Molecular Profiling of Inflammatory Bowel Disease: Is It Ready for Use in Clinical Decision-Making?



Abstract
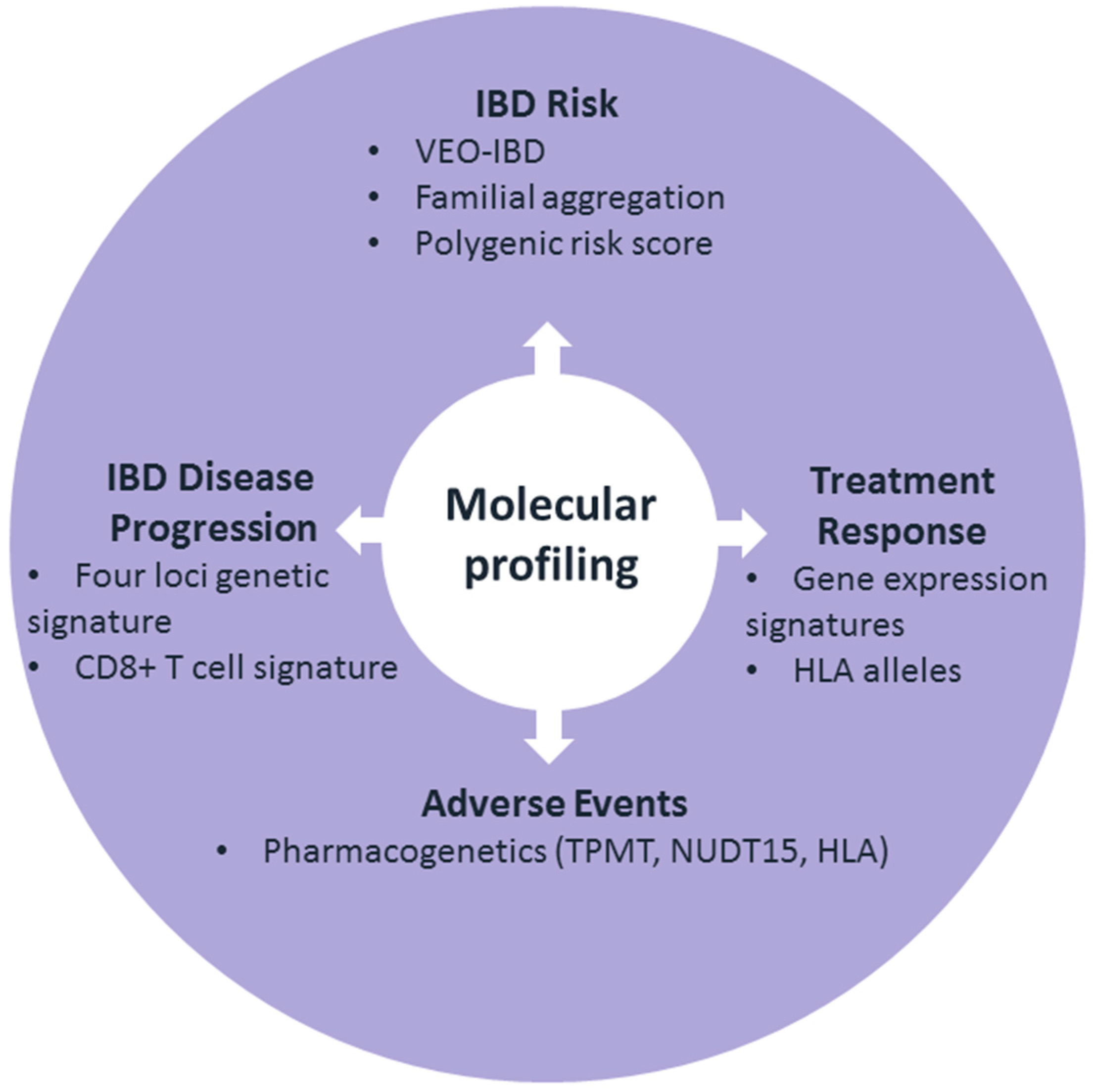
1. Introduction
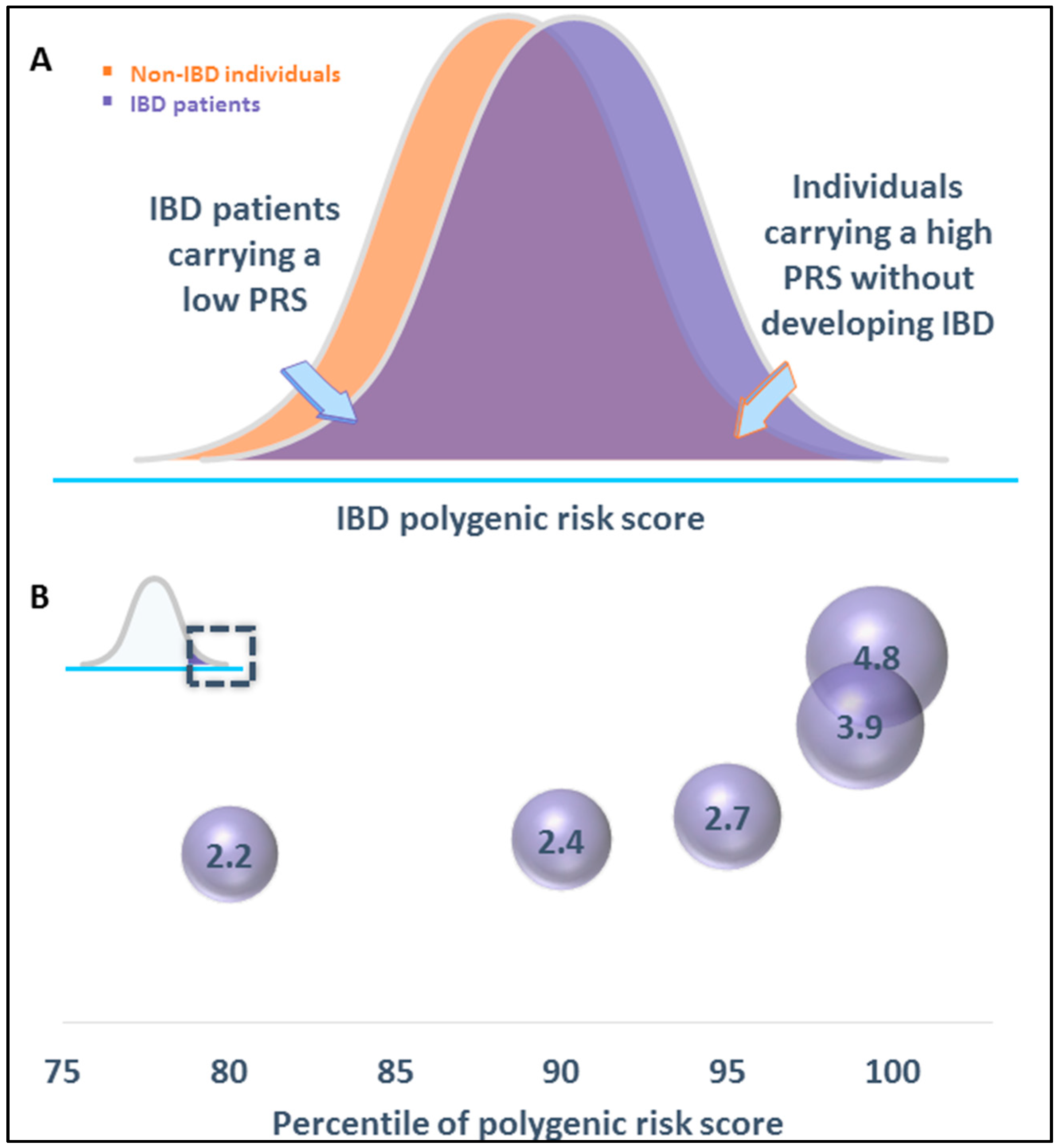
2. The Predictability of IBD Risk
2.1. Case 1: 34-Year-Old Female with a 10-Year History of Ileal CD
2.2. Case 2: Two-Year-Old Child Presents with Very Early Onset (VEO) IBD
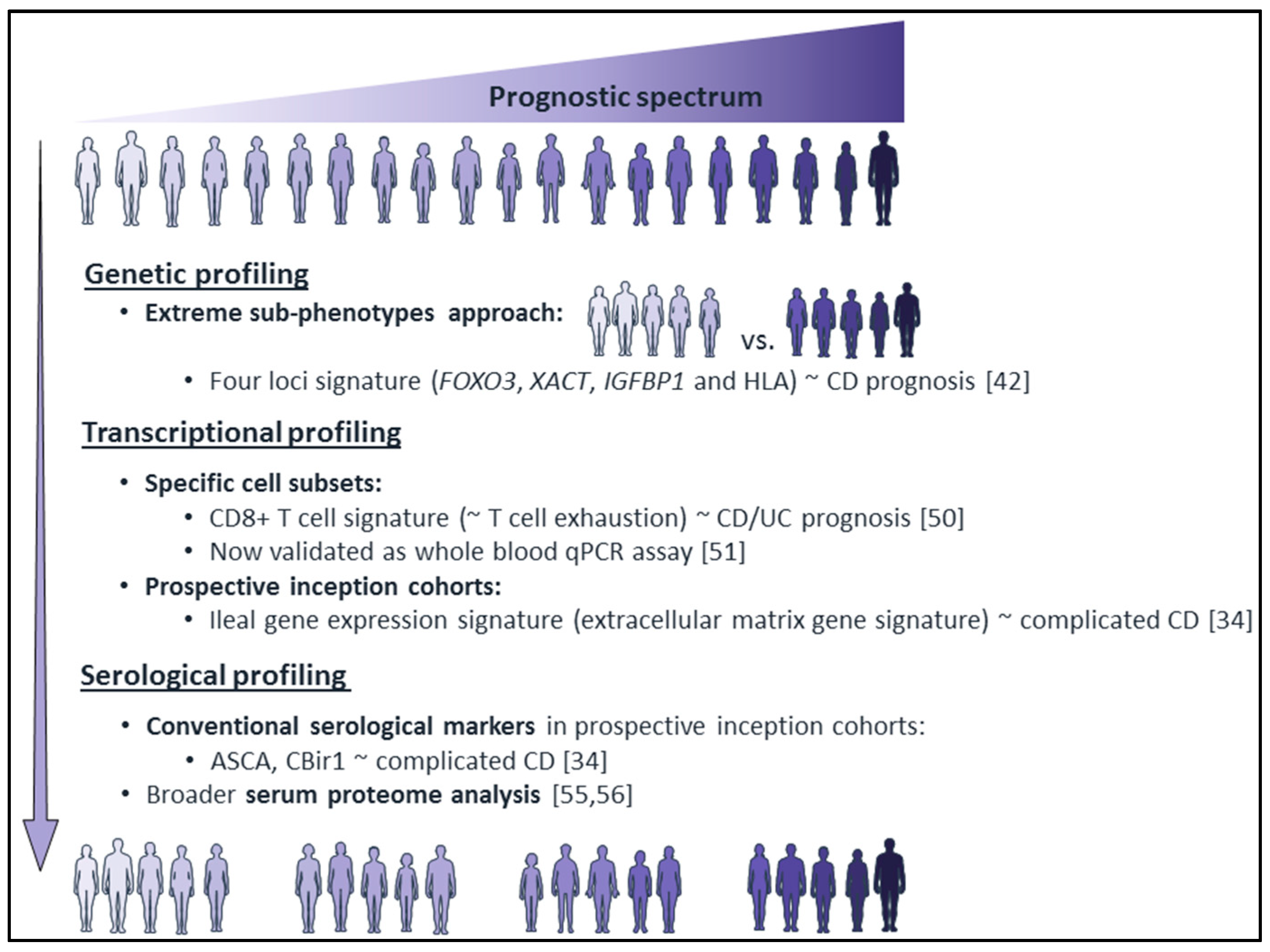
3. The Predictability of IBD Disease Progression
3.1. Case 3: 24-Year-Old Female with a One-Year History of Ileal CD
3.2. Genetic Profiling
3.3. Transcriptional Profiling
3.4. Serological Profiling
4. The Predictability of Treatment Response
4.1. Case 4: 25-Year-Old Male with a Two-Year History of Ileocolonic CD
4.2. Genetic Profiling
4.3. Transcriptional Profiling
5. Predictability of Adverse Events
5.1. Case 5: 28-Year-Old Female with a Three-Year History of UC
5.2. Thiopurines
5.3. 5-Aminosalicylate (5-ASA)
5.4. Anti-TNF Therapy
6. Conclusions
Funding
Conflicts of Interest
References
- Silverberg, M.S.; Satsangi, J.; Ahmad, T.; Arnott, I.D.; Bernstein, C.N.; Brant, S.R.; Caprilli, R.; Colombel, J.F.; Gasche, C.; Geboes, K.; et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005, 19, 5a–36a. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Spekhorst, L.M.; Visschedijk, M.C.; Alberts, R.; Festen, E.A.; van der Wouden, E.J.; Dijkstra, G.; Weersma, R.K. Performance of the Montreal classification for inflammatory bowel diseases. World J. Gastroenterol. 2014, 20, 15374–15381. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Mirkov, M.U.; Verstockt, B.; Cleynen, I. Genetics of inflammatory bowel disease: Beyond NOD2. Lancet Gastroenterol. Hepatol. 2017, 2, 224–234. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef]
- Ng, S.C.; Tang, W.; Ching, J.Y.; Wong, M.; Chow, C.M.; Hui, A.J.; Wong, T.C.; Leung, V.K.; Tsang, S.W.; Yu, H.H.; et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013, 145, 158–165.e152. [Google Scholar] [CrossRef]
- M’Koma, A.E. Inflammatory bowel disease: An expanding global health problem. Clin. Med. Insights Gastroenterol. 2013, 6, 33–47. [Google Scholar] [CrossRef]
- Xu, F.; Dahlhamer, J.M.; Zammitti, E.P.; Wheaton, A.G.; Croft, J.B. Health-Risk Behaviors and Chronic Conditions Among Adults with Inflammatory Bowel Disease - United States, 2015 and 2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 190–195. [Google Scholar] [CrossRef]
- Orholm, M.; Munkholm, P.; Langholz, E.; Nielsen, O.H.; Sorensen, T.I.; Binder, V. Familial occurrence of inflammatory bowel disease. N. Engl. J. Med. 1991, 324, 84–88. [Google Scholar] [CrossRef]
- Halme, L.; Paavola-Sakki, P.; Turunen, U.; Lappalainen, M.; Farkkila, M.; Kontula, K. Family and twin studies in inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 3668–3672. [Google Scholar] [CrossRef]
- Park, J.B.; Yang, S.K.; Byeon, J.S.; Park, E.R.; Moon, G.; Myung, S.J.; Park, W.K.; Yoon, S.G.; Kim, H.S.; Lee, J.G.; et al. Familial occurrence of inflammatory bowel disease in Korea. Inflamm. Bowel. Dis. 2006, 12, 1146–1151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moller, F.T.; Andersen, V.; Wohlfahrt, J.; Jess, T. Familial risk of inflammatory bowel disease: A population-based cohort study 1977-2011. Am. J. Gastroenterol. 2015, 110, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.A.; Rubin, P.H.; Present, D.H. Frequency of inflammatory bowel disease in offspring of couples both presenting with inflammatory bowel disease. Gastroenterology 1991, 100, 1638–1643. [Google Scholar] [CrossRef]
