c-Cbl: An Important Regulator and a Target in Angiogenesis and Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
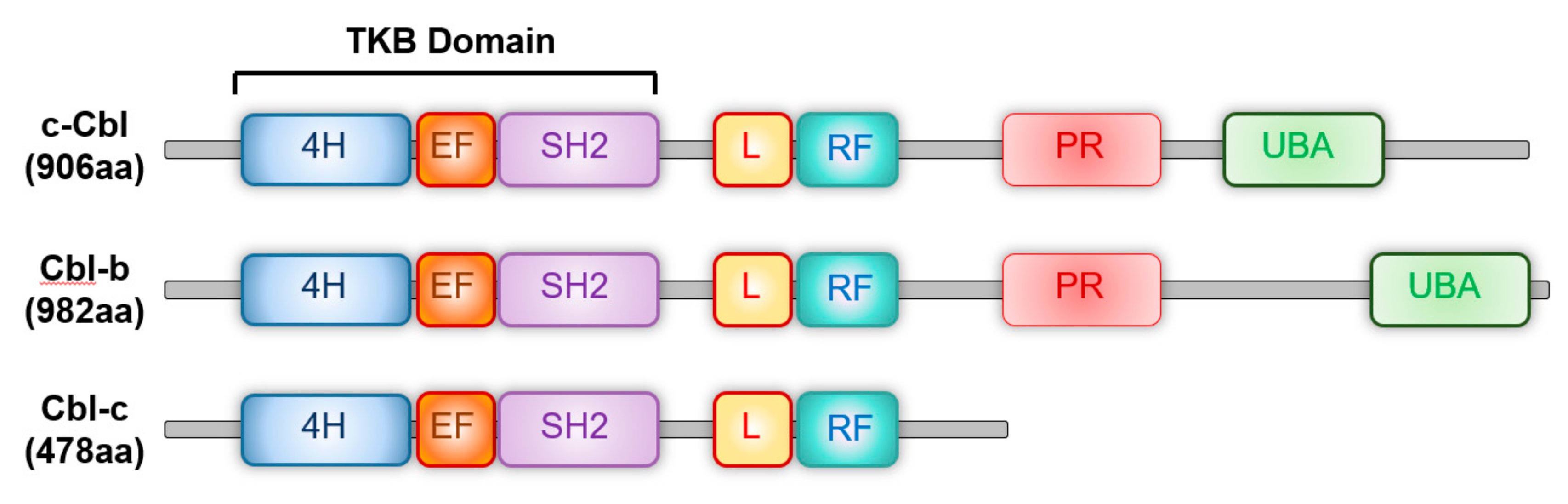
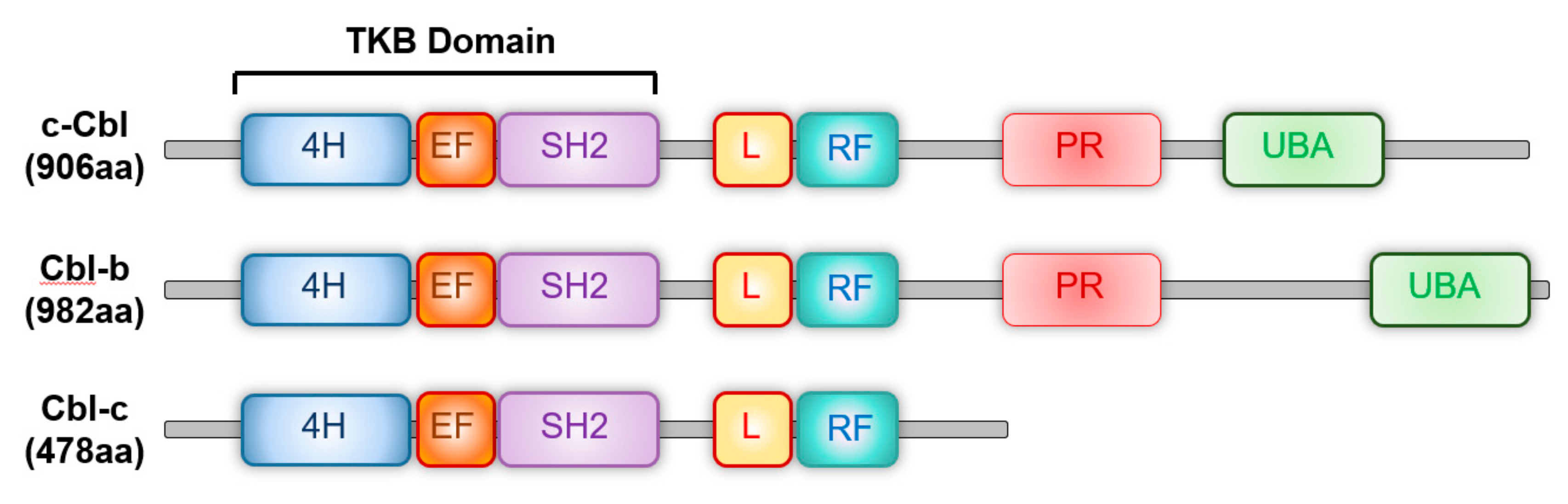
2. c-Cbl Family of Ubiquitin E3 Ligases
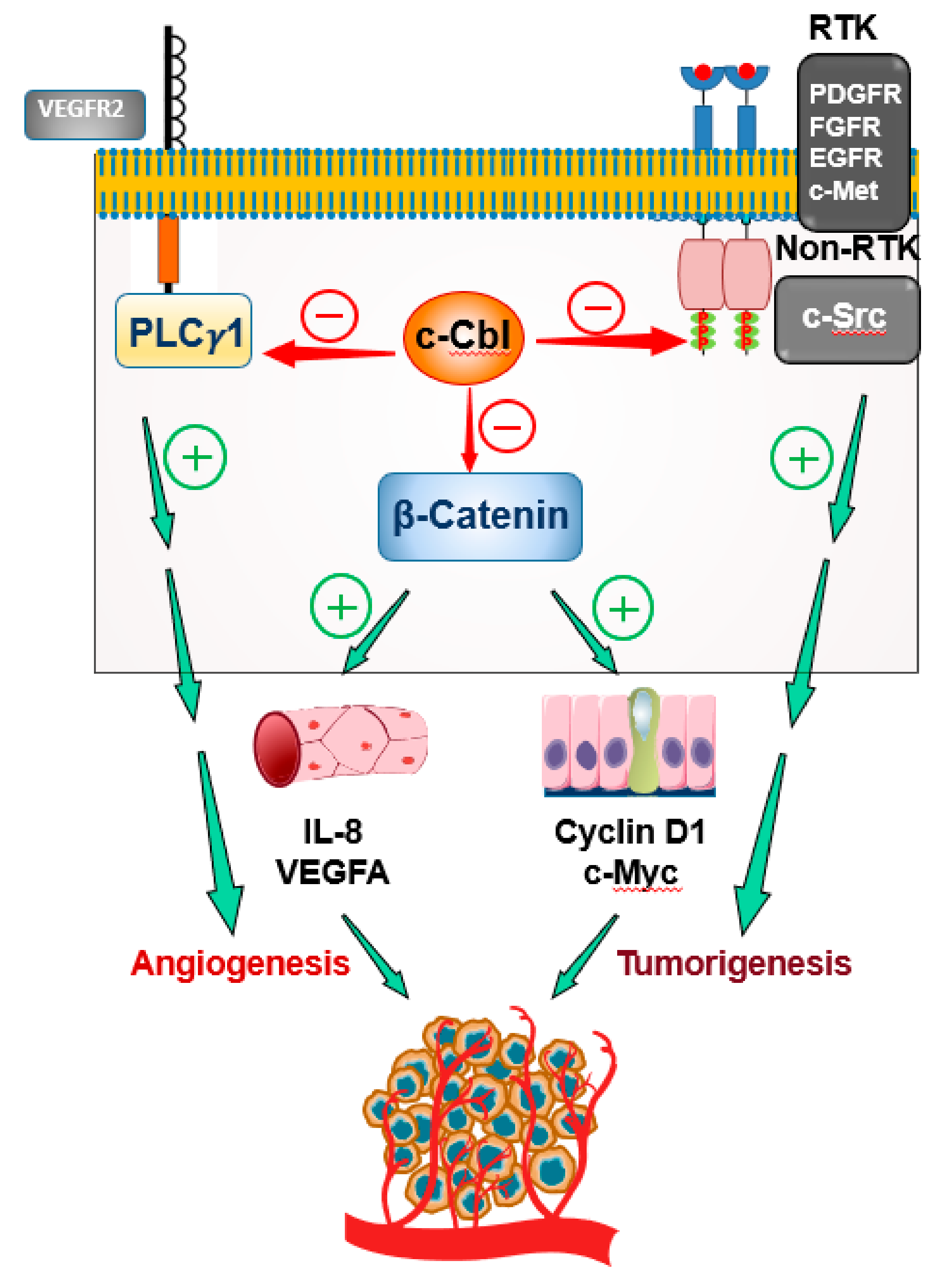
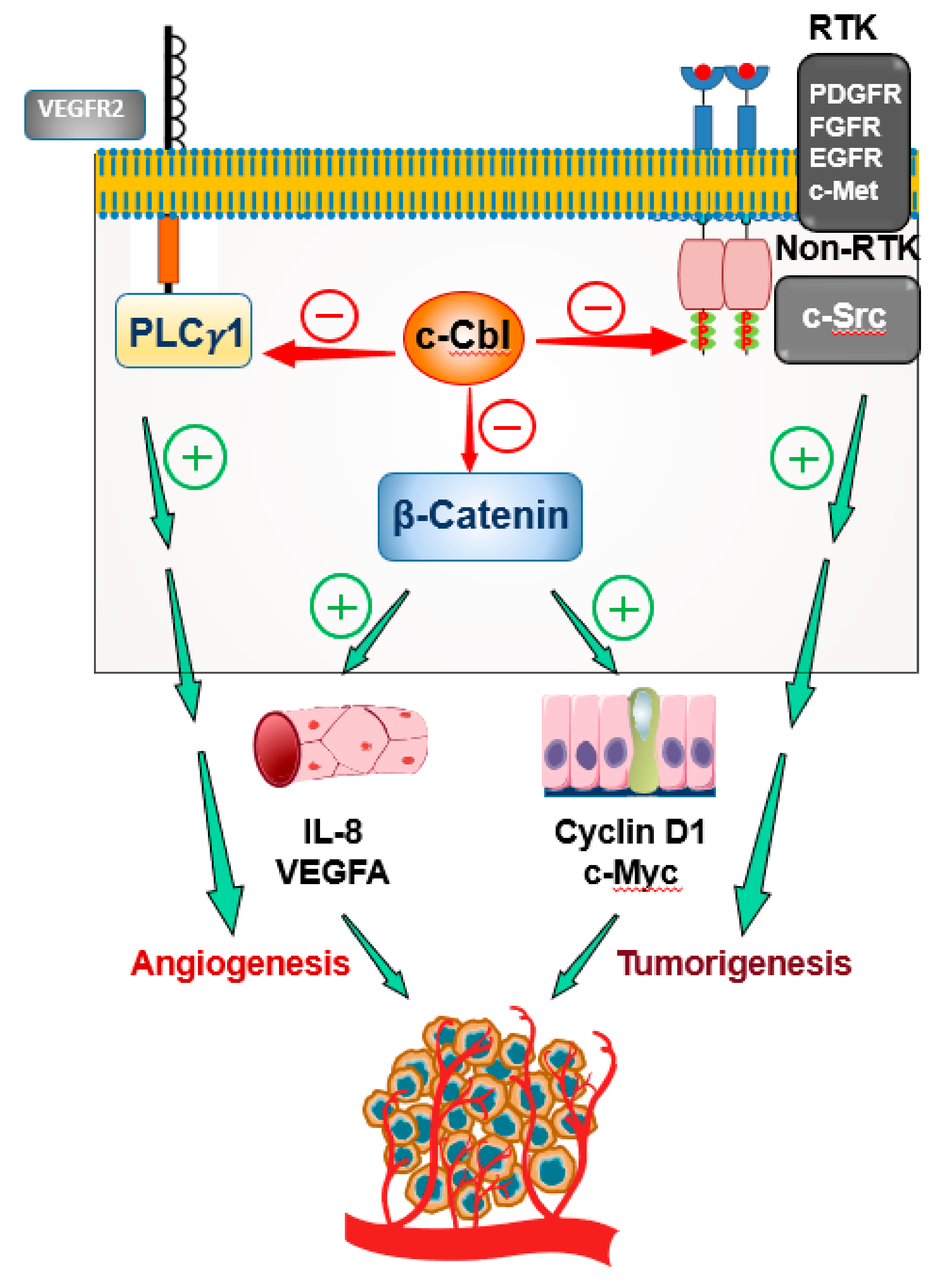
3. c-Cbl as a Major Regulator of Angiogenesis
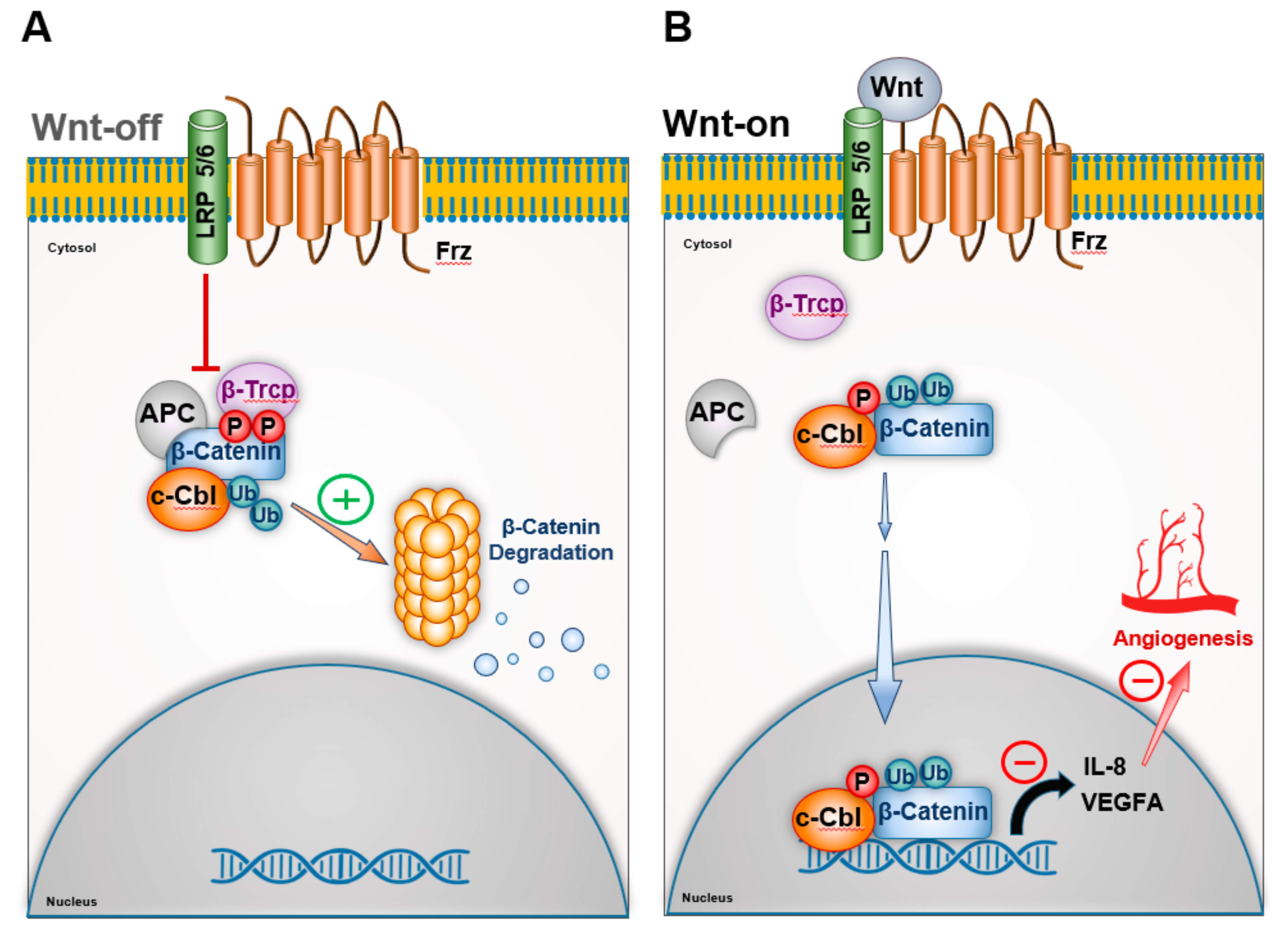
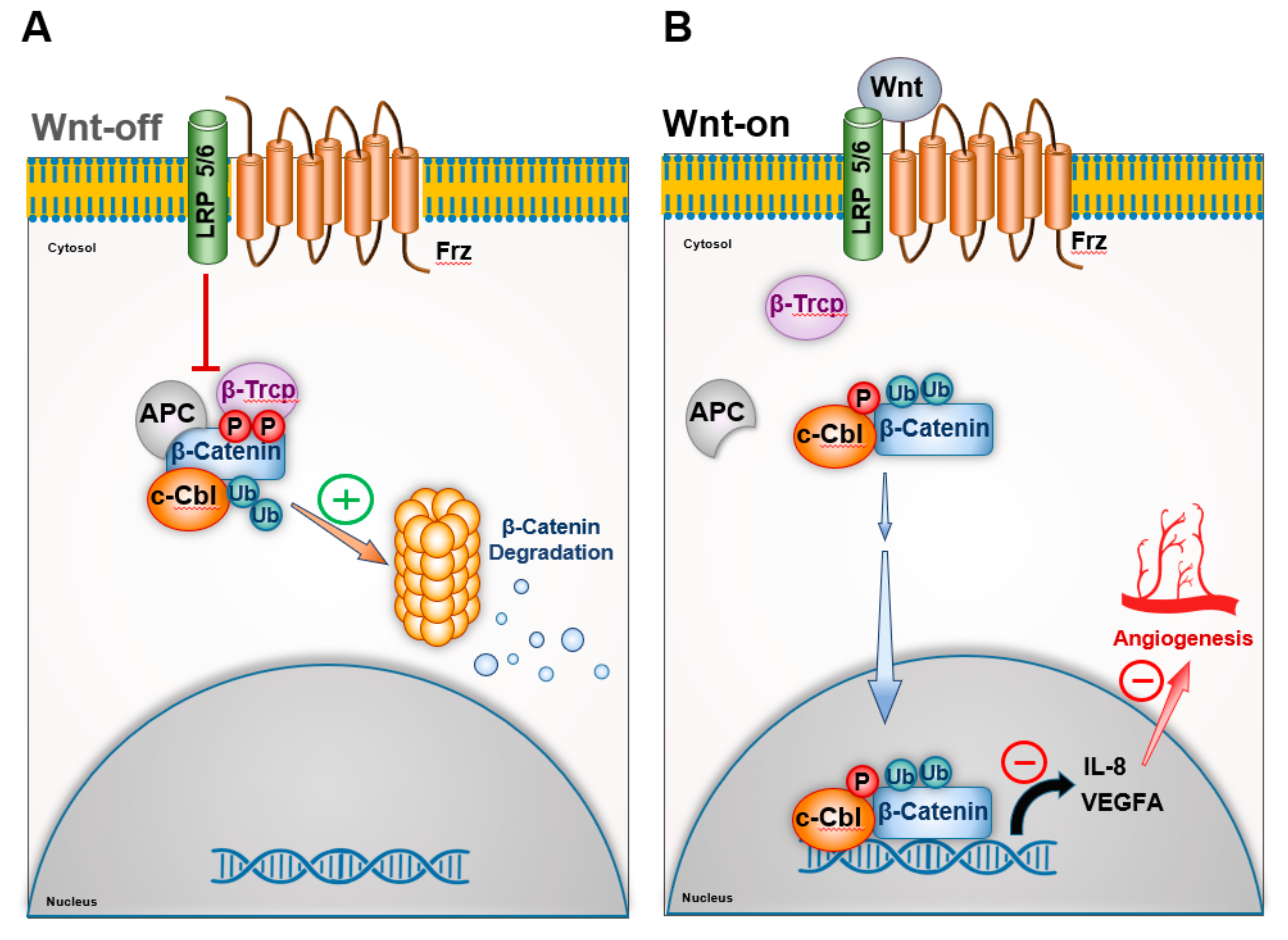
3.1. c-Cbl and β-Catenin
3.2. c-Cbl and VEGF Signaling
3.3. c-Cbl Regulation of VEGFR-2 through Epsin
3.4. c-Cbl and Other Receptor Tyrosine Kinases
3.4.1. c-Cbl and Angiopoietin(1/2)-Tie2 Axis
3.4.2. c-Cbl and Eph Receptors
3.4.3. c-Cbl and c-Src
4. c-Cbl in Tumor-Mediated Angiogenesis
5. c-Cbl in Myocardial Ischemia
6. c-Cbl in Retinal Neovascularization
7. Therapeutic Potential of c-Cbl
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simons, M.; Ware, J.A. Therapeutic angiogenesis in cardiovascular disease. Nat. Rev. Drug Discov. 2003, 2, 863. [Google Scholar] [CrossRef] [PubMed]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Angiogenesis Dysregulation in the Pathogenesis of Systemic Sclerosis. Biomed. Res. Int. 2017, 2017, 5345673. [Google Scholar] [CrossRef]
- Devi, L.; Pothana, L.; Goel, S. Dysregulation of angiogenesis-specific signalling in adult testis results in xenograft degeneration. Sci. Rep. 2017, 7, 2605. [Google Scholar] [CrossRef] [PubMed]
- Patan, S. Vasculogenesis and angiogenesis. Cancer Treat. Res. 2004, 117, 3–32. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Kim, H.-J.; Jang, S.Y.; Park, J.-I.; Byun, J.; Kim, D.-I.; Do, Y.-S.; Kim, J.-M.; Kim, S.; Kim, B.-M.; Kim, W.-B.; et al. Vascular endothelial growth factor-induced angiogenic gene therapy in patients with peripheral artery disease. Exp. Amp; Mol. Med. 2004, 36, 336. [Google Scholar] [CrossRef]
- Talia, N.C.; Alfaro, V.D., III; John, B.K.; Eric, P.J. Diabetic Retinopathy and Angiogenesis. Curr. Diabetes Rev. 2009, 13, 8–13. [Google Scholar] [CrossRef]
- Koch, A.E. Angiogenesis as a target in rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, ii60. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; de la Motte, C.; Graziani, C.; West, G.; Phillips, M.H.; Pola, R.; Rutella, S.; Willis, J.; Gasbarrini, A.; et al. Angiogenesis as a Novel Component of Inflammatory Bowel Disease Pathogenesis. Gastroenterology 2006, 130, 2060–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of Fibroblast Growth Factor Receptors in Carcinogenesis. Mol. Cancer Res. 2010, 8, 1439. [Google Scholar] [CrossRef]
