Amyloid Precursor Protein (APP) and GABAergic Neurotransmission


{kind=link}
Abstract
1. Introduction
2. Amyloid Precursor Protein (APP) and Gamma-Aminobutyric Acid (GABA)ergic Neurotransmission
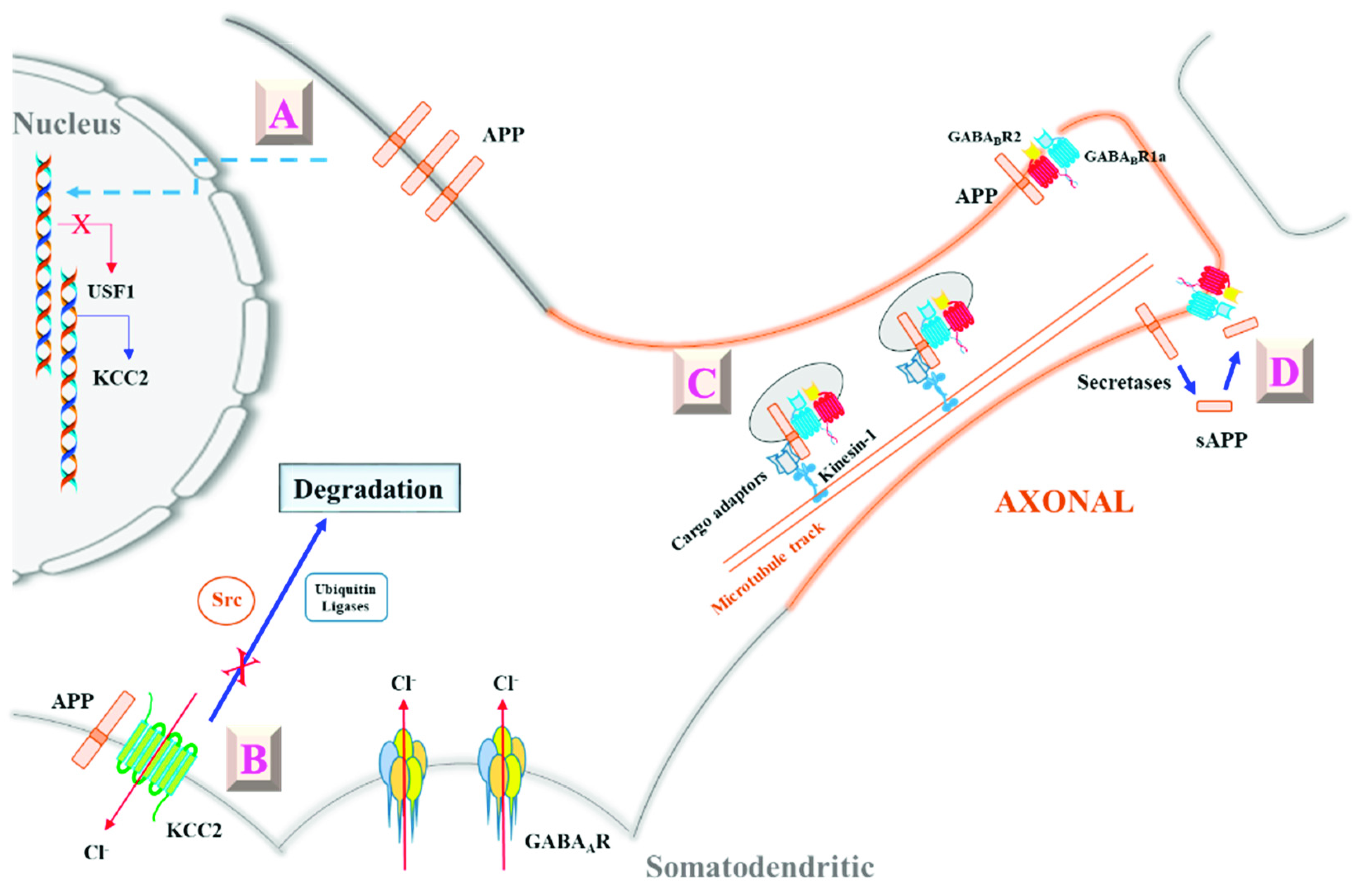
3. APP’s Modulation of GABAergic Neurotransmission through Potassium Chloride Cotransporter 2 (KCC2)
4. APP’s Modulation of Presynaptic GABAB Receptor (GABABR) Activity
5. New Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Alzheimer’s Association. Alzheimer’s Association 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.J.; Kellett, K.A.B.; Thinakaran, G.; Hooper, N.M. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.B.; Moore, S.; Daria, A.; et al. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs. 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Dibello, V.; Greco, A.; Daniele, A.; Seripa, D.; Logroscino, G.; Imbimbo, B.P. Are antibodies directed against amyloid-β (Aβ) oligomers the last call for the Aβ hypothesis of Alzheimer’s disease? Immunotherapy 2019, 11, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.; Dolgin, E. Failed Alzheimer’s trial does not kill leading theory of disease. Nature 2016, 540, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Van der Kant, R.; Goldstein, L.S.B. Cellular functions of the amyloid precursor protein from development to dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Lopez Sanchez, M.I.G.; van Wijngaarden, P.; Trounce, I.A. Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer’s disease. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef] [PubMed]
- Shariati, S.A.M.; De Strooper, B. Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 2013, 587, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jiang, M.; Trumbauer, M.E.; Sirinathsinghji, D.J.; Hopkins, R.; Smith, D.W.; Heavens, R.P.; Dawson, G.R.; Boyce, S.; Conner, M.W.; et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995, 81, 525–531. [Google Scholar] [CrossRef]
- Herms, J.; Anliker, B.; Heber, S.; Ring, S.; Fuhrmann, M.; Kretzschmar, H.; Sisodia, S.; Müller, U. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004, 23, 4106–4115. [Google Scholar] [CrossRef] [PubMed]
- Heber, S.; Herms, J.; Gajic, V.; Hainfellner, J.; Aguzzi, A.; Rülicke, T.; von Kretzschmar, H.; von Koch, C.; Sisodia, S.; Tremml, P.; et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 2000, 20, 7951–7963. [Google Scholar] [CrossRef] [PubMed]
- Hoareau, C.; Borrell, V.; Soriano, E.; Krebs, M.O.; Prochiantz, A.; Allinquant, B. Amyloid precursor protein cytoplasmic domain antagonizes reelin neurite outgrowth inhibition of hippocampal neurons. Neurobiol. Aging 2008, 29, 542–553. [Google Scholar] [CrossRef]
- Rama, N.; Goldschneider, D.; Corset, V.; Lambert, J.; Pays, L.; Mehlen, P. Amyloid precursor protein regulates netrin-1-mediated commissural axon outgrowth. J. Biol. Chem. 2012, 287, 30014–30023. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, H.; Gao, T.; Xue, X.; Wang, C.; Miao, F. Synergic interaction between amyloid precursor protein and neural cell adhesion molecule promotes neurite outgrowth. Oncotarget 2016, 7, 14199–14206. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, H.; Mutlu, S.A.; Bowser, D.A.; Moore, M.J.; Wang, M.C.; Zheng, H. The Amyloid Precursor Protein is a conserved receptor for Slit to mediate axon guidance. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Soba, P.; Eggert, S.; Wagner, K.; Zentgraf, H.; Siehl, K.; Kreger, S.; Löwer, A.; Langer, A.; Merdes, G.; Paro, R.; et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005, 24, 3624–3634. [Google Scholar] [CrossRef] [PubMed]