- Laharie, D.; Debeugny, S.; Peeters, M.; Van Gossum, A.; Gower-Rousseau, C.; Belaiche, J.; Fiasse, R.; Dupas, J.L.; Lerebours, E.; Piotte, S.; et al. Inflammatory bowel disease in spouses and their offspring. Gastroenterology 2001, 120, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, M.; Jahnsen, J.; Lygren, I.; Vatn, M.H.; Moum, B. Are there any differences in phenotype or disease course between familial and sporadic cases of inflammatory bowel disease? Results of a population-based follow-up study. Am. J. Gastroenterol. 2007, 102, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Van Steen, K.; Branche, J.; Sendid, B.; Rutgeerts, P.; Vasseur, F.; Poulain, D.; Broly, F.; Colombel, J.F.; Vermeire, S.; et al. Familial aggregation and antimicrobial response dose-dependently affect the risk for Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Mahid, S.S.; Minor, K.S.; Soto, R.E.; Hornung, C.A.; Galandiuk, S. Smoking and inflammatory bowel disease: A meta-analysis. Mayo Clin. Proc. 2006, 81, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Weersma, R.K.; Stokkers, P.C.; Cleynen, I.; Wolfkamp, S.C.; Henckaerts, L.; Schreiber, S.; Dijkstra, G.; Franke, A.; Nolte, I.M.; Rutgeerts, P.; et al. Confirmation of multiple Crohn’s disease susceptibility loci in a large Dutch-Belgian cohort. Am. J. Gastroenterol. 2009, 104, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.B.; Lee, S.H.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M.; Gearry, R.B.; Lawrance, I.C.; Andrews, J.M.; Bampton, P.; Mahy, G.; et al. Performance of risk prediction for inflammatory bowel disease based on genotyping platform and genomic risk score method. BMC Med. Genet. 2017, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Kevans, D.; Silverberg, M.S.; Borowski, K.; Griffiths, A.; Xu, W.; Onay, V.; Paterson, A.D.; Knight, J.; Croitoru, K. IBD Genetic Risk Profile in Healthy First-Degree Relatives of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Borren, N.Z.; Conway, G.; Garber, J.J.; Khalili, H.; Budree, S.; Mallick, H.; Yajnik, V.; Xavier, R.J.; Ananthakrishnan, A.N. Differences in Clinical Course, Genetics, and the Microbiome Between Familial and Sporadic Inflammatory Bowel Diseases. J. Crohns Colitis 2018, 12, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014, 147, 990–1007.e1003. [Google Scholar] [CrossRef]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Schwerd, T. From Genes to Mechanisms: The Expanding Spectrum of Monogenic Disorders Associated with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 202–212. [Google Scholar] [CrossRef]
- Shouval, D.S.; Biswas, A.; Kang, Y.H.; Griffith, A.E.; Konnikova, L.; Mascanfroni, I.D.; Redhu, N.S.; Frei, S.M.; Field, M.; Doty, A.L.; et al. Interleukin 1beta Mediates Intestinal Inflammation in Mice and Patients With Interleukin 10 Receptor Deficiency. Gastroenterology 2016, 151, 1100–1104. [Google Scholar] [CrossRef]
- Nijman, I.J.; van Montfrans, J.M.; Hoogstraat, M.; Boes, M.L.; van de Corput, L.; Renner, E.D.; van Zon, P.; van Lieshout, S.; Elferink, M.G.; van der Burg, M.; et al. Targeted next-generation sequencing: A novel diagnostic tool for primary immunodeficiencies. J. Allergy Clin. Immunol. 2014, 133, 529–534. [Google Scholar] [CrossRef]
- de Koning, T.J.; Jongbloed, J.D.; Sikkema-Raddatz, B.; Sinke, R.J. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: The opportunities and challenges. Expert Rev. Mol. Diagn. 2015, 15, 61–70. [Google Scholar] [CrossRef]
- Fazeli, W.; Karakaya, M.; Herkenrath, P.; Vierzig, A.; Dotsch, J.; von Kleist-Retzow, J.C.; Cirak, S. Mendeliome sequencing enables differential diagnosis and treatment of neonatal lactic acidosis. Mol. Cell. Pediatr. 2016, 3, 22. [Google Scholar] [CrossRef]
- Beaugerie, L.; Sokol, H. Clinical, serological and genetic predictors of inflammatory bowel disease course. World J. Gastroenterol. 2012, 18, 3806–3813. [Google Scholar] [CrossRef]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef]
- Abreu, M.T.; Taylor, K.D.; Lin, Y.C.; Hang, T.; Gaiennie, J.; Landers, C.J.; Vasiliauskas, E.A.; Kam, L.Y.; Rojany, M.; Papadakis, K.A.; et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 2002, 123, 679–688. [Google Scholar] [CrossRef]
