Inhibition of Hedgehog Signaling in Fibroblasts, Pancreatic, and Lung Tumor Cells by Oxy186, an Oxysterol Analogue with Drug-Like Properties

Abstract
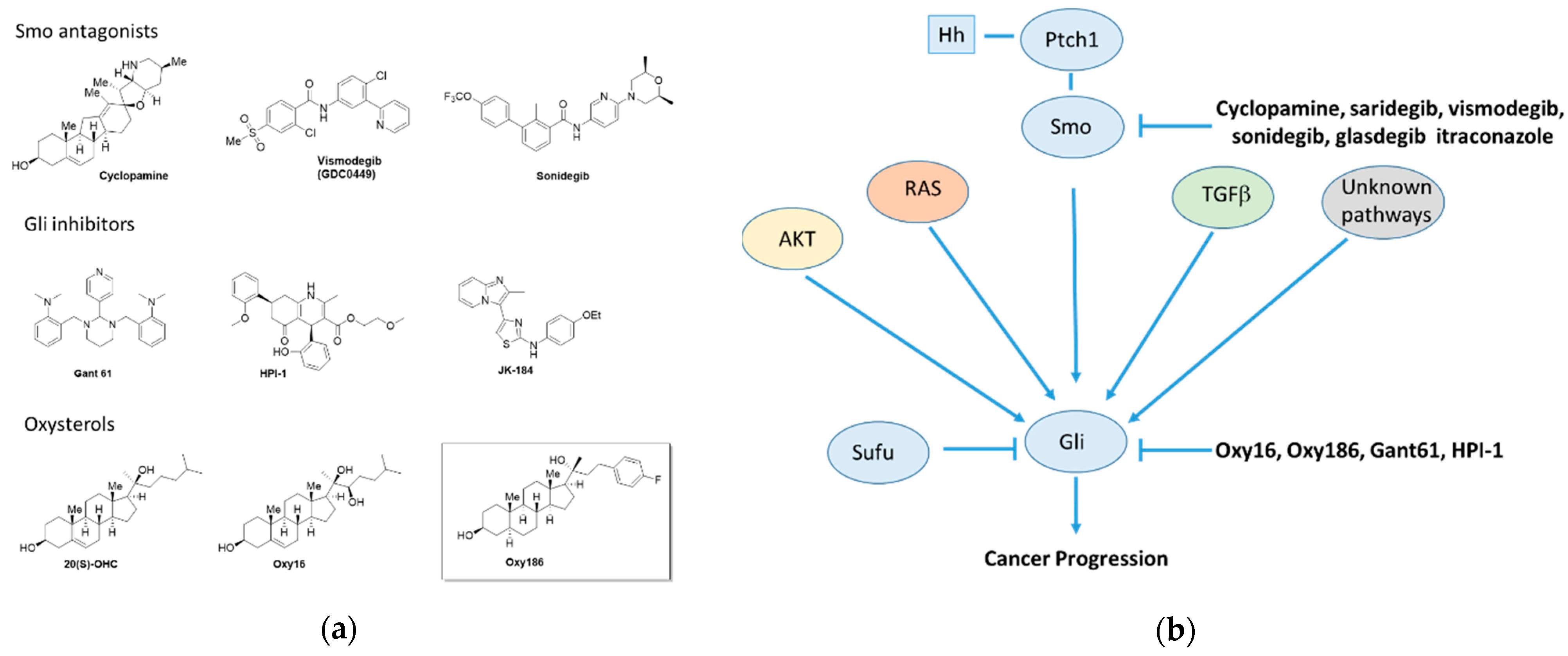
1. Introduction
2. Materials and Methods
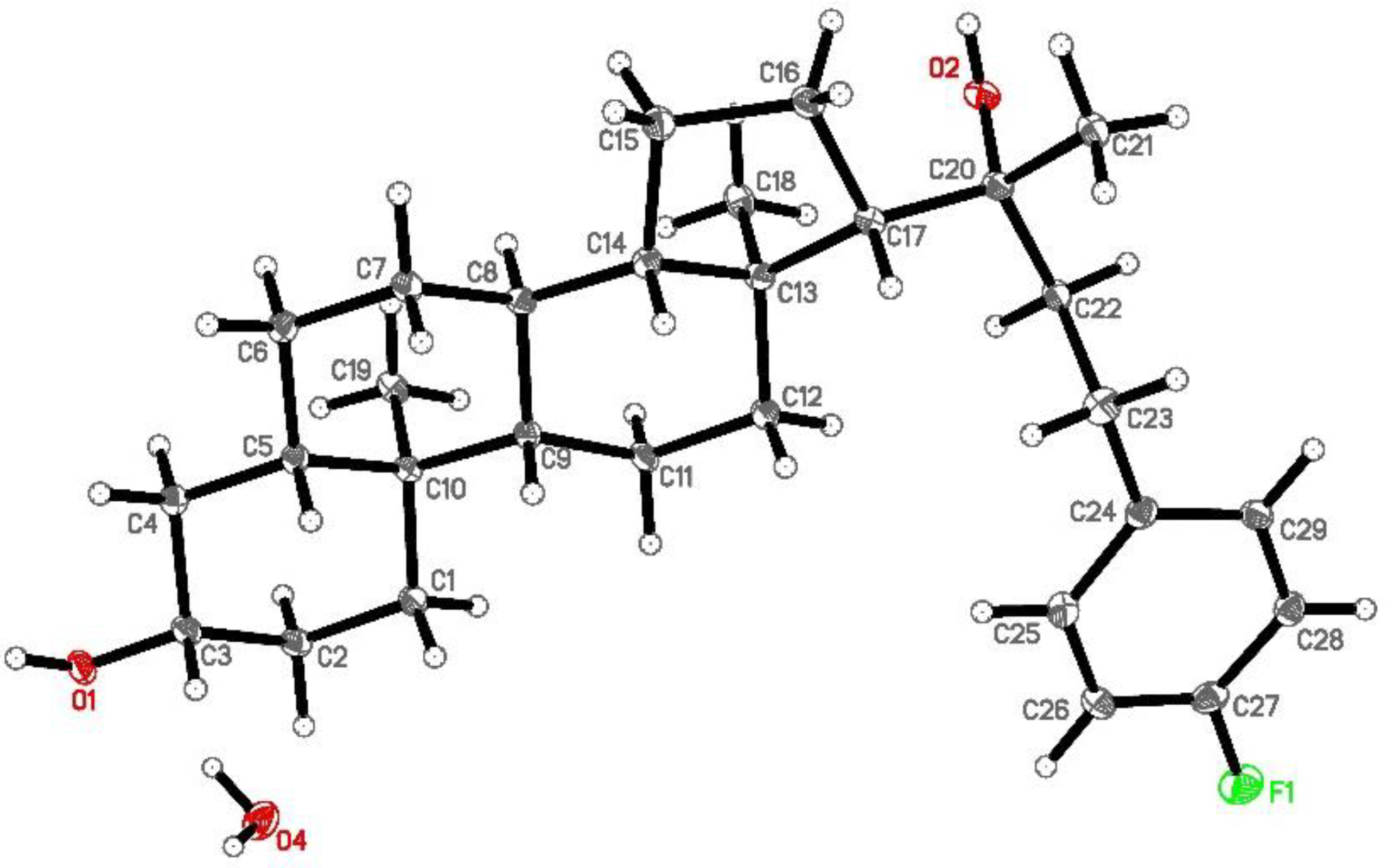
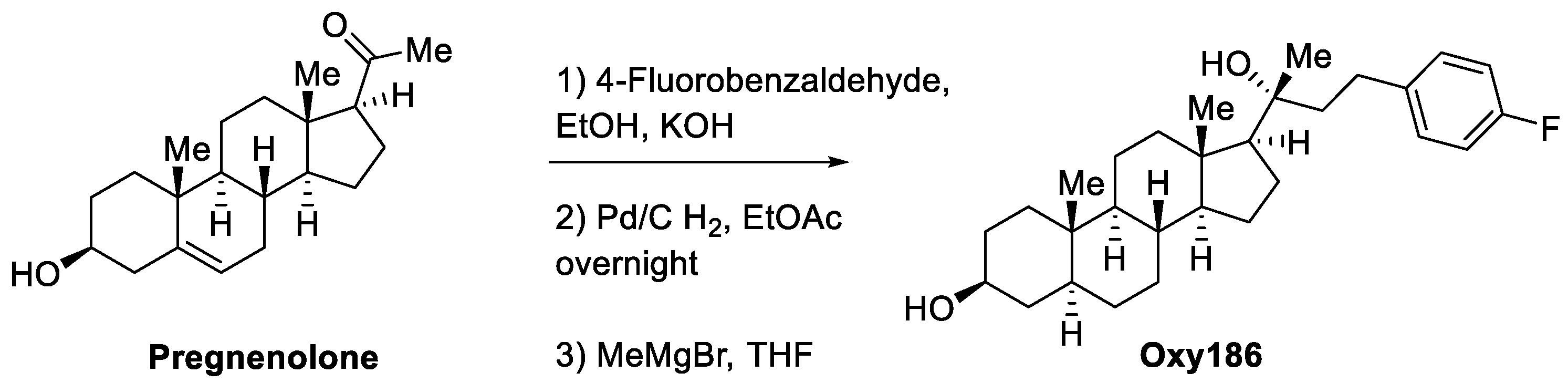
2.1. Synthesis and Molecular Characterization of Oxy186
2.2. Cell Culture and Reagents
2.3. CAPAN-1 Conditioned Medium
2.4. Quantitative RT-PCR
2.5. Transient Transfection Assay
2.6. Cell Counting Assay
2.7. Statistical Analysis
3. Results
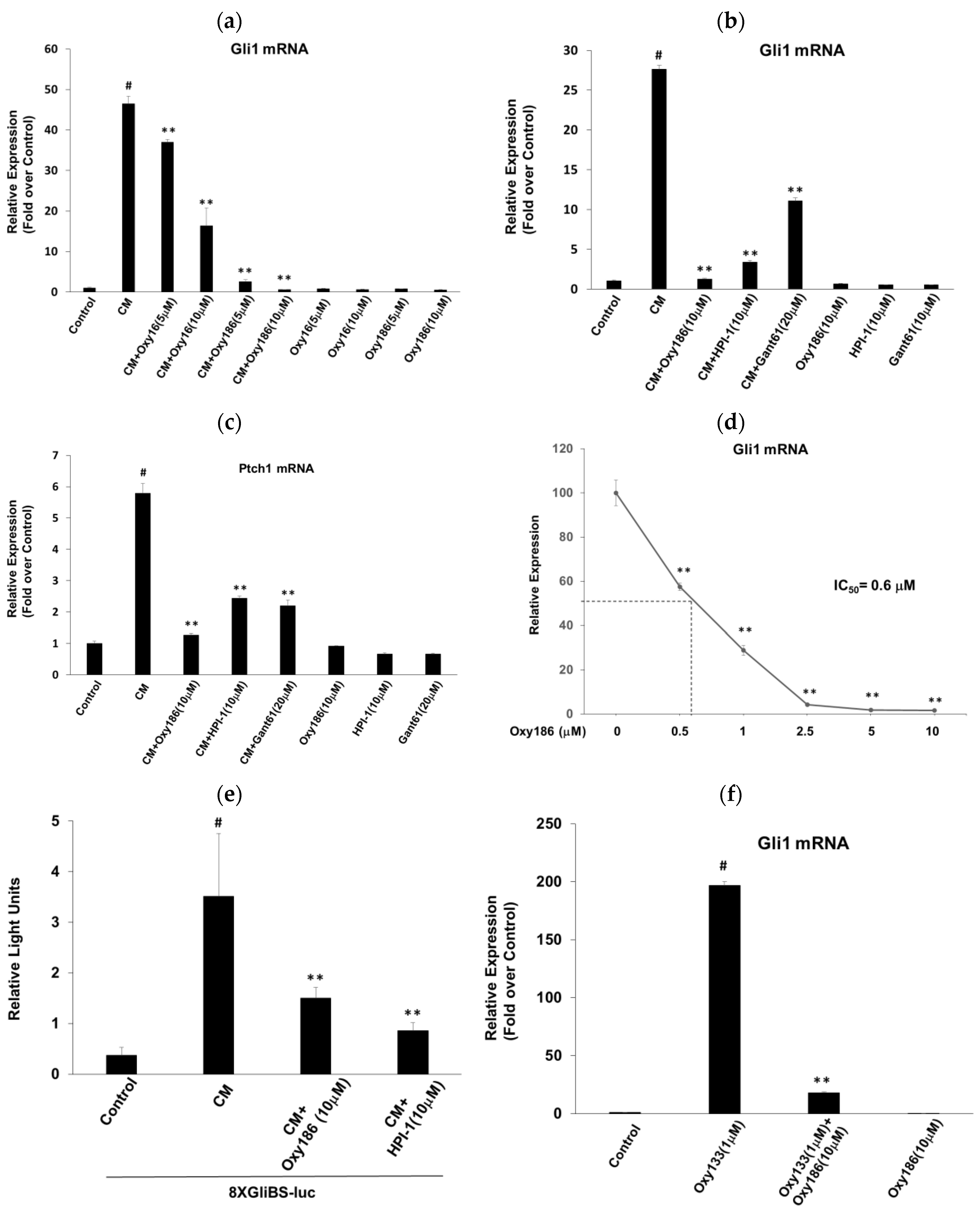
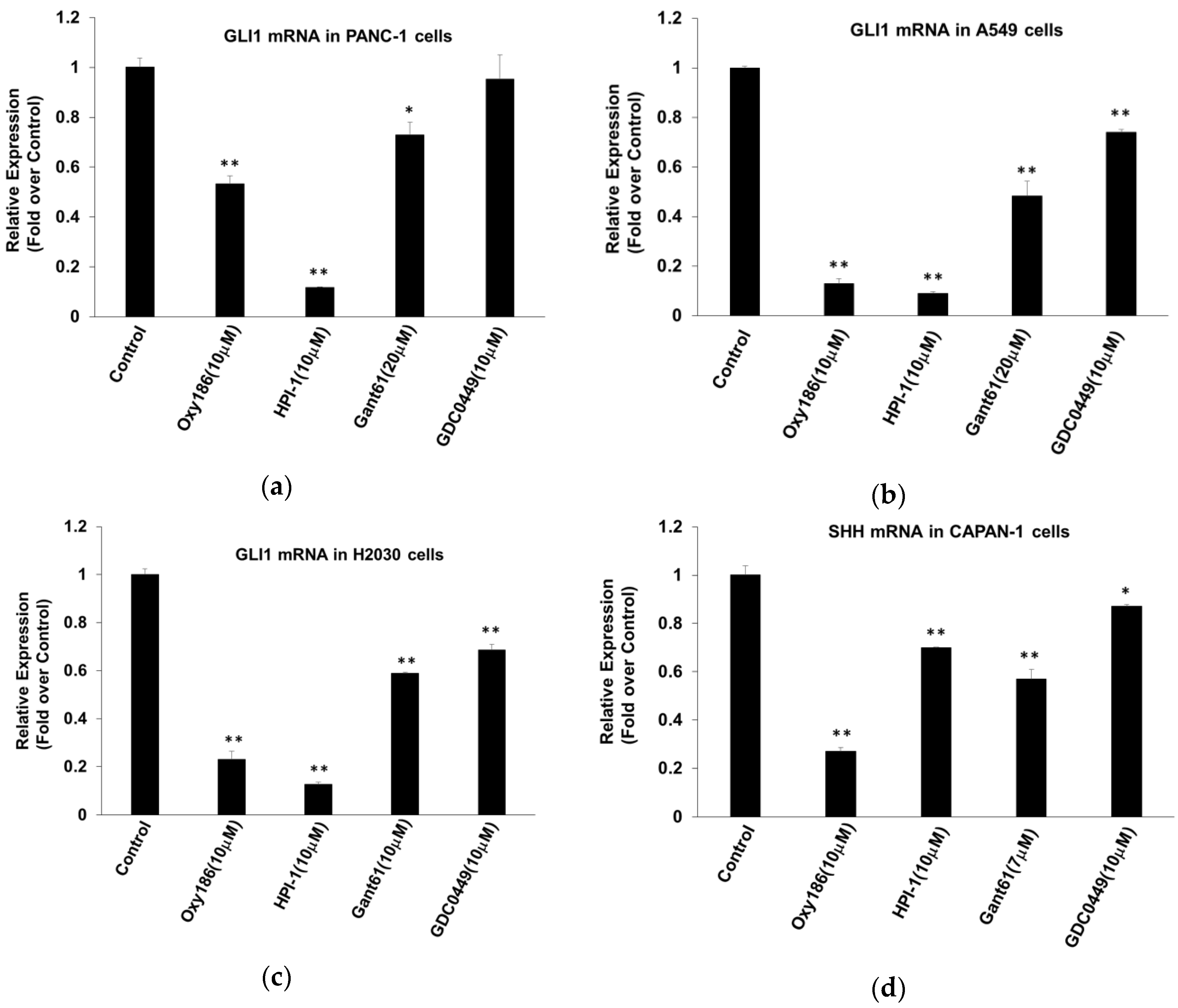
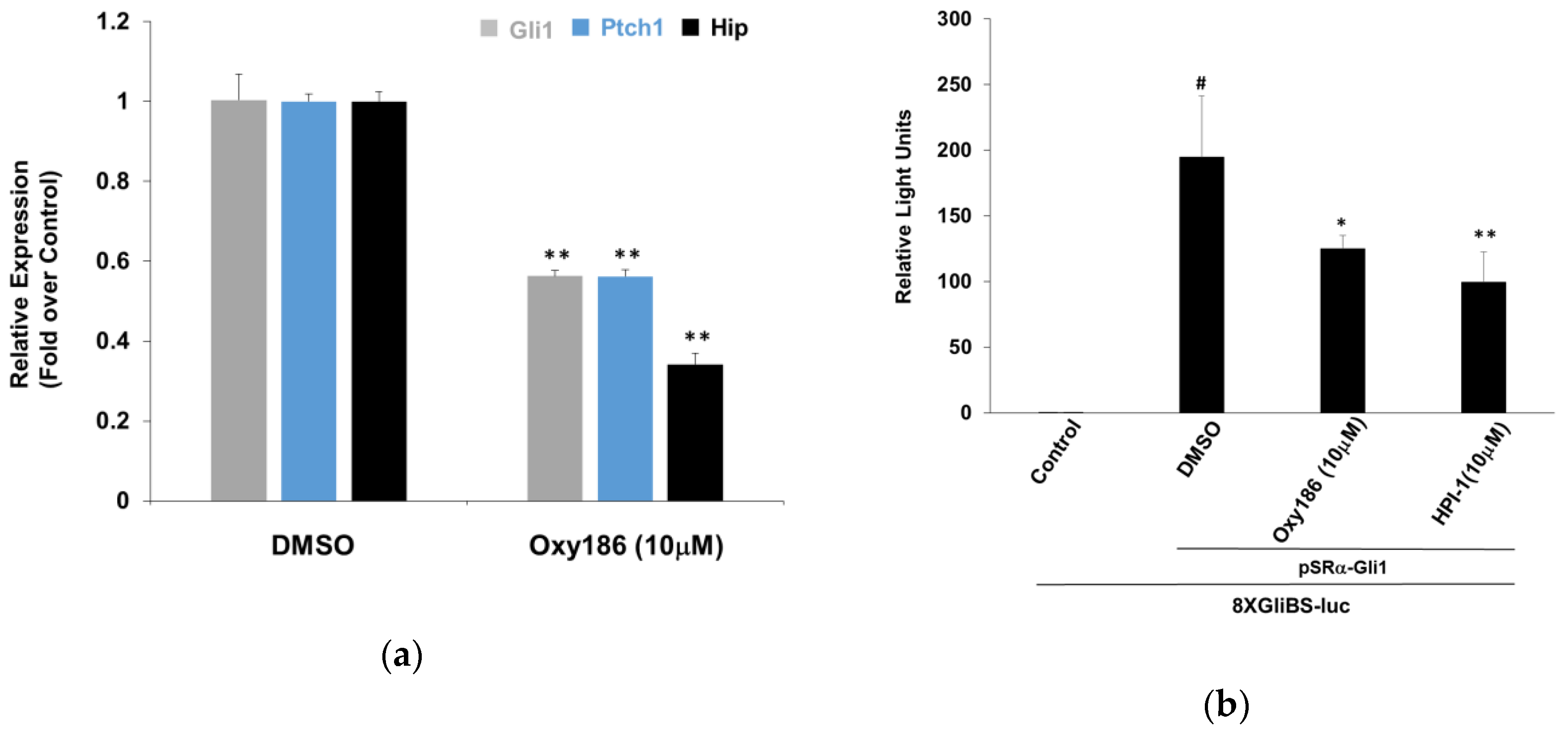
3.1. Oxy186 Inhibits Hh Signaling in Mouse Fibroblasts and Human Cancer Cells
3.2. Oxy186 Inhibits Hh Signaling Epistatic to Sufu−
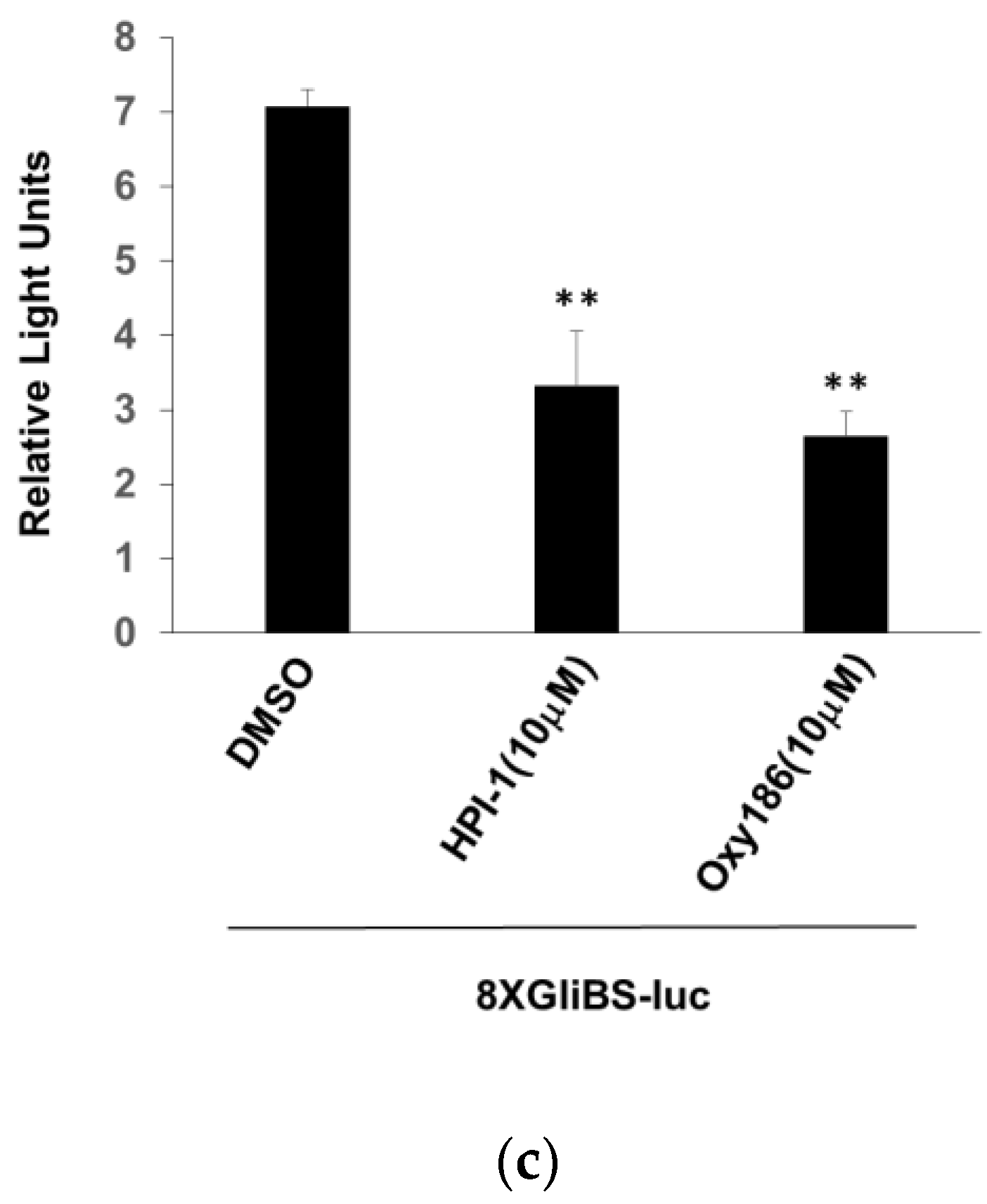
3.3. Oxy186 Inhibits the Transcriptional Activity of Gli1
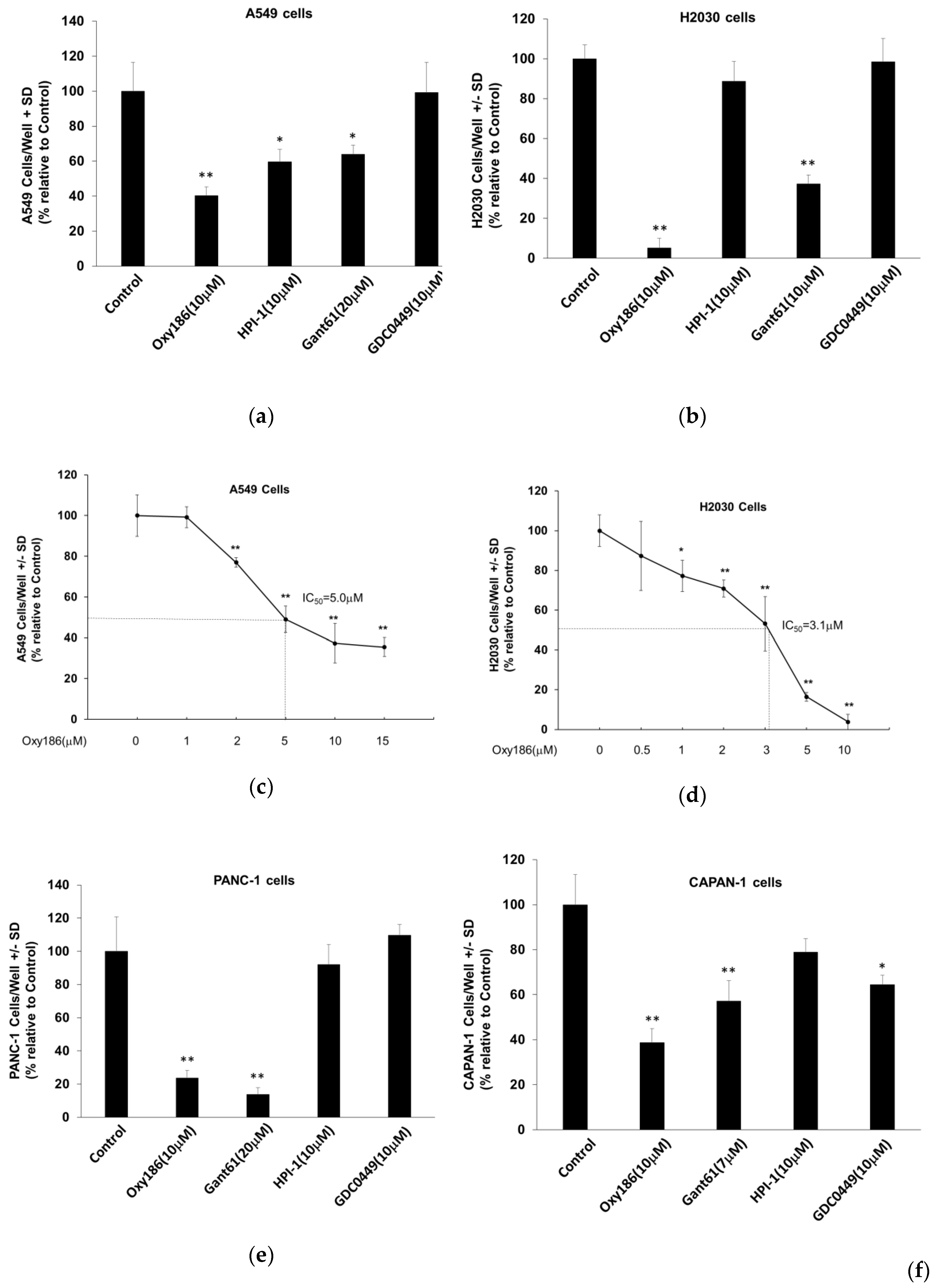
3.4. Oxy186 Inhibits Proliferation of Human Cancer Cells
3.5. Oxy186 is a Weak LXR Activator
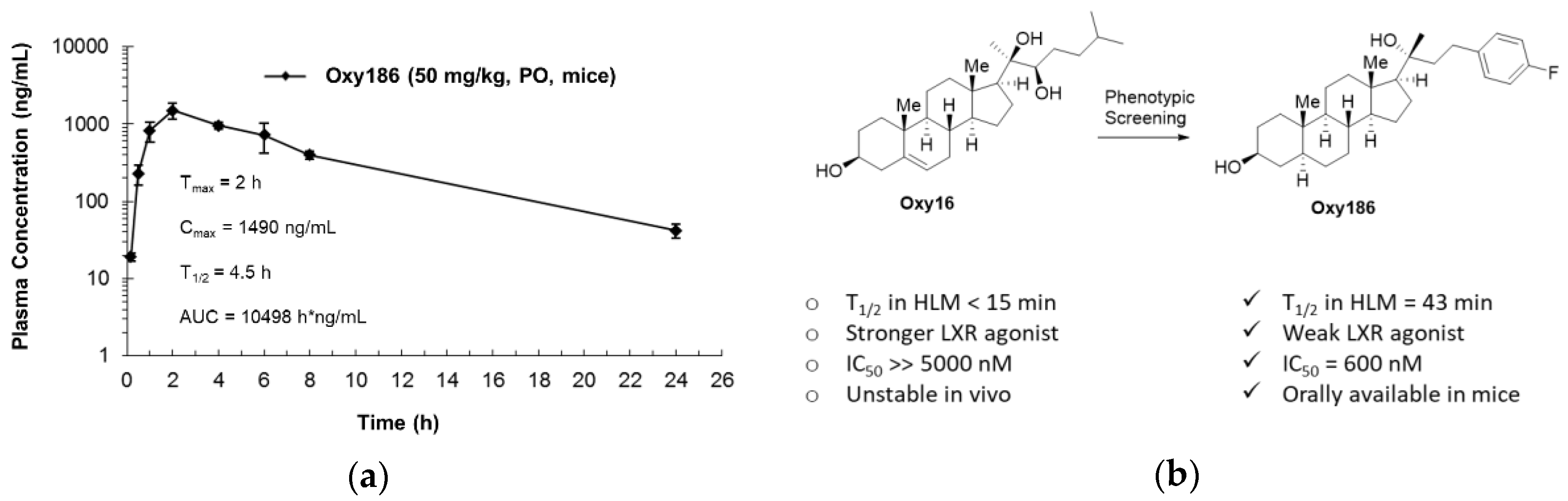
3.6. Drug-Like Properties of Oxy186