- Tiong, K.H.; Mah, L.Y.; Leong, C.-O. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Cell Death 2013, 18, 1447–1468. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, A.B.; Winkler, M.E.; Derynck, R. Transforming growth factor-alpha: A more potent angiogenic mediator than epidermal growth factor. Science 1986, 232, 1250. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Derynck, R. TGF-β signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117. [Google Scholar] [CrossRef]
- Fajardo, L.F.; Kwan, H.H.; Kowalski, J.; Prionas, S.D.; Allison, A.C. Dual role of tumor necrosis factor-alpha in angiogenesis. Am. J. Pathol. 1992, 140, 539–544. [Google Scholar] [PubMed]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-α. Nature 1987, 329, 630–632. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ema, M. Roles of VEGF-A signalling in development, regeneration, and tumours. J. Biochem. 2014, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Bosca, A.B.; Susman, S.; Marginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)–key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. Rev. Roum. De Morphol. Et Embryol. 2018, 59, 455–467. [Google Scholar]
- Chitalia, V.; Shivanna, S.; Martorell, J.; Meyer, R.; Edelman, E.; Rahimi, N. c-Cbl, a ubiquitin E3 ligase that targets active β-catenin: A novel layer of Wnt signaling regulation. J. Biol. Chem. 2013, 288, 23505–23517. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Harrold, I.; Shashar, M.; Meyer, R.; Kiang, C.; Francis, J.; Zhao, Q.; Feng, H.; Edelman, E.R.; Rahimi, N.; et al. The c-Cbl ubiquitin ligase regulates nuclear β-catenin and angiogenesis by its tyrosine phosphorylation mediated through the Wnt signaling pathway. J. Biol. Chem. 2015, 290, 12537–12546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Husain, D.; Meyer, R.D.; Mehta, M.; Pfeifer, W.M.; Chou, E.; Navruzbekov, G.; Ahmed, E.; Rahimi, N. Role of c-Cbl-dependent regulation of phospholipase Cgamma1 activation in experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2010, 51, 6803–6809. [Google Scholar] [CrossRef]
- Meyer, R.D.; Husain, D.; Rahimi, N. c-Cbl inhibits angiogenesis and tumor growth by suppressing activation of PLCgamma1. Oncogene 2011, 30, 2198–2206. [Google Scholar] [CrossRef]