- Ahmad, T.; Armuzzi, A.; Bunce, M.; Mulcahy-Hawes, K.; Marshall, S.E.; Orchard, T.R.; Crawshaw, J.; Large, O.; de Silva, A.; Cook, J.T.; et al. The molecular classification of the clinical manifestations of Crohn’s disease. Gastroenterology 2002, 122, 854–866. [Google Scholar] [CrossRef]
- Seiderer, J.; Schnitzler, F.; Brand, S.; Staudinger, T.; Pfennig, S.; Herrmann, K.; Hofbauer, K.; Dambacher, J.; Tillack, C.; Sackmann, M.; et al. Homozygosity for the CARD15 frameshift mutation 1007fs is predictive of early onset of Crohn’s disease with ileal stenosis, entero-enteral fistulas, and frequent need for surgical intervention with high risk of re-stenosis. Scand. J. Gastroenterol. 2006, 41, 1421–1432. [Google Scholar] [CrossRef]
- Alonso, A.; Domènech, E.; Julià, A.; Panés, J.; García-Sánchez, V.; Mateu, P.N.; Gutiérrez, A.; Gomollón, F.; Mendoza, J.L.; Garcia-Planella, E.; et al. Identification of Risk Loci for Crohn’s Disease Phenotypes Using a Genome-Wide Association Study. Gastroenterology 2015, 148, 794–805. [Google Scholar] [CrossRef]
- Cleynen, I.; Gonzalez, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlik, M.; Crusius, B.J.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: Results from the IBDchip European Project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef]
- Henckaerts, L.; Van Steen, K.; Verstreken, I.; Cleynen, I.; Franke, A.; Schreiber, S.; Rutgeerts, P.; Vermeire, S. Genetic risk profiling and prediction of disease course in Crohn’s disease patients. Clin. Gastroenterol. Hepatol. 2009, 7, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Biasci, D.; Roberts, R.; Gearry, R.B.; Mansfield, J.C.; Ahmad, T.; Prescott, N.J.; Satsangi, J.; Wilson, D.C.; Jostins, L.; et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat. Genet. 2017, 49, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Visschedijk, M.C.; Spekhorst, L.M.; Cheng, S.C.; van Loo, E.S.; Jansen, B.D.; Blokzijl, T.; Kil, H.; De Jong, D.J.; Pierik, M.; Maljaars, J.P.; et al. Genomic and Expression Analyses Identify a Disease-Modifying Variant for Fibrostenotic Crohn’s Disease. J. Crohns Colitis 2018, 12, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Boucher, G.; Waterman, M.; Rivers, C.R.; Patel, M.; Cho, J.H.; Colombel, J.F.; Duerr, R.H.; Binion, D.; McGovern, D.P.; et al. Genetic Predictors of Benign Course of Ulcerative Colitis-A North American Inflammatory Bowel Disease Genetics Consortium Study. Inflamm. Bowel Dis. 2016, 22, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Yang, S.K.; Hong, M.; Jung, S.; Kim, B.M.; Moon, J.W.; Park, S.H.; Ye, B.D.; Oh, S.H.; Kim, K.M.; et al. An intergenic variant rs9268877 between HLA-DRA and HLA-DRB contributes to the clinical course and long-term outcome of ulcerative colitis. J. Crohns Colitis 2018, 12, 1113–1121. [Google Scholar] [CrossRef]
- Jakobsen, C.; Cleynen, I.; Andersen, P.S.; Vermeire, S.; Munkholm, P.; Paerregaard, A.; Wewer, V. Genetic susceptibility and genotype–phenotype association in 588 Danish children with inflammatory bowel disease. J. Crohns Colitis 2014, 8, 678–685. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Huang, H.; Nguyen, D.D.; Sauk, J.; Yajnik, V.; Xavier, R.J. Differential effect of genetic burden on disease phenotypes in Crohn’s disease and ulcerative colitis: Analysis of a North American cohort. Am. J. Gastroenterol. 2014, 109, 395–400. [Google Scholar] [CrossRef]
- Aziz, M.A.; Yousef, Z.; Saleh, A.M.; Mohammad, S.; Al Knawy, B. Towards personalized medicine of colorectal cancer. Crit. Rev. Oncol. Hematol. 2017, 118, 70–78. [Google Scholar] [CrossRef]
- Reck, M.; Rabe, K.F. Precision Diagnosis and Treatment for Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 849–861. [Google Scholar] [CrossRef]
- Lee, J.C.; Lyons, P.A.; McKinney, E.F.; Sowerby, J.M.; Carr, E.J.; Bredin, F.; Rickman, H.M.; Ratlamwala, H.; Hatton, A.; Rayner, T.F.; et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J. Clin. Investig. 2011, 121, 4170–4179. [Google Scholar] [CrossRef]