- Sosa, L.J.; Cáceres, A.; Dupraz, S.; Oksdath, M.; Quiroga, S.; Lorenzo, A. The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 2017, 143, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.; Vink, R.; Blumbergs, P.C.; Masters, C.L.; Cappai, R.; van den Heuvel, C. sAPPα rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J. Neurochem. 2012, 122, 208–220. [Google Scholar] [CrossRef]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef]
- Milosch, N.; Tanriöver, G.; Kundu, A.; Rami, A.; François, J.C.; Baumkötter, F.; Weyer, S.W.; Samanta, A.; Jäschke, A.; Brod, F.; et al. Holo-APP and G-protein-mediated signaling are required for sAPPα-induced activation of the Akt survival pathway. Cell Death Dis. 2014, 5, e1391. [Google Scholar] [CrossRef]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muñiz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its derivatives in the biology and cell fate specification of neural stem cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef]
- Coronel, R.; Lachgar, M.; Bernabeu-Zornoza, A.; Palmer, C.; Domínguez-Alvaro, M.; Revilla, A.; Ocaña, I.; Fernández, A.; Martínez-Serrano, A.; Cano, E.; et al. Neuronal and glial differentiation of human neural stem cells is regulated by Amyloid Precursor Protein (APP) levels. Mol. Neurobiol. 2019, 56, 1248–1261. [Google Scholar] [CrossRef]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, I.; Capetillo-Zarate, E.; Faideau, M.; Willén, K.; Esteras, N.; Frykman, S.; Tjernberg, L.O.; Gouras, G.K. APP depletion alters selective pre- and post-synaptic proteins. Mol. Cell Neurosci. 2019, 95, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Mehr, A.; Ludewig, S.; Stephan, J.; Zimmermann, M.; August, A.; Strecker, P.; Korte, M.; Koo, E.H.; Müller, U.C.; et al. APLP1 Is a synaptic cell adhesion molecule, supporting maintenance of dendritic spines and basal synaptic transmission. J. Neurosci. 2017, 37, 5345–5365. [Google Scholar] [CrossRef] [PubMed]
- Stahl, R.; Schilling, S.; Soba, P.; Rupp, C.; Hartmann, T.; Wagner, K.; Merdes, G.; Eggert, S.; Kins, S. Shedding of APP limits its synaptogenic activity and cell adhesion properties. Front. Cell Neurosci. 2014, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Laßek, M.; Weingarten, J.; Einsfelder, U.; Brendel, P.; Müller, U.; Volknandt, W. Amyloid precursor proteins are constituents of the presynaptic active zone. J. Neurochem. 2013, 127, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fanutza, T.; Del Prete, D.; Ford, M.J.; Castillo, P.E.; D’Adamio, L. APP and APLP2 interact with the synaptic release machinery and facilitate transmitter release at hippocampal synapses. eLife 2015, 4, e09743. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, B.; Yang, L.; Guo, Q.; Aithmitti, N.; Songyang, Z.; Zheng, H. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 2009, 29, 10788–10801. [Google Scholar] [CrossRef] [PubMed]
- Tyan, S.H.; Shih, A.Y.J.; Walsh, J.J.; Maruyama, H.; Sarsoza, F.; Ku, L.; Eggert, S.; Hof, P.R.; Koo, E.H.; Dickstein, D.L. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell Neurosci. 2012, 51, 43–52. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, W.; Yan, Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Cousins, S.L.; Hoey, S.E.A.; Anne Stephenson, F.; Perkinton, M.S. Amyloid precursor protein 695 associates with assembled NR2A- and NR2B-containing NMDA receptors to result in the enhancement of their cell surface delivery. J. Neurochem. 