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signaling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shimo, T.; Kurio, N.; Okui, T.; Obata, K.; Masui, M.; Pang, P.; Horikiri, Y.; Sasaki, A. Expression and Role of Sonic Hedgehog in the Process of Fracture Healing with Aging. In Vivo 2016, 30, 99–105. [Google Scholar] [PubMed]
- Petrova, R.; Joyner, A.L. Roles of Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef]
- Smelkinson, M.G. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J. Dev. Biol. 2017, 4, 14. [Google Scholar] [CrossRef]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players; Oncogenic Drivers; and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properties and tumor mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signaling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Wessler, S.; Krisch, L.M.; Elmer, D.P.; Aberger, F. From inflammation to gastric cancer - the importance of Hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun. Signal. 2017, 15, 15. [Google Scholar] [CrossRef]
- Javelaud, D.; Pierrat, M.J.; Mauviel, A. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 2012, 586, 2016–2025. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, E208. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog--a cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef]
- Aszterbaum, M.; Rothman, A.; Johnson, R.L.; Fisher, M.; Xie, J.; Bonifas, J.M.; Zhang, X.; Scott, M.P.; Epstein, E.H., Jr. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J. Invest. Derm. 1998, 110, 885–888. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926; a Hedgehog Pathway Inhibitor; for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II; randomized; placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA Approval Summary: Glasdegib for newly-diagnosed acute myeloid leukemia. Clin. Cancer Res. 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. Hedgehog pathway and GLI1 isoforms in human cancer. Discov. Med. 2012, 69, 105–113. [Google Scholar]
- Shevde, L.A.; Samant, R.S. Nonclassical hedgehog-GLI signaling and its clinical implications. Int. J. Cancer 2014, 135, 1–6. [Google Scholar] [CrossRef]
- Kota, J.; Hancock, J.; Kwon, J.; Korc, M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Kasai, K. GLI1; a master regulator of the hallmark of pancreatic cancer. Pathol. Int. 2016, 66, 653–660. [Google Scholar] [CrossRef]
- Theunissen, J.; de Sauvage, F.J. Paracrine Hedgehog Signaling in Cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef] [PubMed]