- Biasci, D.; Lee, J.C.; Noor, N.M.; Pombal, D.R.; Hou, M.; Lewis, N.; Ahmad, T.; Hart, A.; Parkes, M.; McKinney, E.F.; et al. A blood-based prognostic biomarker in IBD. Gut 2019. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.; Noor, N.M.; Dowling, F.; Leung, H.; Bond, S.; Whitehead, L.; Upponi, S.; Kinnon, P.; Sandham, A.P.; Lyons, P.A.; et al. PRedicting Outcomes For Crohn’s dIsease using a moLecular biomarkEr (PROFILE): Protocol for a multicentre, randomised, biomarker-stratified trial. BMJ Open 2018, 8, e026767. [Google Scholar] [CrossRef]
- Marigorta, U.M.; Denson, L.A.; Hyams, J.S.; Mondal, K.; Prince, J.; Walters, T.D.; Griffiths, A.; Noe, J.D.; Crandall, W.V.; Rosh, J.R.; et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat. Genet. 2017, 49, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Kennedy, N.A.; Hjelm, F.N.; Modig, E.; Sundell, M.; Söderholm, J.; Andreassen, B.K.; Bergemalm, D.; Ventham, N.T.; Hjortswang, H.; et al. DOP082 Proximity extension assay immunoassay technology identifies novel serum biomarkers that can diagnose and classify inflammatory bowel diseases: IBD Character Consortium. J. Crohns Colitis 2016, 10, S82. [Google Scholar]
- Kalla, R.; Adams, A.; Vatn, S.; Bergemalm, D.; Ricanek, P.; Lindstrøm, J.; Ocklind, A.; Nordberg, N.; Kennedy, N.; Ventham, N.; et al. OP022 Proximity extension assay based proteins show immune cell specificity and can diagnose and predict outcomes in inflammatory bowel diseases: IBD Character study. J. Crohns Colitis 2017, 11, S13. [Google Scholar] [CrossRef]
- Gonczi, L.; Vegh, Z.; Golovics, P.A.; Rutka, M.; Gecse, K.B.; Bor, R.; Farkas, K.; Szamosi, T.; Bene, L.; Gasztonyi, B.; et al. Prediction of Short- and Medium-term Efficacy of Biosimilar Infliximab Therapy. Do Trough Levels and Antidrug Antibody Levels or Clinical And Biochemical Markers Play the More Important Role? J. Crohns Colitis 2017, 11, 697–705. [Google Scholar] [CrossRef][Green Version]
- Reinisch, W.; Colombel, J.F.; Sandborn, W.J.; Mantzaris, G.J.; Kornbluth, A.; Adedokun, O.J.; Miller, M.; Tang, K.L.; Rutgeerts, P.; Cornillie, F. Factors associated with short- and long-term outcomes of therapy for Crohn’s disease. Clin. Gastroenterol. Hepatol. 2015, 13, 539–547.e2. [Google Scholar] [CrossRef] [PubMed]
- Billiet, T.; Papamichael, K.; de Bruyn, M.; Verstockt, B.; Cleynen, I.; Princen, F.; Singh, S.; Ferrante, M.; Van Assche, G.; Vermeire, S. A Matrix-based Model Predicts Primary Response to Infliximab in Crohn’s Disease. J. Crohns Colitis 2015, 9, 1120–1126. [Google Scholar] [CrossRef]
- Arnott, I.D.; Landers, C.J.; Nimmo, E.J.; Drummond, H.E.; Smith, B.K.; Targan, S.R.; Satsangi, J. Sero-reactivity to microbial components in Crohn’s disease is associated with disease severity and progression, but not NOD2/CARD15 genotype. Am. J. Gastroenterol. 2004, 99, 2376–2384. [Google Scholar] [CrossRef]
- Esters, N.; Pierik, M.; van Steen, K.; Vermeire, S.; Claessens, G.; Joossens, S.; Vlietinck, R.; Rutgeerts, P. Transmission of CARD15 (NOD2) variants within families of patients with inflammatory bowel disease. Am. J. Gastroenterol. 2004, 99, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.D.; Plevy, S.E.; Yang, H.; Landers, C.J.; Barry, M.J.; Rotter, J.I.; Targan, S.R. ANCA pattern and LTA haplotype relationship to clinical responses to anti-TNF antibody treatment in Crohn’s disease. Gastroenterology 2001, 120, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Mascheretti, S.; Hampe, J.; Kuhbacher, T.; Herfarth, H.; Krawczak, M.; Folsch, U.R.; Schreiber, S. Pharmacogenetic investigation of the TNF/TNF-receptor system in patients with chronic active Crohn’s disease treated with infliximab. Pharmacogenom. J. 2002, 2, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Pierik, M.; Vermeire, S.; Steen, K.V.; Joossens, S.; Claessens, G.; Vlietinck, R.; Rutgeerts, P. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment. Pharmacol. Ther. 2004, 20, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Billiet, T.; Dreesen, E.; Cleynen, I.; Wollants, W.J.; Ferrante, M.; Van Assche, G.; Gils, A.; Vermeire, S. A Genetic Variation in the Neonatal Fc-Receptor Affects Anti-TNF Drug Concentrations in Inflammatory Bowel Disease. Am. J. Gastroenterol. 2016, 111, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.J.; Watier, H.E.; Schreiber, S.; Hampe, J.; Taillard, F.; Olson, A.; Thorne, N.; Zhang, H.; Colombel, J.F. Polymorphism in IgG Fc receptor gene FCGR3A and response to infliximab in Crohn’s disease: A subanalysis of the ACCENT I study. Pharm. Genom. 2006, 16, 911–914. [Google Scholar] [CrossRef]