2009, 111, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Cousins, S.L.; Dai, W.; Stephenson, F.A. APLP1 and APLP2, members of the APP family of proteins, behave similarly to APP in that they associate with NMDA receptors and enhance NMDA receptor surface expression. J. Neurochem. 2015, 133, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Mockett, B.G.; Guévremont, D.; Elder, M.K.; Parfitt, K.D.; Peppercorn, K.; Morrissey, J.; Singh, A.; Hintz, T.J.; Kochen, L.; Tom Dieck, S.; et al. Glutamate receptor trafficking and protein synthesis mediate the facilitation of ltp by secreted amyloid precursor protein-alpha. J. Neurosci. 2019, 39, 3188–3203. [Google Scholar] [CrossRef] [PubMed]
- Hick, M.; Herrmann, U.; Weyer, S.W.; Mallm, J.P.; Tschäpe, J.A.; Borgers, M.; Mercken, M.; Roth, F.C.; Draguhn, A.; Slomianka, L.; et al. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 2015, 129, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Ireland, D.R.; Ballagh, I.; Bourne, K.; Marechal, N.M.; Turner, P.R.; Bilkey, D.K.; Tate, W.P.; Abraham, W.C. Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol. Dis. 2008, 31, 250–260. [Google Scholar] [CrossRef]
- Marik, S.A.; Olsen, O.; Tessier-Lavigne, M.; Gilbert, C.D. Physiological role for amyloid precursor protein in adult experience-dependent plasticity. Proc. Natl. Acad. Sci. USA 2016, 113, 7912–7917. [Google Scholar] [CrossRef] [PubMed]
- Korte, M.; Herrmann, U.; Zhang, X.; Draguhn, A. The role of APP and APLP for synaptic transmission, plasticity, and network function: Lessons from genetic mouse models. Exp. Brain Res. 2012, 217, 435–440. [Google Scholar] [CrossRef]
- Preat, T.; Goguel, V. Role of Drosophila Amyloid Precursor Protein in memory formation. Front. Mol Neurosci. 2016, 9, 142. [Google Scholar] [CrossRef]
- Xiong, M.; Jones, O.D.; Peppercorn, K.; Ohline, S.M.; Tate, W.P.; Abraham, W.C. Secreted amyloid precursor protein-alpha can restore novel object location memory and hippocampal LTP in aged rats. Neurobiol. Learn Mem. 2017, 138, 291–299. [Google Scholar] [CrossRef]
- Furukawa, K.; Barger, S.W.; Blalock, E.M.; Mattson, M.P. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature 1996, 379, 74–78. [Google Scholar] [CrossRef]
- Ishida, A.; Furukawa, K.; Keller, J.N.; Mattson, M.P. Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. Neuroreport 1997, 8, 2133–2137. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.T.Y.; Mockett, B.G.; Ohline, S.M.; Parfitt, K.D.; Wicky, H.E.; Peppercorn, K.; Schoderboeck, L.; Yahaya, M.F.B.; Tate, W.P.; Hughes, S.M.; et al. Lentivirus-mediated expression of human secreted amyloid precursor protein-alpha prevents development of memory and plasticity deficits in a mouse model of Alzheimer’s disease. Mol. Brain 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, J.S.; Scanziani, M. How inhibition shapes cortical activity. Neuron 2011, 72, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.C.; Macdonald, R.L. A structural look at GABA A receptor mutations linked to epilepsy syndromes. Brain Res. 2019. [Google Scholar] [CrossRef]
- Chua, H.C.; Chebib, M. GABAA receptors and the diversity in their structure and pharmacology. Adv. Pharmacol. 2017, 79, 1–34. [Google Scholar] [PubMed]
- Terunuma, M. Diversity of structure and function of GABAB receptors: A Complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef]
- Tang, B.L. K+-Cl− co-transporter 2 (KCC2)—A membrane trafficking perspective. Mol. Membr. Biol. 2016, 33, 100–110. [Google Scholar] [CrossRef]