- Prenen, H.; Smeets, D.; Mazzone, M.; Lambrechts, D.; Sagaert, X.; Sciot, R.; Debiec-Rychter, M. Phospholipase C gamma 1 (PLCG1) R707Q mutation is counterselected under targeted therapy in a patient with hepatic angiosarcoma. Oncotarget 2015, 6, 36418–36425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Wang, Z. Akt binds to and phosphorylates phospholipase C-gamma1 in response to epidermal growth factor. Mol. Biol. Cell 2006, 17, 2267–2277. [Google Scholar] [CrossRef]
- Swaminathan, G.; Tsygankov, A.Y. The Cbl family proteins: Ring leaders in regulation of cell signaling. J. Cell Physiol. 2006, 209, 21–43. [Google Scholar] [CrossRef]
- Dou, H.; Buetow, L.; Hock, A.; Sibbet, G.J.; Vousden, K.H.; Huang, D.T. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat. Struct. Mol. Biol. 2012, 19, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Langdon, W.; W Hartley, J.; Klinken, S.; K Ruscetti, S.; Morse, H. v-cbl, an Oncogene from a Dual-Recombinant Murine Retrovirus that Induces Early B-Lineage Lymphomas. Proc. Natl. Acad. Sci. USA 1989, 86, 1168–1172. [Google Scholar] [CrossRef]
- J Blake, T.; G Heath, K.; Langdon, W. The truncation that generated the v-cbl oncogene reveals an ability for nuclear transport, DNA binding and acute transformation. EMBO J. 1993, 12, 2017–2026. [Google Scholar] [CrossRef]
- Blake, T.J.; Shapiro, M.; Morse, H.C., 3rd; Langdon, W.Y. The sequences of the human and mouse c-cbl proto-oncogenes show v-cbl was generated by a large truncation encompassing a proline-rich domain and a leucine zipper-like motif. Oncogene 1991, 6, 653–657. [Google Scholar]
- Chiang, Y.J.; Kole, H.K.; Brown, K.; Naramura, M.; Fukuhara, S.; Hu, R.J.; Jang, I.K.; Gutkind, J.S.; Shevach, E.; Gu, H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature 2000, 403, 216–220. [Google Scholar] [CrossRef]
- Lutz-Nicoladoni, C.; Wolf, D.; Sopper, S. Modulation of Immune Cell Functions by the E3 Ligase Cbl-b. Front. Oncol. 2015, 5, 58. [Google Scholar] [CrossRef]
- Mukhopadhyay, C.; Triplett, A.; Bargar, T.; Heckman, C.; Wagner, K.-U.; Naramura, M. Casitas B-cell lymphoma (Cbl) proteins protect mammary epithelial cells from proteotoxicity of active c-Src accumulation. Proc. Natl. Acad. Sci. USA 2016, 113, E8228–E8237. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, B.; Zutshi, N.; An, W.; Goetz, B.; Arya, P.; Bielecki, T.A.; Mushtaq, I.; Storck, M.D.; Meza, J.L.; Band, V. An essential role of CBL and CBL-B ubiquitin ligases in mammary stem cell maintenance. Development 2017, 144, 1072–1086. [Google Scholar] [CrossRef] [Green Version]
- Smit, L.; Borst, J. The Cbl family of signal transduction molecules. Crit. Rev. Oncog. 1997, 8, 359–379. [Google Scholar] [CrossRef]
