Peficitinib Inhibits the Chemotactic Activity of Monocytes via Proinflammatory Cytokine Production in Rheumatoid Arthritis Fibroblast-Like Synoviocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Cell Culture
2.3. Immunohistochemical Analysis
2.4. Western Blot Analysis
2.5. Proliferation Assays
2.6. Chemotaxis Assays
2.7. Enzyme-Linked Immunosorbent Assays (ELISA)
2.8. Statistical Analyses
3. Results
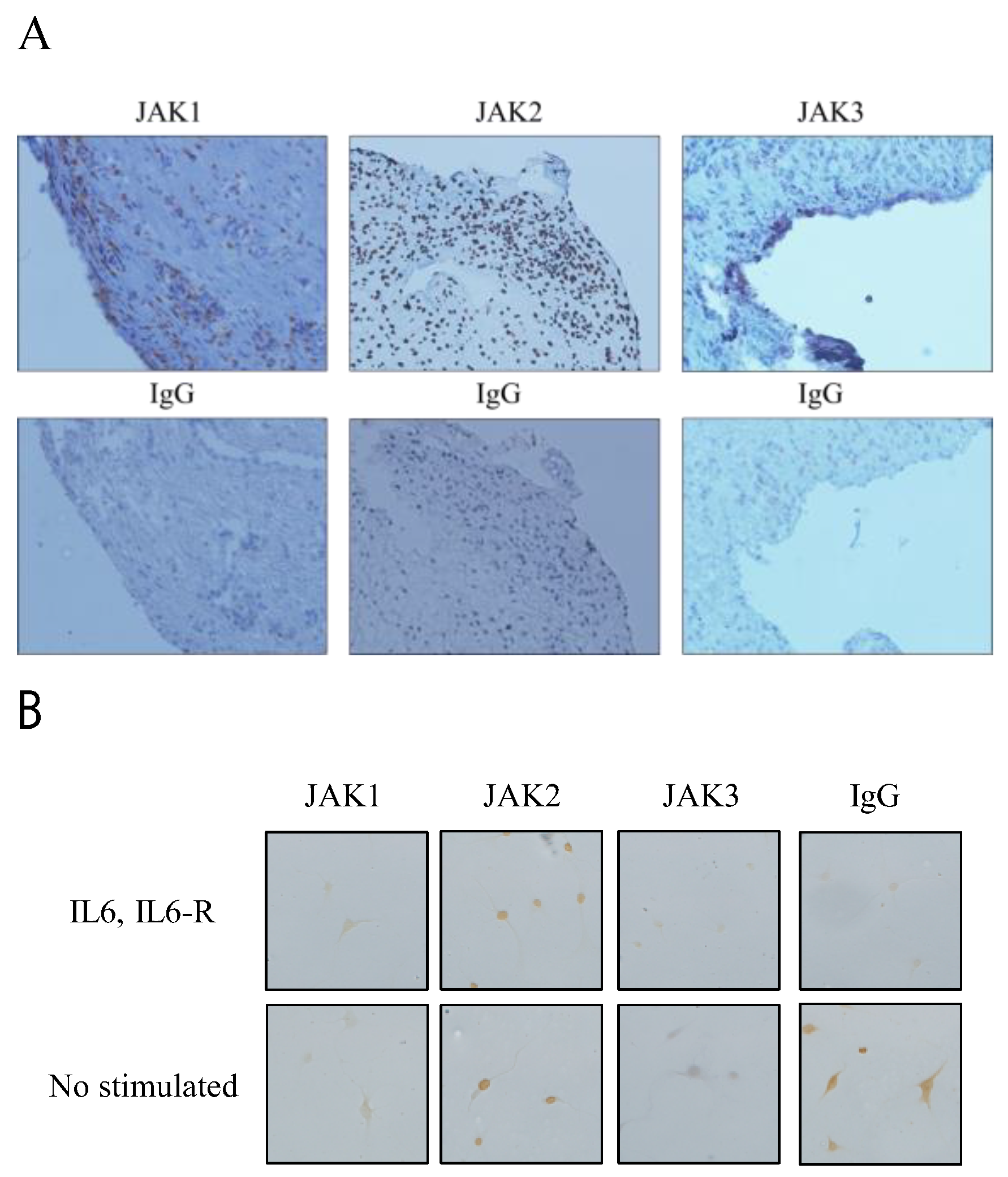
3.1. Expression of JAK1, JAK2, and JAK3 in RA STs and FLSs
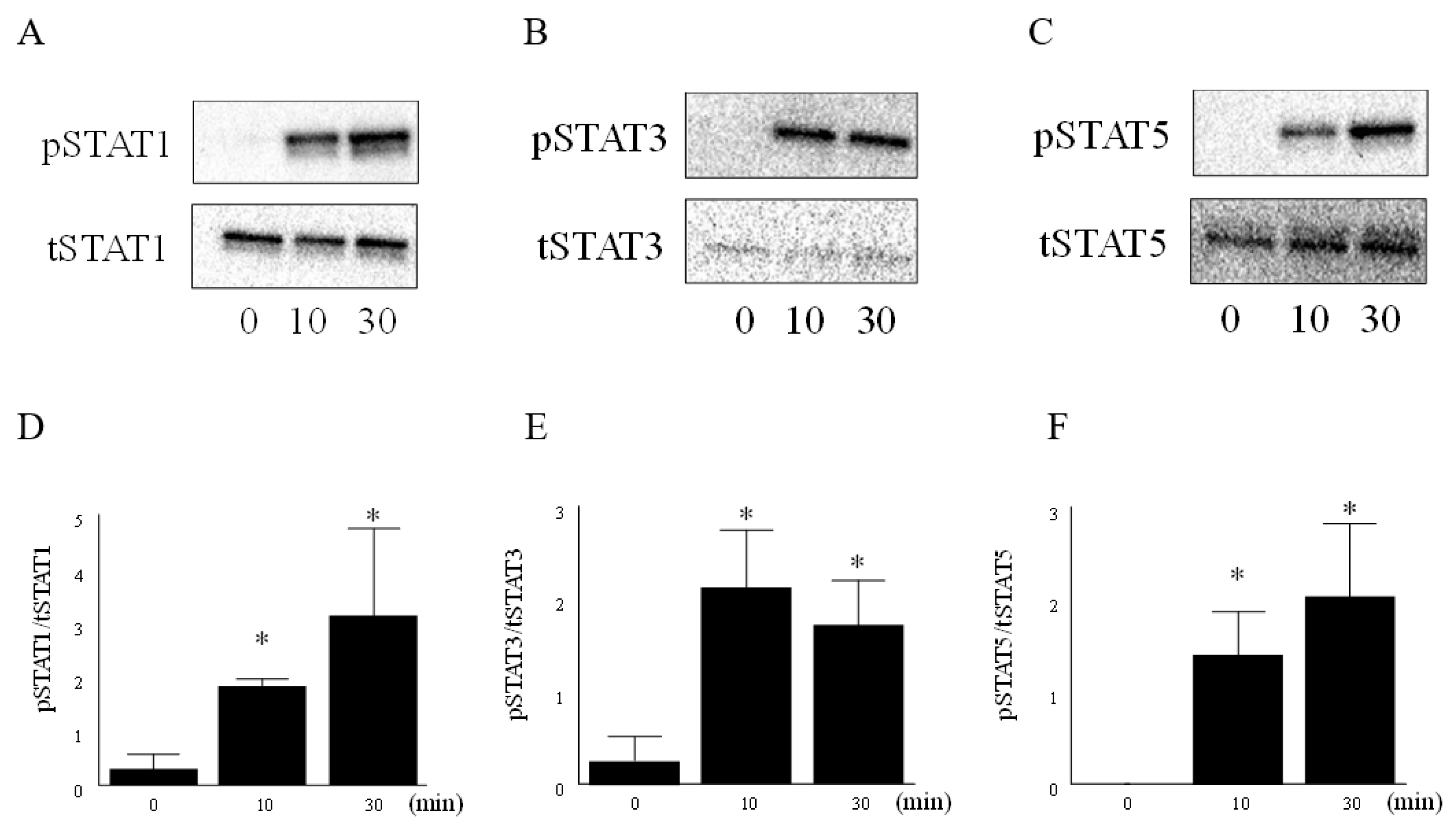
3.2. IL-6 and IL-6R Activated the JAK-STAT Pathway in RA FLS
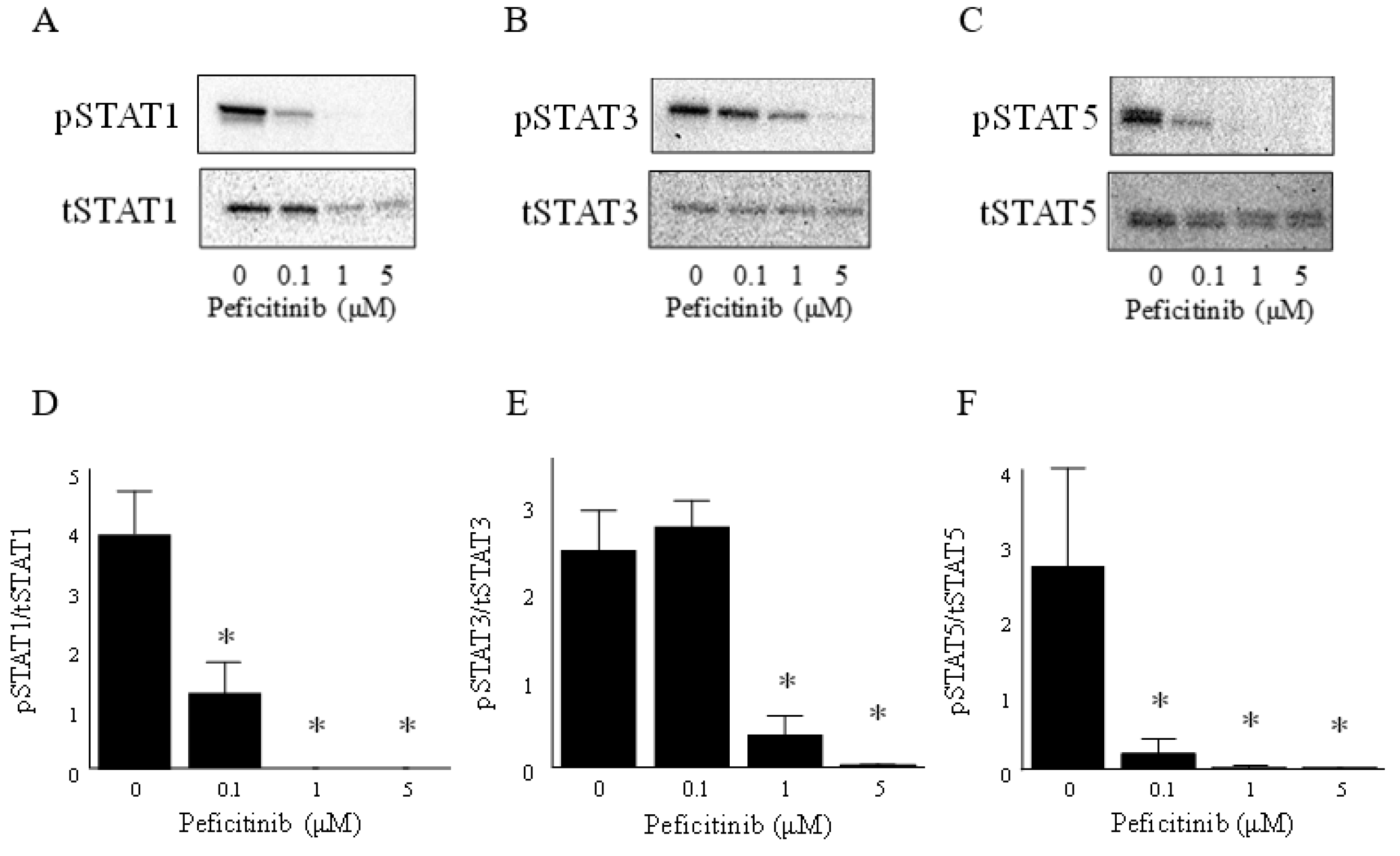
3.3. Peficitinib Inhibited the JAK-STAT Pathway in RA FLS
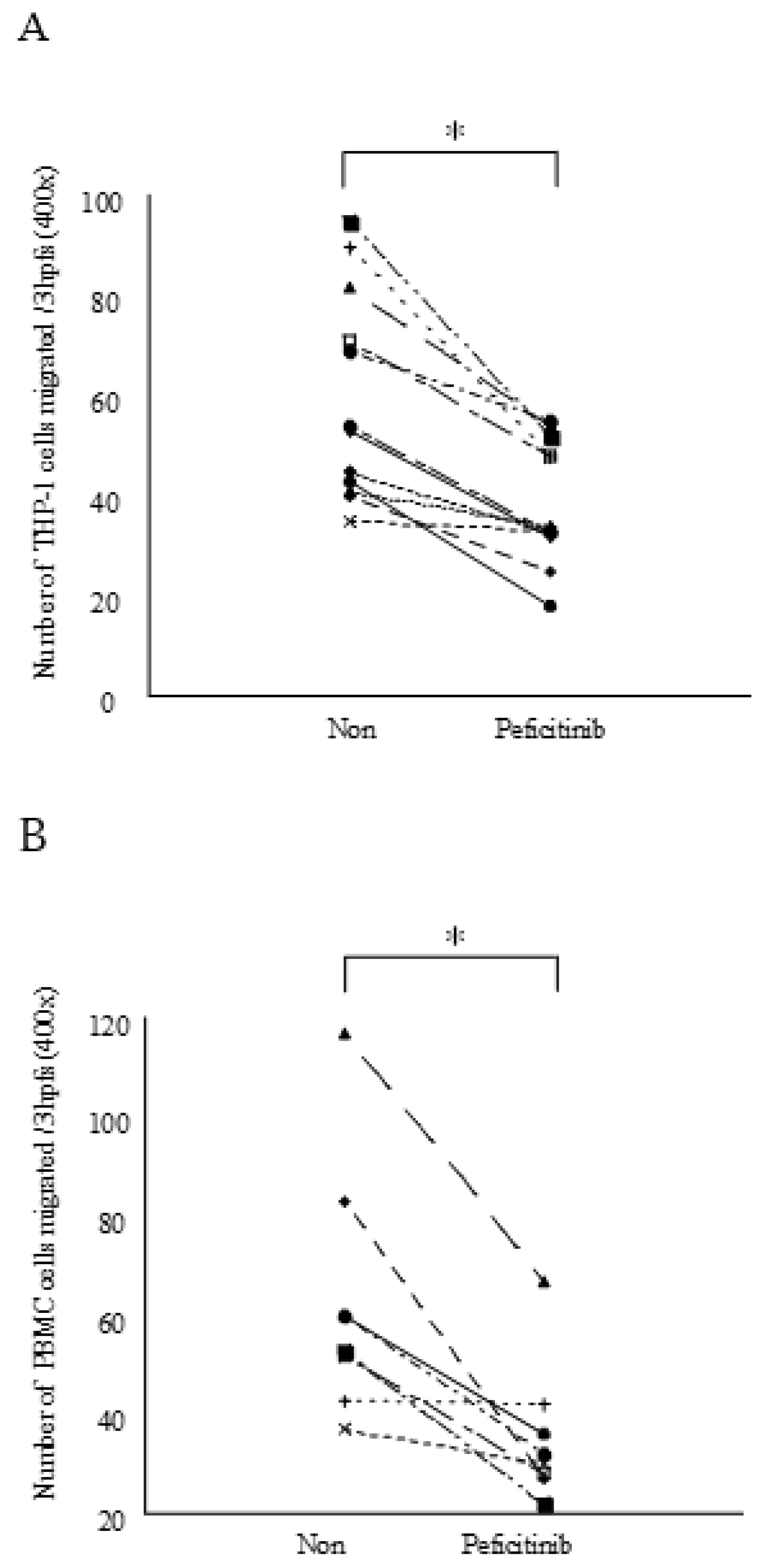
3.4. Peficitinib Inhibited the Monocyte Chemotactic Activity
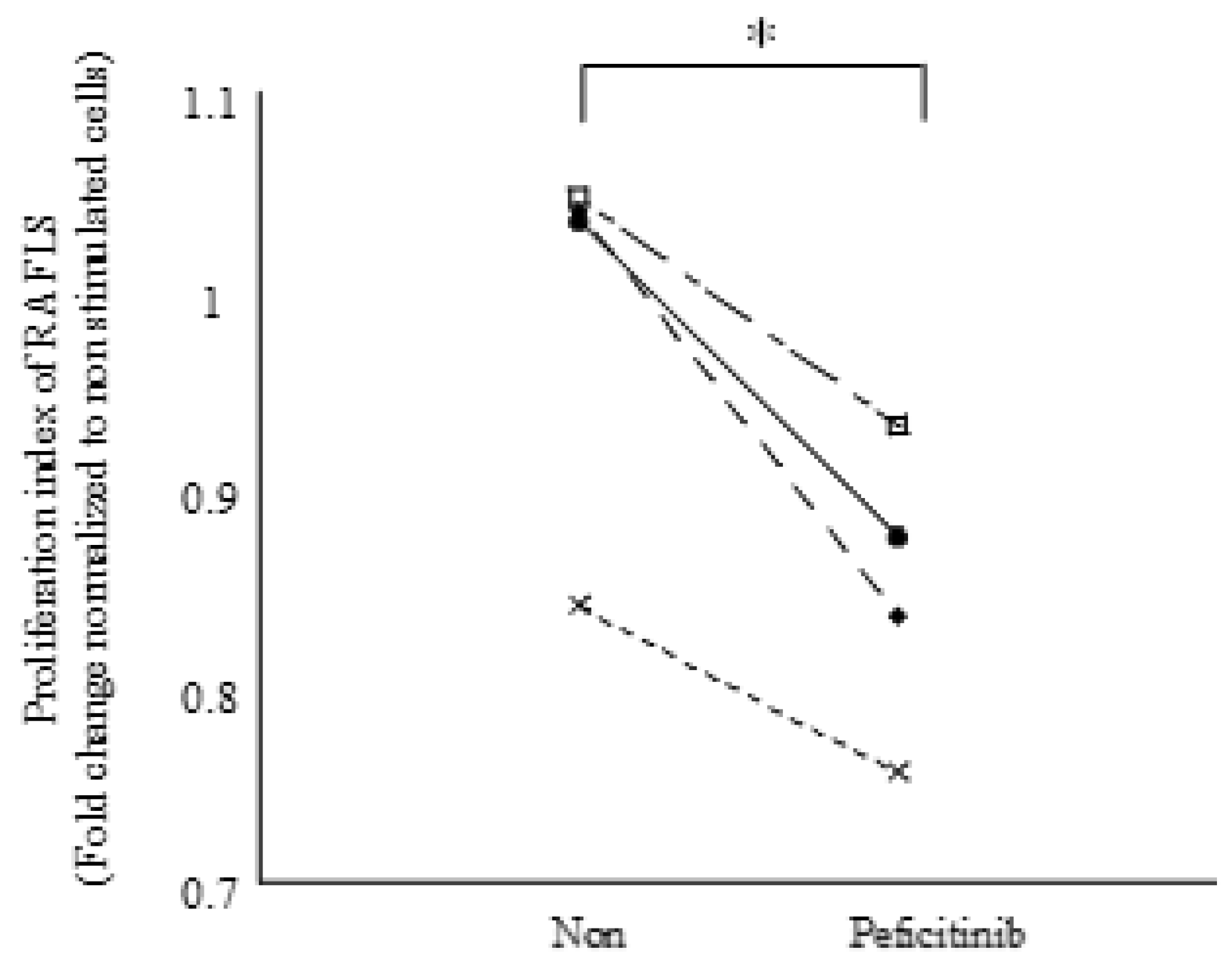
3.5. Peficitinib Inhibited the Proliferation of RA FLSs
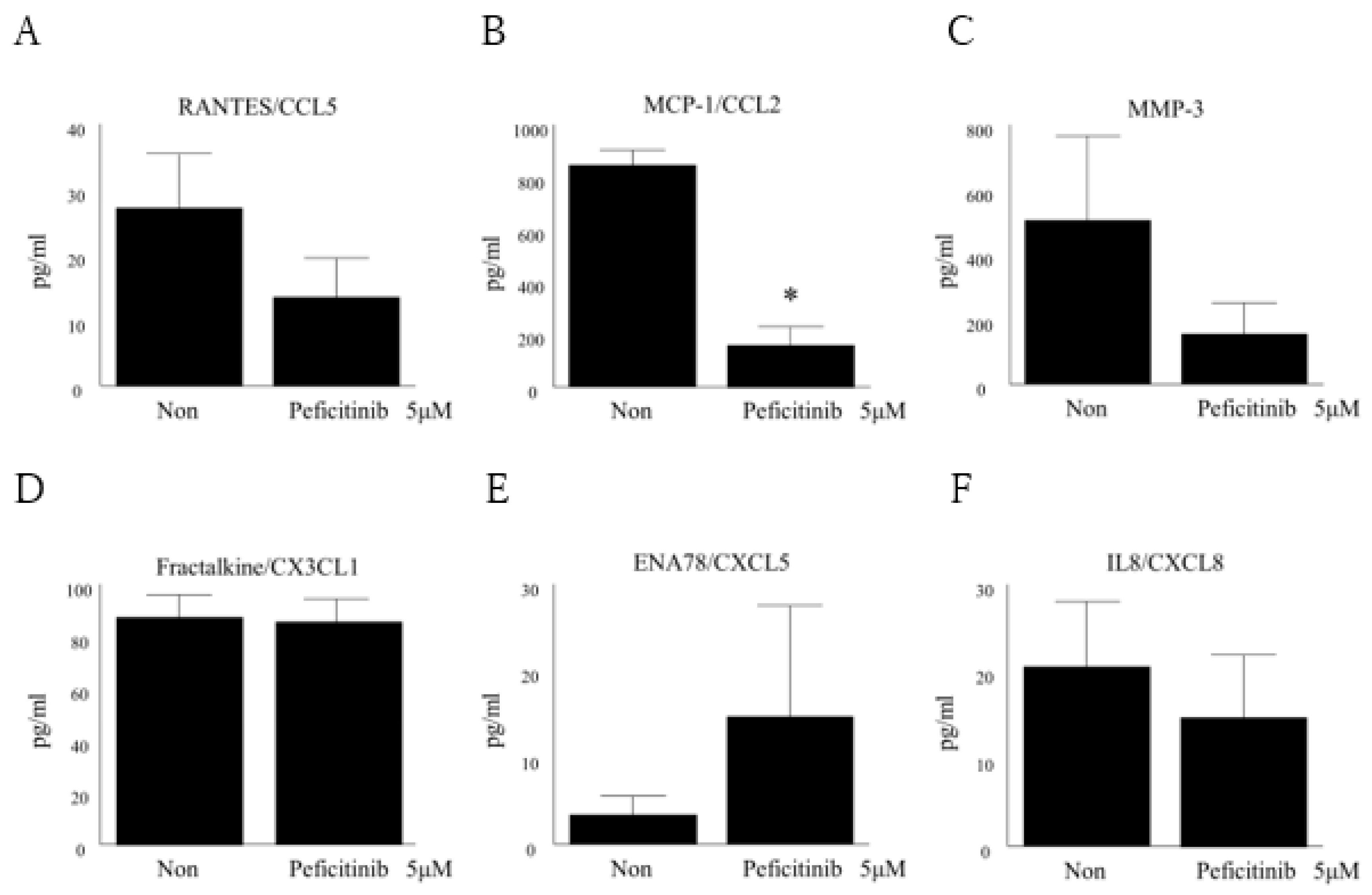
3.6. Peficitinib Suppressed the Secretion of Inflammatory Mediators in RA FLS