- Damhofer, H.; Medema, J.P.; Veenstra, V.L.; Badea, L.; Popescu, I.; Roelink, H.; Bijlsma, M.F. Assessment of the stromal contribution to Sonic Hedgehog dependent pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 1031–1042. [Google Scholar] [CrossRef]
- Gu, J.; Saiyin, H.; Fu, D.; Li, J. Stroma—A Double-Edged Sword in Pancreatic Cancer: A Lesson from Targeting Stroma in Pancreatic Cancer with Hedgehog Signaling Inhibitors. Pancreas 2018, 47, 382–389. [Google Scholar] [CrossRef]
- Gerling, M.; Büller, N.V.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergström, Å.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.; et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef]
- Javelaud, D.; Alexaki, V.I.; Dennler, S.; Mohammad, K.S.; Guise, T.A.; Mauvie, A. The TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011, 71, 5606–5610. [Google Scholar] [CrossRef]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef]
- Lee, J.; Wu, X.; Pasca di Magliano, M.; Peters, E.C.; Wang, Y.; Hong, J.; Hebrok, M.; Ding, S.; Cho, C.Y.; Schultz, P.G. A small-molecule antagonist of the hedgehog signaling pathway. Chembiochem 2007, 8, 1916–1919. [Google Scholar] [CrossRef]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Stappenbeck, F.; Xiao, W.; Epperson, M.; Riley, M.; Priest, A.; Huang, D.; Nguyen, K.; Jung, M.E.; Thies, R.S.; Farouz, F. Novel oxysterols activate the Hedgehog pathway and induce osteogenesis. Bioorg. Med. Chem. Lett. 2012, 22, 5893–5897. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.R.; Sever, N.; Carlson, M.; Nelson, S.F.; Beachy, P.A.; Parhami, F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J. Biol. Chem. 2007, 282, 8959–8968. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.A.; Amantea, C.M.; Kianmahd, B.; Tetradis, S.; Lieberman, J.R.; Hahn, T.J.; Parhami, F. Oxysterol-induced osteoblastic differentiation of pluripotent mesenchymal cells is mediated through a PKC- and PKA dependent pathway. J. Cell Biochem. 2007, 100, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stappenbeck, F.; Matsui, W.; Parhami, F. Inhibition of Pancreatic Cancer Cell-Induced Paracrine Hedgehog Signaling by Liver X Receptor Agonists and Oxy16, a Naturally Occurring Oxysterol. J. Cell Biochem. 2017, 118, 499–509. [Google Scholar] [CrossRef]