- Singh, A.J.; Meyer, R.D.; Navruzbekov, G.; Shelke, R.; Duan, L.; Band, H.; Leeman, S.E.; Rahimi, N. A critical role for the E3-ligase activity of c-Cbl in VEGFR-2-mediated PLCgamma1 activation and angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 5413–5418. [Google Scholar] [CrossRef]
- Rahimi, N. The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer 2012, 11, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N.; Costello, C.E. Emerging roles of post-translational modifications in signal transduction and angiogenesis. Proteomics 2015, 15, 300–309. [Google Scholar] [CrossRef]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Taher, T.E.; Tjin, E.P.; Beuling, E.A.; Borst, J.; Spaargaren, M.; Pals, S.T. c-Cbl is involved in Met signaling in B cells and mediates hepatocyte growth factor-induced receptor ubiquitination. J. Immunol. 2002, 169, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Leestemaker, Y.; Ovaa, H. Tools to investigate the ubiquitin proteasome system. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef] [PubMed]
- El Hokayem, J.; Amadei, C.; Obeid, J.P.; Nawaz, Z. Ubiquitination of nuclear receptors. Clin. Sci. 2017, 131, 917–934. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.H.; Ko, J.; Donoghue, D.J. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 2017, 16, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Tria, G.; Ahmed, S.F.; Hock, A.; Dou, H.; Sibbet, G.J.; Svergun, D.I.; Huang, D.T. Casitas B-lineage lymphoma linker helix mutations found in myeloproliferative neoplasms affect conformation. Bmc Biol. 2016, 14, 76. [Google Scholar] [CrossRef]
- Shashar, M.; Siwak, J.; Tapan, U.; Lee, S.Y.; Meyer, R.D.; Parrack, P.; Tan, J.; Khatami, F.; Francis, J.; Zhao, Q.; et al. c-Cbl mediates the degradation of tumorigenic nuclear beta-catenin contributing to the heterogeneity in Wnt activity in colorectal tumors. Oncotarget 2016, 7, 71136–71150. [Google Scholar] [CrossRef]
- Kumaradevan, S.; Lee, S.Y.; Richards, S.; Lyle, C.; Zhao, Q.; Tapan, U.; Jiangliu, Y.; Ghumman, S.; Walker, J.; Belghasem, M.; et al. c-Cbl Expression Correlates with Human Colorectal Cancer Survival and Its Wnt/beta-Catenin Suppressor Function Is Regulated by Tyr371 Phosphorylation. Am. J. Pathol. 2018, 188, 1921–1933. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Reis, M.; Liebner, S. Wnt signaling in the vasculature. Exp. Cell Res. 2013, 319, 1317–1323. [Google Scholar] [CrossRef]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Orte, E.; Saenz-Narciso, B.; Moreno, S.; Cabello, J. Multiple functions of the noncanonical Wnt pathway. Trends Genet. 2013, 29, 545–553. [Google Scholar] [CrossRef]