- Owens, D.F.; Boyce, L.H.; Davis, M.B.; Kriegstein, A.R. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 1996, 16, 6414–6423. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Boulenguez, P.; Liabeuf, S.; Bos, R.; Bras, H.; Jean-Xavier, C.; Brocard, C.; Stil, A.; Darbon, P.; Cattaert, D.; Delpire, E.; et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 2010, 16, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.N.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, J.; Jiang, J.; Zheng, X.; Justice, N.J.; Wang, K.; Ran, X.; Li, Y.; Huo, Q.; Zhang, J.; et al. APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Doshina, A.; Gourgue, F.; Onizuka, M.; Opsomer, R.; Wang, P.; Ando, K.; Tasiaux, B.; Dewachter, I.; Kienlen-Campard, P.; Brion, J.P.; et al. Cortical cells reveal APP as a new player in the regulation of GABAergic neurotransmission. Sci. Rep. 2017, 7, 370. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.C.; Raveh, A.; Schneider, A.; Fritzius, T.; Früh, S.; Rem, P.D.; Stawarski, M.; Lalanne, T.; Turecek, R.; Choo, M.; et al. Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat Commun. 2019, 10, 1331. [Google Scholar] [CrossRef]
- Rice, H.C.; de Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science. 2019, 363. [Google Scholar] [CrossRef]
- Dawson, G.R.; Seabrook, G.R.; Zheng, H.; Smith, D.W.; Graham, S.; O’Dowd, G.; Bowery, B.J.; Boyce, S.; Trumbauer, M.E.; Chen, H.Y.; et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 1999, 90, 1–13. [Google Scholar] [CrossRef]
- Seabrook, G.R.; Smith, D.W.; Bowery, B.J.; Easter, A.; Reynolds, T.; Fitzjohn, S.M.; Morton, R.A.; Zheng, H.; Dawson, G.R.; Sirinathsinghji, D.J.; et al. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 1999, 38, 349–359. [Google Scholar] [CrossRef]
- Senechal, Y.; Kelly, P.H.; Dev, K.K. Amyloid precursor protein knockout mice show age-dependent deficits in passive avoidance learning. Behav. Brain Res. 2008, 186, 126–132. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, W.; Brankačk, J.; Weyer, S.W.; Müller, U.C.; Tort, A.B.L.; Draguhn, A. Impaired theta-gamma coupling in APP-deficient mice. Sci. Rep. 2016, 6, 21948. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Z.; Sun, L.; Yang, L.; Li, H.; Cole, A.L.; Rodriguez-Rivera, J.; Lu, H.C.; Zheng, H. The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J. Neurosci. 2014, 34, 13314–13325. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Goh, E.L.K.; Sailor, K.A.; Kitabatake, Y.; Ming, G.L.; Song, H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 2006, 439, 589–593. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z.; Wang, B.; Justice, N.J.; Zheng, H. Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J. Neurosci. 2009, 29, 15660–15668. [Google Scholar] [CrossRef] [PubMed]
- Born, H.A.; Kim, J.Y.; Savjani, R.R.; Das, P.; Dabaghian, Y.A.; Guo, Q.; Yoo, J.W.; Schuler, D.R.; Cirrito, J.R.; Zheng, H.; et al. Genetic suppression of transgenic APP rescues hypersynchronous network activity in a mouse model of Alzheimer’s disease. J. Neurosci. 2014, 34, 3826–3840. [Google Scholar] [CrossRef] [PubMed]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Fenoglio, K.A.; Simeone, T.A.; Coons, S.W.; Wu, J.; Chang, Y.; Kerrigan, J.F.; Rho, J.M. GABAA receptor-mediated activation of L-type calcium channels induces neuronal excitation in surgically resected human hypothalamic hamartomas. Epilepsia 2008, 49, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol. 2008, 85, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Markkanen, M.; Uvarov, P.; Airaksinen, M.S. Role of upstream stimulating factors in the transcriptional regulation of the neuron-specific K-Cl cotransporter KCC2. Brain Res. 2008, 1236, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.; Bergeron, M.J.; Lavertu, G.; Castonguay, A.; Tripathy, S.; Bonin, R.P.; Perez-Sanchez, J.; Boudreau, D.; Wang, B.; Dumas, L.; et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 2013, 19, 1524–1528. [Google Scholar] [CrossRef]