- Montgomery, S.R.; Nargizyan, T.; Meliton, V.; Nachtergaele, S.; Rohatgi, R.; Stappenbeck, F.; Jung, M.E.; Johnson, J.S.; Aghdasi, B.; Tian, H.; et al. A novel osteogenic oxysterol compound for therapeutic development to promote bone growth: Activation of hedgehog signaling and osteogenesis through smoothened binding. J. Bone Min. Res. 2014, 29, 1872–1885. [Google Scholar] [CrossRef]
- Watanabe, B.; Nakagawa, Y.; Ogura, T.; Miyagawa, H. Stereoselective synthesis of (22R)- and (22S)-castasterone/ponasterone A hybrid compounds and evaluation of their molting hormone activity. Steroids 2004, 69, 483–493. [Google Scholar] [CrossRef]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. Biomed Res Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog–GLI Signaling Inhibition Suppresses Tumor Growth in Squamous Lung Cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharm. Exp. 1997, 283, 46–58. [Google Scholar]
- Strushkevich, N.; MacKenzie, F.; Cherkesova, T.; Grabovec, I.; Usanov, S.; Park, H.W. Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. USA 2011, 108, 10139–10143. [Google Scholar] [CrossRef]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Jeng, C.C.; Sheen, I.S.; Wu, S.H.; Lu, S.J.; Wang, C.H.; Chang, C.F. Glioma-Associated Oncogene Homolog Inhibitors Have the Potential of Suppressing Cancer Stem Cells of Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1375. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, J.; Shen, H.; Dong, W.; Ni, Y.; Du, J. Tumor-stroma ratio is an independent predictor for survival in NSCLC. Int. J. Clin. Exp. Pathol. 2015, 8, 11348–11355. [Google Scholar]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Lunardia, S.; Muschela, R.J.; Brunnera, T.B. The stromal compartments in pancreatic cancer: Are there any therapeutic targets? Cancer Lett. 2014, 343, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain; rather than support; pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Gore, J.; Korc, M. Pancreatic cancer stroma: Friend or foe? Cancer Cell 2014, 25, 711–712. [Google Scholar] [CrossRef]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXR alpha and LXR beta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef]
- Chisholm, J.W.; Hong, J.; Mills, S.A.; Lawn, R.M. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J. Lipid Res. 2003, 44, 2039–2048. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | HLM, % Remaining after 60 min | HLM, Half-Life | MLM, % Remaining after 60 min | MLM, Half-Life |
|---|---|---|---|---|
| Oxy16 | 6% | 15 min | 5% | 14min |
| Oxy186 | 40% | 45 min | 50% | 60min |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Stappenbeck, F.; Parhami, F. Inhibition of Hedgehog Signaling in Fibroblasts, Pancreatic, and Lung Tumor Cells by Oxy186, an Oxysterol Analogue with Drug-Like Properties. Cells 2019, 8, 509. https://doi.org/10.3390/cells8050509
Wang F, Stappenbeck F, Parhami F. Inhibition of Hedgehog Signaling in Fibroblasts, Pancreatic, and Lung Tumor Cells by Oxy186, an Oxysterol Analogue with Drug-Like Properties. Cells. 2019; 8(5):509. https://doi.org/10.3390/cells8050509
Chicago/Turabian StyleWang, Feng, Frank Stappenbeck, and Farhad Parhami. 2019. "Inhibition of Hedgehog Signaling in Fibroblasts, Pancreatic, and Lung Tumor Cells by Oxy186, an Oxysterol Analogue with Drug-Like Properties" Cells 8, no. 5: 509. https://doi.org/10.3390/cells8050509
APA StyleWang, F., Stappenbeck, F., & Parhami, F. (2019). Inhibition of Hedgehog Signaling in Fibroblasts, Pancreatic, and Lung Tumor Cells by Oxy186, an Oxysterol Analogue with Drug-Like Properties. Cells, 8(5), 509. https://doi.org/10.3390/cells8050509