel resistance in cancer therapy. Cancer Cell Int. 2016. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Yeung, Y.; Lau, D.K.; Chionh, F.; Tran, H.; Tse, J.W.T.; Weickhardt, A.J.; Nikfarjam, M.; Scott, A.M.; Tebbutt, N.C.; Mariadason, J.M. K-Ras mutation and amplification status is predictive of resistance and high basal pAKT is predictive of sensitivity to everolimus in biliary tract cancer cell lines. Mol. Oncol. 2017, 11, 1130–1142. [Google Scholar] [CrossRef]
- Park, J.H.; Ryu, M.H.; Park, Y.S.; Park, S.R.; Na, Y.S.; Rhoo, B.Y.; Kang, Y.K. Successful control of heavily pretreated metastatic gastric cancer with the mTOR inhibitor everolimus (RAD001) in a patient with PIK3CA mutation and pS6 overexpression. BMC Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; Pham, N.A.; Kim, L.; Sakashita, S.; Li, M.; Ng, C.; Wang, Y.; Loparco, P.; Sierra, R.; Wang, L.; et al. Novel combinations of PI3K-mTOR inhibitors with dacomitinib or chemotherapy in PTEN-deficient patient-derived tumor xenografts. Oncotarget 2017, 8, 84659–84670. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gu, W.; Zhu, X. NEAT1 mediates paclitaxel-resistance of non-small cell of lung cancer through activation of Akt/mTOR signaling pathway. J. Drug Target. 2019, 1–23. [Google Scholar]
- Christopoulos, P.; Engel-Riedel, W.; Grohe, C.; Kropf-Sanchen, C.; von Pawel, J.; Gutz, S.; Kollmeier, J.; Eberhardt, W.; Ukena, D.; Baum, V.; et al. Everolimus with paclitaxel and carboplatin as first-line treatment for metastatic large-cell neuroendocrine lung carcinoma: A multicenter phase II trial. Ann. Oncol. 2017, 28, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Frankel, P.; Lim, D.; Yeon, C.; Leong, L.; Chao, J.; Ruel, N.; Luevanos, E.; Koehler, S.; Chung, S.; et al. Phase Ib Trial of mFOLFOX6 and Everolimus (NSC-733504) in Patients with Metastatic Gastroesophageal Adenocarcinoma. Oncology 2016, 90, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Kordes, S.; Klumpen, H.J.; Weterman, M.J.; Schellens, J.H.; Richel, D.J.; Wilmink, J.W. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2015, 75, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Carvajal, D.; Tovar, C.; Yang, H.; Vu, B.T.; Heimbrook, D.C.; Vassilev, L.T. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005, 65, 1918–1924. [Google Scholar] [CrossRef]
- Drakos, E.; Atsaves, V.; Schlette, E.; Li, J.; Papanastasi, I.; Rassidakis, G.Z.; Medeiros, L.J. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009, 23, 2290–2299. [Google Scholar] [CrossRef]
- Zanjirband, M.; Edmondson, R.J.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134. [Google Scholar] [CrossRef]
- Deben, C.; Wouters, A.; Op de Beeck, K.; van Den Bossche, J.; Jacobs, J.; Zwaenepoel, K.; Peeters, M.; Van Meerbeeck, J.; Lardon, F.; Rolfo, C.; et al. The MDM2-inhibitor Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget 2015, 6, 22666–22679. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Punzi, S.; Meliksetian, M.; Riva, L.; Marocchi, F.; Pruneri, G.; Criscitiello, C.; Orsi, F.; Spaggiari, L.; Casiraghi, M.; Della Vigna, P.; et al. Development of Personalized Therapeutic Strategies by Targeting Actionable Vulnerabilities in Metastatic and Chemotherapy-Resistant Breast Cancer PDXs. Cells 2019, 8, 605. https://doi.org/10.3390/cells8060605
Punzi S, Meliksetian M, Riva L, Marocchi F, Pruneri G, Criscitiello C, Orsi F, Spaggiari L, Casiraghi M, Della Vigna P, et al. Development of Personalized Therapeutic Strategies by Targeting Actionable Vulnerabilities in Metastatic and Chemotherapy-Resistant Breast Cancer PDXs. Cells. 2019; 8(6):605. https://doi.org/10.3390/cells8060605
Chicago/Turabian StylePunzi, Simona, Marine Meliksetian, Laura Riva, Federica Marocchi, Giancarlo Pruneri, Carmen Criscitiello, Franco Orsi, Lorenzo Spaggiari, Monica Casiraghi, Paolo Della Vigna, and et al. 2019. "Development of Personalized Therapeutic Strategies by Targeting Actionable Vulnerabilities in Metastatic and Chemotherapy-Resistant Breast Cancer PDXs" Cells 8, no. 6: 605. https://doi.org/10.3390/cells8060605
APA StylePunzi, S., Meliksetian, M., Riva, L., Marocchi, F., Pruneri, G., Criscitiello, C., Orsi, F., Spaggiari, L., Casiraghi, M., Della Vigna, P., Luzi, L., Curigliano, G., Pelicci, P. G., & Lanfrancone, L. (2019). Development of Personalized Therapeutic Strategies by Targeting Actionable Vulnerabilities in Metastatic and Chemotherapy-Resistant Breast Cancer PDXs. Cells, 8(6), 605. https://doi.org/10.3390/cells8060605