- Wake, H.; Watanabe, M.; Moorhouse, A.J.; Kanematsu, T.; Horibe, S.; Matsukawa, N.; Asai, K.; Ojika, K.; Hirata, M.; Nabekura, J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 2007, 27, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.C.; Jurd, R.; Moss, S.J. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell Neurosci. 2010, 45, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.C.; Deeb, T.Z.; Walker, J.A.; Davies, P.A.; Moss, S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011, 14, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, M. Presynaptic metabotropic glutamate and GABAB receptors. Handb. Exp. Pharmacol. 2008, 184, 373–407. [Google Scholar]
- Sakaba, T.; Neher, E. Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature 2003, 424, 775–778. [Google Scholar] [CrossRef]
- Chalifoux, J.R.; Carter, A.G. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron 2010, 66, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Chalifoux, J.R.; Carter, A.G. GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 2011, 21, 339–344. [Google Scholar] [CrossRef]
- Vigot, R.; Barbieri, S.; Bräuner-Osborne, H.; Turecek, R.; Shigemoto, R.; Zhang, Y.P.; Luján, R.; Jacobson, L.H.; Biermann, B.; Fritschy, J.M.; et al. Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron 2006, 50, 589–601. [Google Scholar] [CrossRef]
- Biermann, B.; Ivankova-Susankova, K.; Bradaia, A.; Abdel Aziz, S.; Besseyrias, V.; Kapfhammer, J.P.; Missler, M.; Gassmann, M.; Bettler, B. The Sushi domains of GABAB receptors function as axonal targeting signals. J. Neurosci. 2010, 30, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Hannan, S.; Wilkins, M.E.; Smart, T.G. Sushi domains confer distinct trafficking profiles on GABAB receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 12171–12176. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Pérez-Garci, E.; Schneider, A.; Kollewe, A.; Gauthier-Kemper, A.; Fritzius, T.; Raveh, A.; Dinamarca, M.C.; Hanuschkin, A.; Bildl, W.; et al. Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat. Neurosci. 2016, 19, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Muresan, Z.; Muresan, V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J. Cell Biol. 2005, 171, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Vagnoni, A.; Perkinton, M.S.; Gray, E.H.; Francis, P.T.; Noble, W.; Miller, C.C.J. Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum. Mol. Genet. 2012, 21, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Coburger, I.; Dahms, S.O.; Roeser, D.; Gührs, K.H.; Hortschansky, P.; Than, M.E. Analysis of the overall structure of the multi-domain amyloid precursor protein (APP). PLoS ONE 2013, 8, e81926. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.W. Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 2004, 84, 1051–1095. [Google Scholar] [CrossRef] [PubMed]
- Vertkin, I.; Styr, B.; Slomowitz, E.; Ofir, N.; Shapira, I.; Berner, D.; Fedorova, T.; Laviv, T.; Barak-Broner, N.; Greitzer-Antes, D.; et al. GABAB receptor deficiency causes failure of neuronal homeostasis in hippocampal networks. Proc. Natl. Acad. Sci. USA 2015, 112, E3291–E3299. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, R.M.; Farber, S.A.; Growdon, J.H.; Wurtman, R.J. Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc. Natl. Acad. Sci. USA 1993, 90, 5191–5193. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Kröhnert, K.; Schäfer, C.; Rammner, B.; Koo, S.J.; Claßen, G.A.; Krauss, M.; Haucke, V.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028. [Google Scholar] [CrossRef]