- Wang, Y. Wnt/Planar cell polarity signaling: A new paradigm for cancer therapy. Mol. Cancer 2009, 8, 2103. [Google Scholar] [CrossRef]
- Kohn, A.D.; Moon, R.T. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef]
- Sugimura, R.; Li, L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Res. Part Cembryo Today Rev. 2010, 90, 243–256. [Google Scholar] [CrossRef]
- Van Amerongen, R. Alternative Wnt pathways and receptors. Cold Spring Harbor Persp. Biol. 2012, a007914. [Google Scholar] [CrossRef]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Duchartre, Y.; Kim, Y.M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef]
- Karimaian, A.; Majidinia, M.; Bannazadeh Baghi, H.; Yousefi, B. The crosstalk between Wnt/beta-catenin signaling pathway with DNA damage response and oxidative stress: Implications in cancer therapy. Dna Repair 2017, 51, 14–19. [Google Scholar] [CrossRef]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharm. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharm.. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Theis, V.; Theiss, C. VEGF - A Stimulus for Neuronal Development and Regeneration in the CNS and PNS. Curr. Protein Pept. Sci. 2018, 19, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.J.; Kume, T.; McKay, C.; Xu, M.J.; Ihle, J.N.; Carpenter, G. Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J. Biol. Chem. 2002, 277, 9335–9341. [Google Scholar] [CrossRef]
- Lawson, N.D.; Mugford, J.W.; Diamond, B.A.; Weinstein, B.M. phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 2003, 17, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.D.; Latz, C.; Rahimi, N. Recruitment and activation of phospholipase Cgamma1 by vascular endothelial growth factor receptor-2 are required for tubulogenesis and differentiation of endothelial cells. J. Biol. Chem. 2003, 278, 16347–16355. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Sheldon, H.; Martincorena, I.; Van Loo, P.; Gundem, G.; Wedge, D.C.; Ramakrishna, M.; Cooke, S.L.; Pillay, N.; et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46, 376–379. [Google Scholar] [CrossRef]
- Jang, H.J.; Yang, Y.R.; Kim, J.K.; Choi, J.H.; Seo, Y.K.; Lee, Y.H.; Lee, J.E.; Ryu, S.H.; Suh, P.G. Phospholipase C-gamma1 involved in brain disorders. Adv. Biol. Regul. 2013, 53, 51–62. [Google Scholar] [CrossRef]