- Louis, E.; El Ghoul, Z.; Vermeire, S.; Dall’Ozzo, S.; Rutgeerts, P.; Paintaud, G.; Belaiche, J.; De Vos, M.; Van Gossum, A.; Colombel, J.F.; et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment Pharmacol. Ther. 2004, 19, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Louis, E.; Rutgeerts, P.; De Vos, M.; Van Gossum, A.; Belaiche, J.; Pescatore, P.; Fiasse, R.; Pelckmans, P.; Vlietinck, R.; et al. NOD2/CARD15 does not influence response to infliximab in Crohn’s disease. Gastroenterology 2002, 123, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Hlavaty, T.; Pierik, M.; Henckaerts, L.; Ferrante, M.; Joossens, S.; van Schuerbeek, N.; Noman, M.; Rutgeerts, P.; Vermeire, S. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment Pharmacol. Ther. 2005, 22, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Bek, S.; Nielsen, J.V.; Bojesen, A.B.; Franke, A.; Bank, S.; Vogel, U.; Andersen, V. Systematic review: Genetic biomarkers associated with anti-TNF treatment response in inflammatory bowel diseases. Aliment Pharmacol. Ther. 2016, 44, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, M.C.; Mei, L.; Friedman, M.; Dhere, T.; Haritunians, T.; Hakonarson, H.; Kim, C.; Glessner, J.; Targan, S.R.; McGovern, D.P.; et al. Genome wide association (GWA) predictors of anti-TNFα therapeutic responsiveness in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.E.; Yajnik, V.; Khalili, H.; Giallourakis, C.; Garber, J.; Xavier, R.; Ananthakrishnan, A.N. Genetic Markers Predict Primary Non-Response and Durable Response To Anti-TNF Biologic Therapies in Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1816–1822. [Google Scholar] [CrossRef]
- Vande Casteele, N.; Gils, A. Pharmacokinetics of anti-TNF monoclonal antibodies in inflammatory bowel disease: Adding value to current practice. J. Clin. Pharmacol. 2015, 55, S39–50. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, E.; Gils, A.; Vermeire, S. Pharmacokinetic Modeling and Simulation of Biologicals in Inflammatory Bowel Disease: The Dawning of a New Era for Personalized Treatment. Curr. Drug Targets 2018, 19, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Cepok, S.; Wolf, C.; Berthele, A.; Uhr, M.; Bettecken, T.; Buck, D.; Hartung, H.P.; Holsboer, F.; Muller-Myhsok, B.; et al. Single-nucleotide polymorphisms in HLA- and non-HLA genes associated with the development of antibodies to interferon-beta therapy in multiple sclerosis patients. Pharmacogenomics J. 2012, 12, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Billiet, T.; Vande Casteele, N.; Van Stappen, T.; Princen, F.; Singh, S.; Gils, A.; Ferrante, M.; Van Assche, G.; Cleynen, I.; Vermeire, S. Immunogenicity to infliximab is associated with HLA-DRB1. Gut 2015, 64, 1344–1345. [Google Scholar] [CrossRef] [PubMed]
- Sazonovs, A.; Kennedy, N.A.; Bewshea, C.; Moutsianas, L.; Walker, G.J.; De Lange, K.; Goodhand, J.; Anderson, C.; Barrett, J.; Consortium, P.I.; et al. OP013 HLA-DQA1 contributes to the development of antibodies to anti-TNF therapy in Crohn’s disease. J. Crohns Colitis 2018, 12, S009–S010. [Google Scholar] [CrossRef]
- Arijs, I.; Li, K.; Toedter, G.; Quintens, R.; Van Lommel, L.; Van Steen, K.; Leemans, P.; De Hertogh, G.; Lemaire, K.; Ferrante, M.; et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009, 58, 1612–1619. [Google Scholar] [CrossRef]