- Ring, S.; Weyer, S.W.; Kilian, S.B.; Waldron, E.; Pietrzik, C.U.; Filippov, M.A.; Herms, J.; Buchholz, C.; Eckman, C.B.; Korte, M.; et al. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 2007, 27, 7817–7826. [Google Scholar] [CrossRef]
- Heaney, C.F.; Kinney, J.W. Role of GABA(B) receptors in learning and memory and neurological disorders. Neurosci. Biobehav. Rev. 2016, 63, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Malcangio, M. GABA receptors and pain. Neuropharmacology 2018, 136, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Benke, D. GABAB receptor trafficking and interacting proteins: Targets for the development of highly specific therapeutic strategies to treat neurological disorders? Biochem. Pharmacol. 2013, 86, 1525–1530. [Google Scholar] [CrossRef]
- Sola Vigo, F.; Kedikian, G.; Heredia, L.; Heredia, F.; Añel, A.D.; Rosa, A.L.; Lorenzo, A. Amyloid-beta precursor protein mediates neuronal toxicity of amyloid beta through Go protein activation. Neurobiol Aging 2009, 30, 1379–1392. [Google Scholar] [CrossRef]
- Kedikian, G.; Heredia, F.; Salvador, V.R.; Raimunda, D.; Isoardi, N.; Heredia, L.; Lorenzo, A. Secreted amyloid precursor protein and holo-APP bind amyloid beta through distinct domains eliciting different toxic responses on hippocampal neurons. J. Neurosci. Res. 2010, 88, 1795–1803. [Google Scholar] [PubMed]
- Wang, Z.; Jackson, R.J.; Hong, W.; Taylor, W.M.; Corbett, G.T.; Moreno, A.; Liu, W.; Li, S.; Frosch, M.P.; Slutsky, I.; et al. Human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J. Neurosci. 2017, 37, 11947–11966. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Miyata, H.; Kametani, F.; Nonaka, T.; Akiyama, H.; Hisanaga, S.I.; Hasegawa, M. Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015, 129, 895–907. [Google Scholar] [CrossRef]
- Puzzo, D.; Piacentini, R.; Fá, M.; Gulisano, W.; Li Puma, D.D.; Staniszewski, A.; Zhang, H.; Tropea, M.R.; Cocco, S.; Palmeri, A.; et al. LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Helm, K.A.; Haberman, R.P.; Dean, S.L.; Hoyt, E.C.; Melcher, T.; Lund, P.K.; Gallagher, M. GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus. Neuropharmacology 2005, 48, 956–964. [Google Scholar] [CrossRef]
- Froestl, W.; Gallagher, M.; Jenkins, H.; Madrid, A.; Melcher, T.; Teichman, S.; Mondadori, C.G.; Pearlman, R. SGS742: The first GABA(B) receptor antagonist in clinical trials. Biochem. Pharmacol. 2004, 68, 1479–1487. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Alzheimer’s disease: Channeling APP to non-amyloidogenic processing. Biochem. Biophys. Res. Commun. 2005, 331, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.L. Amyloid Precursor Protein (APP) and GABAergic Neurotransmission. Cells 2019, 8, 550. https://doi.org/10.3390/cells8060550
Tang BL. Amyloid Precursor Protein (APP) and GABAergic Neurotransmission. Cells. 2019; 8(6):550. https://doi.org/10.3390/cells8060550
Chicago/Turabian StyleTang, Bor Luen. 2019. "Amyloid Precursor Protein (APP) and GABAergic Neurotransmission" Cells 8, no. 6: 550. https://doi.org/10.3390/cells8060550
APA StyleTang, B. L. (2019). Amyloid Precursor Protein (APP) and GABAergic Neurotransmission. Cells, 8(6), 550. https://doi.org/10.3390/cells8060550