- Meyer, R.D.; Rahimi, N. Comparative structure-function analysis of VEGFR-1 and VEGFR-2: What have we learned from chimeric systems? Ann. NY Acad. Sci. 2003, 995, 200–207. [Google Scholar] [CrossRef]
- Rahimi, N. A role for protein ubiquitination in VEGFR-2 signalling and angiogenesis. Biochem Soc Trans 2009, 37, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Madhivanan, K.; Mukherjee, D.; Aguilar, R.C. The epsin protein family: coordinators of endocytosis and signaling. Biomol. Concepts 2012, 3, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, C.E.; Mohney, R.P.; Miller, S.L.; Hanes, R.N.; O'Bryan, J.P. The ubiquitin-interacting motifs target the endocytic adaptor protein epsin for ubiquitination. Curr. Biol. 2002, 12, 1112–1116. [Google Scholar] [CrossRef]
- Song, H.; Pasula, S.; Brophy, M.; Tessneer, K.L.; Wu, H.; Dong, Y.; Chen, H. Abstract 15705: Novel Epsin-VEGFR2 Interactions Facilitated by c-Cbl Ubiquitination of Epsin and VEGFR2 Regulate VEGFR2 Signaling and Physiological and Pathological Angiogenesis. Circulation 2014, 130, A15705. [Google Scholar] [CrossRef]
- Tessneer, K.L.; Pasula, S.; Cai, X.; Dong, Y.; McManus, J.; Liu, X.; Yu, L.; Hahn, S.; Chang, B.; Chen, Y.; et al. Genetic reduction of vascular endothelial growth factor receptor 2 rescues aberrant angiogenesis caused by epsin deficiency. Arter. Thromb. Vasc. Biol. 2014, 34, 331–337. [Google Scholar] [CrossRef]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite Role of Angiopoietin-1, a Ligand for the TIE2 Receptor, during Embryonic Angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in vivo Angiogenesis. Science 1997, 277, 55. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Chen, D.; Takahashi, T.; Fujikawa, K.; Kearney, M.; Magner, M.; Yancopoulos George, D.; Isner Jeffrey, M. Tie2 Receptor Ligands, Angiopoietin-1 and Angiopoietin-2, Modulate VEGF-Induced Postnatal Neovascularization. Circ. Res. 1998, 83, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Naik, U.P. Pericyte-endothelial cell interaction. Cell Adhes. Migr. 2012, 6, 157–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milam, K.E.; Parikh, S.M. The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue Barriers 2015, 3, e957508. [Google Scholar] [CrossRef]
- Witzenbichler, B.; Maisonpierre, P.C.; Jones, P.; Yancopoulos, G.D.; Isner, J.M. Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. J. Biol. Chem. 1998, 273, 18514–18521. [Google Scholar] [CrossRef]