- Arijs, I.; Quintens, R.; Van Lommel, L.; Van Steen, K.; De Hertogh, G.; Lemaire, K.; Schraenen, A.; Perrier, C.; Van Assche, G.; Vermeire, S.; et al. Predictive value of epithelial gene expression profiles for response to infliximab in Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Verstockt, S.; Creyns, B.; Tops, S.; Van Assche, G.; Gils, A.; Ceuppens, J.L.; Vermeire, S.; Ferrante, M.; Breynaert, C. Mucosal IL13RA2 expression predicts nonresponse to anti-TNF therapy in Crohn’s disease. Aliment Pharmacol. Ther. 2019, 49, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Perrier, C.; De Hertogh, G.; Cremer, J.; Creyns, B.; Van Assche, G.; Ferrante, M.; Ceuppens, J.L.; Vermeire, S.; Breynaert, C. Effects of Epithelial IL-13Ralpha2 Expression in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2983. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Kitani, A.; Fichtner-Feigl, S.; Fuss, I.J. The signaling function of the IL-13Ralpha2 receptor in the development of gastrointestinal fibrosis and cancer surveillance. Curr. Mol. Med. 2009, 9, 740–750. [Google Scholar] [CrossRef]
- Telesco, S.E.; Brodmerkel, C.; Zhang, H.; Kim, L.L.; Johanns, J.; Mazumder, A.; Li, K.; Baribaud, F.; Curran, M.; Strauss, R.; et al. Gene Expression Signature for Prediction of Golimumab Response in a Phase 2a Open-Label Trial of Patients With Ulcerative Colitis. Gastroenterology 2018, 155, 1008–1011.e8. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Billmeier, U.; Dieterich, W.; Rath, T.; Sonnewald, S.; Reid, S.; Hirschmann, S.; Hildner, K.; Waldner, M.J.; Mudter, J.; et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 2019, 68, 814–828. [Google Scholar] [CrossRef]
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFalpha non-response in biopsy and blood of patients with IBD. Gut 2019, 68, 604–614. [Google Scholar] [CrossRef]
- Verstockt, B.; Verstockt, S.; Blevi, H.; Cleynen, I.; de Bruyn, M.; Van Assche, G.; Vermeire, S.; Ferrante, M. TREM-1, the ideal predictive biomarker for endoscopic healing in anti-TNF-treated Crohn’s disease patients? Gut 2018. [Google Scholar] [CrossRef]
- Verstockt, B.; Verstockt, S.; Dehairs, J.; Ballet, V.; Blevi, H.; Wollants, W.J.; Breynaert, C.; Van Assche, G.; Vermeire, S.; Ferrante, M. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019, 40, 733–742. [Google Scholar] [CrossRef]
- Tew, G.W.; Hackney, J.A.; Gibbons, D.; Lamb, C.A.; Luca, D.; Egen, J.G.; Diehl, L.; Eastham Anderson, J.; Vermeire, S.; Mansfield, J.C.; et al. Association Between Response to Etrolizumab and Expression of Integrin αE and Granzyme A in Colon Biopsies of Patients With Ulcerative Colitis. Gastroenterology 2016, 150, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.T.; Mansfield, J.C.; Lamb, C.A.; Feagan, B.G.; Panes, J.; Salas, A.; et al. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet 2014, 384, 309–318. [Google Scholar] [CrossRef]
- Arijs, I.; De Hertogh, G.; Lemmens, B.; Van Lommel, L.; de Bruyn, M.; Vanhove, W.; Cleynen, I.; Machiels, K.; Ferrante, M.; Schuit, F.; et al. Effect of vedolizumab (anti-alpha4beta7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut 2018, 67, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Tantisira, K.G.; Lasky-Su, J.; Harada, M.; Murphy, A.; Litonjua, A.A.; Himes, B.E.; Lange, C.; Lazarus, R.; Sylvia, J.; Klanderman, B.; et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N. Engl. J. Med. 2011, 365, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Sladek, S.L. Mercaptopurine pharmacogenetics: Monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am. J. Hum. Genet. 1980, 32, 651–662. [Google Scholar] [PubMed]