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Mcinnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, M.F.; Garcia-carbonell, R.; Whisenant, K.D.; Guma, M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis. Res. Ther. 2017, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.M.; Garvey, J.F.; Ng, C.T.; Veale, D.J.; Fearon, U.; Cummins, E.P.; Taylor, C.T. Hypoxia activates NF-kappaB-dependent gene expression through the canonical signaling pathway. Antioxid. Redox Signal. 2009, 11, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamazaki, S.; Yamagami, K.; Kuno, M.; Morita, Y.; Okuma, K.; Nakamura, K.; Chida, N.; Inami, M.; Inoue, T.; et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J. Pharmacol. Sci. 2017, 133, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Orr, C.; Fearon, U. Cellular and molecular perspectives in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Zwerina, J.; Firestein, G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 909–916. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tanaka, Y.; Iwasaki, M.; Ishikura, H.; Saeki, S.; Kaneko, Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: A 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann. Rheum. Dis. 2016, 75, 1057–1064. [Google Scholar] [CrossRef]
- Genovese, M.C.; Greenwald, M.; Codding, C.; Kivitz, A.J.; Wang, A.; Shay, K.; Wang, X.; Garg, J.P.; Cardiel, M.H.; Zubrzycka-Sienkiewicz, A.; et al. Peficitinib, a JAK Inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2017, 69, 932–942. [Google Scholar] [CrossRef]
- Kivitz, A.J.; Gutierrez-Ureña, S.R.; Poiley, J.; Genovese, M.C.; Kristy, R.; Shay, K.; Wang, X.; Garg, J.P.; Zubrzycka-Sienkiewicz, A. Peficitinib, a JAK Inhibitor, in the treatment of moderate-to-severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol. 2017, 69, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.G.; Ahern, M.J.; Coleman, M.; Weedon, H.; Papangelis, V.; Beroukas, D.; Roberts-Thomson, P.J.; Smith, M.D. Expression of Jak3, STAT1, STAT4, and STAT6 in inflammatory arthritis: Unique Jak3 and STAT4 expression in dendritic cells in seropositive rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kasperkovitz, P.V.; Verbeet, N.L.; Smeets, T.J.; van Rietschoten, J.G.; Kraan, M.C.; van der Pouw Kraan, T.C.; Tak, P.P.; Verweij, C.L. Activation of the STAT1 pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2004, 63, 233–239. [Google Scholar] [CrossRef]
- Walker, J.G.; Ahern, M.J.; Coleman, M.; Weedon, H.; Papangelis, V.; Beroukas, D.; Roberts-Thomson, P.J.; Smith, M.D. Changes in synovial tissue Jak-STAT expression in rheumatoid arthritis in response to successful DMARD treatment. Ann. Rheum. Dis. 2006, 65, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, S.; Corr, M.; Firestein, G.S.; Boyle, D.L. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression infibroblast-like synoviocytes: Autocrine role of type I interferon. Ann. Rheum. Dis. 2012, 71, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Karonitsch, T.; Beckmann, D.; Dalwigk, K.; Niederreiter, B.; Studenic, P.; Byrne, R.A.; Holinka, J.; Sevelda, F.; Korb-Pap, A.; Steiner, G.; et al. Targeted inhibition of Janus kinases abates interfon gamma-induced invasive behaviour of fibroblast-like synoviocytes. Rheumatology 2018, 57, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Mcgarry, T.; Orr, C.; Mccormick, J.; Veale, D.J.; Fearon, U. Tofacitinib regulates synovial inflammation in psoriatic arthritis, inhibiting STAT activation and induction of negative feedback inhibitors. Ann. Rheum. Dis. 2016, 75, 311–315. [Google Scholar] [CrossRef]
- Nakayamada, S.; Kubo, S.; Iwata, S.; Tanaka, Y. Recent progress in JAK inhibitors for the treatment of rheumatoid arthritis. BioDrugs 2016, 30, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.D.; Flanagan, M.E.; Telliez, J.B. Discovery and development of janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023–5038. [Google Scholar] [CrossRef]
- Shi, J.G.; Chen, X.; Lee, F.; Emm, T.; Scherle, P.A.; Lo, Y.; Punwani, N.; Williams, W.V.; Yeleswaram, S. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Kremer, J.M.; E Kartman, C.; E Schlichting, D.; Xie, L.; Carmack, T.; Pantojas, C.; Burson, J.S.; Tony, H.