- Wehrle, C.; Van Slyke, P.; Dumont, D.J. Angiopoietin-1-induced ubiquitylation of Tie2 by c-Cbl is required for internalization and degradation. Biochem. J. 2009, 423, 375. [Google Scholar] [CrossRef]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483. [Google Scholar] [CrossRef]
- Ferrara, L.A.D.E.; Fassbender, E.; Shen, S.; Woolfenden, A.; Qiu, Y.; Poor, S.; Anderson, K.; Jaffee, B. EphA2 Stimulation of Angiogenesis is Dependent on VEGFR2. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5613. [Google Scholar]
- Wang, Y.j.; Ota, S.; Kataoka, H.; Kanamori, M.; Li, Z.y.; Band, H.; Tanaka, M.; Sugimura, H. Negative regulation of EphA2 receptor by Cbl. Biochem. Biophys. Res. Commun. 2002, 296, 214–220. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef]
- Donnini, S.; Monti, M.; Castagnini, C.; Solito, R.; Botta, M.; Schenone, S.; Giachetti, A.; Ziche, M. Pyrazolo-pyrimidine-derived c-Src inhibitor reduces angiogenesis and survival of squamous carcinoma cells by suppressing vascular endothelial growth factor production and signaling. Int. J. Cancer 2007, 120, 995–1004. [Google Scholar] [CrossRef]
- Yokouchi, M.; Kondo, T.; Sanjay, A.; Houghton, A.; Yoshimura, A.; Komiya, S.; Zhang, H.; Baron, R. Src-catalyzed Phosphorylation of c-Cbl Leads to the Interdependent Ubiquitination of Both Proteins. J. Biol. Chem. 2001, 276, 35185–35193. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.W.; Park, J.B.; Park, S.Y.; Seo, J.; Shin, S.H.; Park, J.W.; Kim, S.J.; Watanabe, M.; Chun, Y.S. The E3 ligase C-CBL inhibits cancer cell migration by neddylating the proto-oncogene c-Src. Oncogene 2018, 37, 5552–5568. [Google Scholar] [CrossRef]
- Marzilli, M.; Merz, C.N.B.; Boden, W.E.; Bonow, R.O.; Capozza, P.G.; Chilian, W.M.; DeMaria, A.N.; Guarini, G.; Huqi, A.; Morrone, D.; et al. Obstructive Coronary Atherosclerosis and Ischemic Heart Disease: An Elusive Link! J. Am. Coll. Cardiol. 2012, 60, 951. [Google Scholar] [CrossRef]
- Rafiq, K.; Kolpakov Mikhail, A.; Seqqat, R.; Guo, J.; Guo, X.; Qi, Z.; Yu, D.; Mohapatra, B.; Zutshi, N.; An, W.; et al. c-Cbl Inhibition Improves Cardiac Function and Survival in Response to Myocardial Ischemia. Circulation 2014, 129, 2031–2043. [Google Scholar] [CrossRef] [Green Version]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef]
- Yoshida, T. Molecular mechanism of choroidal neovascularization in age-related macular degeneration. Nippon Ganka Gakkai Zasshi 2007, 111, 881–891. [Google Scholar]
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: A comprehensive review. Drug Des. Dev. 2016, 10, 1857–1867. [Google Scholar] [CrossRef]