- Yang, S.K.; Hong, M.; Baek, J.; Choi, H.; Zhao, W.; Jung, Y.; Haritunians, T.; Ye, B.D.; Kim, K.J.; Park, S.H.; et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat. Genet. 2014, 46, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Nishii, R.; Perez-Andreu, V.; Yang, W.; Klussmann, F.A.; Zhao, X.; Lin, T.N.; Hoshitsuki, K.; Nersting, J.; Kihira, K.; et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat. Genet. 2016, 48, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.F.; Ferrari, N.; Debuysere, H.; Marteau, P.; Gendre, J.P.; Bonaz, B.; Soule, J.C.; Modigliani, R.; Touze, Y.; Catala, P.; et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology 2000, 118, 1025–1030. [Google Scholar] [CrossRef]
- Cuffari, C.; Hunt, S.; Bayless, T. Utilisation of erythrocyte 6-thioguanine metabolite levels to optimise azathioprine therapy in patients with inflammatory bowel disease. Gut 2001, 48, 642–646. [Google Scholar] [CrossRef]
- Dubinsky, M.C.; Yang, H.; Hassard, P.V.; Seidman, E.G.; Kam, L.Y.; Abreu, M.T.; Targan, S.R.; Vasiliauskas, E.A. 6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel disease. Gastroenterology 2002, 122, 904–915. [Google Scholar] [CrossRef]
- Schwab, M.; Schaeffeler, E.; Marx, C.; Zanger, U.; Aulitzky, W.; Eichelbaum, M. Shortcoming in the diagnosis of TPMT deficiency in a patient with Crohn’s disease using phenotyping only. Gastroenterology 2001, 121, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Asada, A.; Nishida, A.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Kito, K.; Sugimoto, M.; Andoh, A. NUDT15 R139C-related thiopurine leukocytopenia is mediated by 6-thioguanine nucleotide-independent mechanism in Japanese patients with inflammatory bowel disease. J. Gastroenterol. 2016, 51, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, X.D.; Chao, K.; Zhi, M.; Zheng, H.; Ruan, H.L.; Xin, S.; Ding, N.; Hu, P.J.; Huang, M.; et al. NUDT15 polymorphisms are better than thiopurine S-methyltransferase as predictor of risk for thiopurine-induced leukopenia in Chinese patients with Crohn’s disease. Aliment Pharmacol. Ther. 2016, 44, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.J.; Harrison, J.W.; Heap, G.A.; Voskuil, M.D.; Andersen, V.; Anderson, C.A.; Ananthakrishnan, A.N.; Barrett, J.C.; Beaugerie, L.; Bewshea, C.M.; et al. Association of Genetic Variants in NUDT15 With Thiopurine-Induced Myelosuppression in Patients With Inflammatory Bowel Disease. JAMA 2019, 321, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Heap, G.A.; Weedon, M.N.; Bewshea, C.M.; Singh, A.; Chen, M.; Satchwell, J.B.; Vivian, J.P.; So, K.; Dubois, P.C.; Andrews, J.M.; et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat. Genet. 2014, 46, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Jansen, L.E.; Rose, R.V.; Gregor, J.C.; Ponich, T.; Chande, N.; Khanna, R.; Yan, B.; Jairath, V.; Khanna, N.; et al. HLA-DQA1-HLA-DRB1 polymorphism is a major predictor of azathioprine-induced pancreatitis in patients with inflammatory bowel disease. Aliment Pharmacol. Ther. 2018, 47, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Heap, G.A.; So, K.; Weedon, M.; Edney, N.; Bewshea, C.; Singh, A.; Annese, V.; Beckly, J.; Buurman, D.; Chaudhary, R.; et al. Clinical Features and HLA Association of 5-Aminosalicylate (5-ASA)-induced Nephrotoxicity in Inflammatory Bowel Disease. J. Crohns Colitis 2016, 10, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Van Moerkercke, W.; Billiet, T.; Vandecandelaere, P.; Vande Casteele, N.; Breynaert, C.; Ballet, V.; Ferrante, M.; Noman, M.; Assche, G.V.; et al. Characteristics of Skin Lesions Associated With Anti-Tumor Necrosis Factor Therapy in Patients With Inflammatory Bowel Disease: A Cohort Study. Ann. Intern. Med. 2016, 164, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Tillack, C.; Ehmann, L.M.; Friedrich, M.; Laubender, R.P.; Papay, P.; Vogelsang, H.; Stallhofer, J.; Beigel, F.; Bedynek, A.; Wetzke, M.; et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-γ-expressing Th1 cells and IL-17A/IL-22-expressing Th17 cells and respond to anti-IL-12/IL-23 antibody treatment. Gut 2014, 63, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Caudle, K.E.; Gong, L.; Whirl-Carrillo, M.; Stein, C.M.; Scott, S.A.; Lee, M.T.; Gage, B.F.; Kimmel, S.E.; Perera, M.A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Pharmacogenetics-Guided