-P.; Macias, W.L.; et al. Response to baricitinib based on prior biologic use in patients with refractory rheumatoid arthritis. Rheumatology 2018, 57, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Kunkel, S.L.; A Harlow, L.; Johnson, B.; Evanoff, H.L.; Haines, G.K.; Burdick, M.D.; Pope, R.M.; Strieter, R.M. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J. Clin. Investig. 1992, 90, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Stuhlmuller, B.; Keyszer, G.; Kinne, R.W. Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 1997, 40, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, S.; Akahoshi, T.; Wada, C.; Kondo, H. Expression of the chemokine superfamily in rheumatoid arthritis. Clin. Exp. Immunol. 1994, 97, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Nanki, T.; Nagasaka, K.; Hayashida, K.; Saita, Y.; Miyasaka, N. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J. Immunol. 2001, 167, 5381–5385. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Nanki, T.; Girschick, H.; Yavuz, S.; Ochi, T.; Lipsky, P.E. Synovial stromal cells from rheumatoid arthritis patients attract monocytes by producing MCP-1 and IL-8. Arthritis Res. 2001, 3, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, A.; Caterino, A.L.; Elefante, E.; Valentini, V.; Vitale, A.; Talarico, R.; Cantarini, L.; Frediani, B. One year in review 2016: Novelties in the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 357–372. [Google Scholar]
- Bellucci, E.; Terenzi, R.; La Paglia, G.M.; Gentileschi, S.; Tripoli, A.; Tani, C.; Alunno, A. One year in review 2016: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 793–801. [Google Scholar]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Hu, D.; Zhang, L.; Tang, P. Role of extracellular vesicles in rheumatoid arthritis. Mol. Immunol. 2018, 93, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Qian, J. Downregulated MEG3 participates in rheumatoid arthritis via promoting proliferation of fibroblast-like synoviocytes. Exp. Ther. Med. 2019, 17, 1637–1642. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikari, Y.; Isozaki, T.; Tsubokura, Y.; Kasama, T. Peficitinib Inhibits the Chemotactic Activity of Monocytes via Proinflammatory Cytokine Production in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Cells 2019, 8, 561. https://doi.org/10.3390/cells8060561
Ikari Y, Isozaki T, Tsubokura Y, Kasama T. Peficitinib Inhibits the Chemotactic Activity of Monocytes via Proinflammatory Cytokine Production in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Cells. 2019; 8(6):561. https://doi.org/10.3390/cells8060561
Chicago/Turabian StyleIkari, Yuzo, Takeo Isozaki, Yumi Tsubokura, and Tsuyoshi Kasama. 2019. "Peficitinib Inhibits the Chemotactic Activity of Monocytes via Proinflammatory Cytokine Production in Rheumatoid Arthritis Fibroblast-Like Synoviocytes" Cells 8, no. 6: 561. https://doi.org/10.3390/cells8060561
APA StyleIkari, Y., Isozaki, T., Tsubokura, Y., & Kasama, T. (2019). Peficitinib Inhibits the Chemotactic Activity of Monocytes via Proinflammatory Cytokine Production in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Cells, 8(6), 561. https://doi.org/10.3390/cells8060561