- Cabral, T.; Mello, L.G.M.; Lima, L.H.; Polido, J.; Regatieri, C.V.; Belfort, R.; Mahajan, V.B. Retinal and choroidal angiogenesis: a review of new targets. Int. J. Retin. Vitr. 2017, 3, 31. [Google Scholar] [CrossRef]
- Kwak, N.; Okamoto, N.; Wood, J.M.; Campochiaro, P.A. VEGF Is Major Stimulator in Model of Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3158–3164. [Google Scholar]
- Weathington, N.M.; Mallampalli, R.K. Emerging therapies targeting the ubiquitin proteasome system in cancer. J. Clin. Investig. 2014, 124, 6–12. [Google Scholar] [CrossRef]
- Drews, O.; Taegtmeyer, H. Targeting the ubiquitin-proteasome system in heart disease: the basis for new therapeutic strategies. Antioxid. Redox Signal. 2014, 21, 2322–2343. [Google Scholar] [CrossRef]
- Lub, S.; Maes, K.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Van Valckenborgh, E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 2015, 7, 6521–6537. [Google Scholar] [CrossRef] [Green Version]
- Kaabeche, K.; Lemonnier, J.; Le Mee, S.; Caverzasio, J.; Marie, P.J. Cbl-mediated degradation of Lyn and Fyn induced by constitutive fibroblast growth factor receptor-2 activation supports osteoblast differentiation. J. Biol. Chem. 2004, 279, 36259–36267. [Google Scholar] [CrossRef]
- Naramura, M.; Kole, H.K.; Hu, R.J.; Gu, H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 15547–15552. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.A.; Schnall, R.G.; Venter, D.J.; Barnett, L.; Bertoncello, I.; Thien, C.B.; Langdon, W.Y.; Bowtell, D.D. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol. Cell. Biol. 1998, 18, 4872–4882. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyle, C.L.; Belghasem, M.; Chitalia, V.C. c-Cbl: An Important Regulator and a Target in Angiogenesis and Tumorigenesis. Cells 2019, 8, 498. https://doi.org/10.3390/cells8050498
Lyle CL, Belghasem M, Chitalia VC. c-Cbl: An Important Regulator and a Target in Angiogenesis and Tumorigenesis. Cells. 2019; 8(5):498. https://doi.org/10.3390/cells8050498
Chicago/Turabian StyleLyle, Chimera L., Mostafa Belghasem, and Vipul C. Chitalia. 2019. "c-Cbl: An Important Regulator and a Target in Angiogenesis and Tumorigenesis" Cells 8, no. 5: 498. https://doi.org/10.3390/cells8050498
APA StyleLyle, C. L., Belghasem, M., & Chitalia, V. C. (2019). c-Cbl: An Important Regulator and a Target in Angiogenesis and Tumorigenesis. Cells, 8(5), 498. https://doi.org/10.3390/cells8050498