Warfarin Dosing: 2017 Update. Clin. Pharmacol. Ther. 2017, 102, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Davila-Fajardo, C.L.; Diaz-Villamarin, X.; Antunez-Rodriguez, A.; Fernandez-Gomez, A.E.; Garcia-Navas, P.; Martinez-Gonzalez, L.J.; Davila-Fajardo, J.A.; Barrera, J.C. Pharmacogenetics in the Treatment of Cardiovascular Diseases and Its Current Progress Regarding Implementation in the Clinical Routine. Genes 2019, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Weersma, R.K.; Xavier, R.J.; Consortium, I.B.D.M.O.; Vermeire, S.; Barrett, J.C. Multiomics Analyses to Deliver the Most Effective Treatment to Every Patient With Inflammatory Bowel Disease. Gastroenterology 2018, 155, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Boschetti, G.; Chang, C.; Ungaro, R.; Giri, M.; Chuang, L.-S.; Nayar, S.; Greenstein, A.; Dubinsky, M.; Walker, L.; et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti TNF therapy. bioRxiv 2018, 503102. [Google Scholar]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e317. [Google Scholar] [CrossRef] [PubMed]
- Uniken Venema, W.T.; Voskuil, M.D.; Vila, A.V.; van der Vries, G.; Jansen, B.H.; Jabri, B.; Faber, K.N.; Dijkstra, G.; Xavier, R.J.; Wijmenga, C.; et al. Single-Cell RNA Sequencing of Blood and Ileal T Cells From Patients With Crohn’s Disease Reveals Tissue-Specific Characteristics and Drug Targets. Gastroenterology 2019, 156, 812–815.e22. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Relative Risk | Absolute Risk | |
|---|---|---|
| Lifetime risk for IBD | 1× | 1.3% |
| Familial aggregation | ||
| IBD in first-degree relatives | 4–15× | 5.2–19.5% |
| Both parents affected | 20–25× | 30% |
| Genetic factors | ||
| NOD2 variant 1 | 2.1–3.0× | 2.7–3.9% |
| Typical susceptibility variants | 1.1–1.5× | 1.4–2.0% |
| PRS—Individuals in the top 1% | 3.9× | 5.1% |
| Environmental factor | ||
| Current smoking 1 | 1.8× | 2.3% |
| Category | Application | Usability 1 |
|---|---|---|
| Risk/diagnosis | VEO-IBD: application of (targeted) next-generation sequencing | ✔ |
| Familial IBD: risk of IBD is increased 4–15-fold in first-degree relatives of patients | ✔ | |
| Sporadic/familial IBD: individual risk variants | ✘ | |
| Sporadic/familial IBD: polygenic risk scores | ✔ | |
| Disease progression | Genetic testing: individual susceptibility variants | ✘ |
| Genetic testing: polygenic risk scores | ✘ | |
| Genetic testing: variants from extreme sub-phenotype approaches [42,43,45] | ✔ | |
| Transcriptional profiling: CD8+ T cell transcription signature [50,51] | ✔ | |
| Treatment response | Genetic testing: polygenic risk scores | ✘ |
| Genetic testing: Immunogenicity (HLA-DQA1*05) [79] | ✔ | |
| Transcriptional profiling: OSM, TREM1, gene signature including IL13RA2 [80,81,82,86,88,89,90] | ✔ | |
| Adverse events | Thiopurine-induced myelotoxicity: TPMT, NUDT15 genetic testing | ✔ |
| Thiopurine-induced pancreatitis: HLA genetic testing [105,106] | ✔ | |
| Skin lesions under anti-TNF therapy: IL23R rs11209026 genetic testing [109] | ✔ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-S.; Cleynen, I. Molecular Profiling of Inflammatory Bowel Disease: Is It Ready for Use in Clinical Decision-Making? Cells 2019, 8, 535. https://doi.org/10.3390/cells8060535
Lee H-S, Cleynen I. Molecular Profiling of Inflammatory Bowel Disease: Is It Ready for Use in Clinical Decision-Making? Cells. 2019; 8(6):535. https://doi.org/10.3390/cells8060535
Chicago/Turabian StyleLee, Ho-Su, and Isabelle Cleynen. 2019. "Molecular Profiling of Inflammatory Bowel Disease: Is It Ready for Use in Clinical Decision-Making?" Cells 8, no. 6: 535. https://doi.org/10.3390/cells8060535
APA StyleLee, H.-S., & Cleynen, I. (2019). Molecular Profiling of Inflammatory Bowel Disease: Is It Ready for Use in Clinical Decision-Making? Cells, 8(6), 535. https://doi.org/10.3390/cells8060535