Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines

 , , ,
, , ,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Growth Curves
2.3. Real-Time Cell Proliferation Monitoring by xCELLigence System
2.4. Apoptosis Assay
2.5. Migration Assay
2.6. Lactate Measurements
2.7. Metabolic Flux Analysis and Mitochondrial Respiratory Complex Enzymatic Activity
2.8. Mitochondrial DNA Quantification
2.9. Live Cell Imaging of mtΔΨ and ROS
2.10. Western Blotting Analysis
2.11. Flow Cytometric Detection of Surface Markers
2.12. Reverse Transcription and Real-Time PCR Analysis
2.13. 3D Culture
2.14. Animal Studies
2.15. Statistical Analysis
3. Results
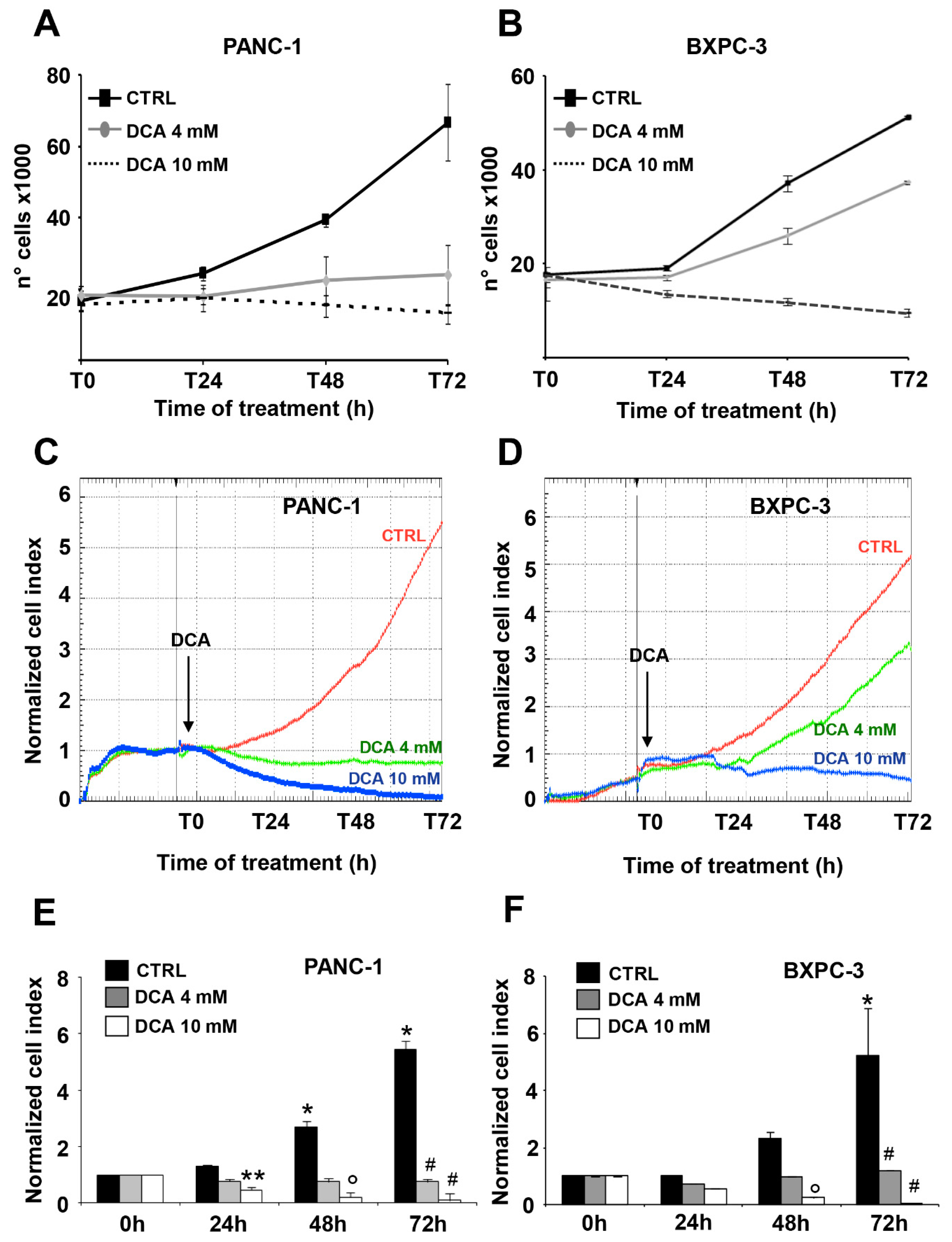
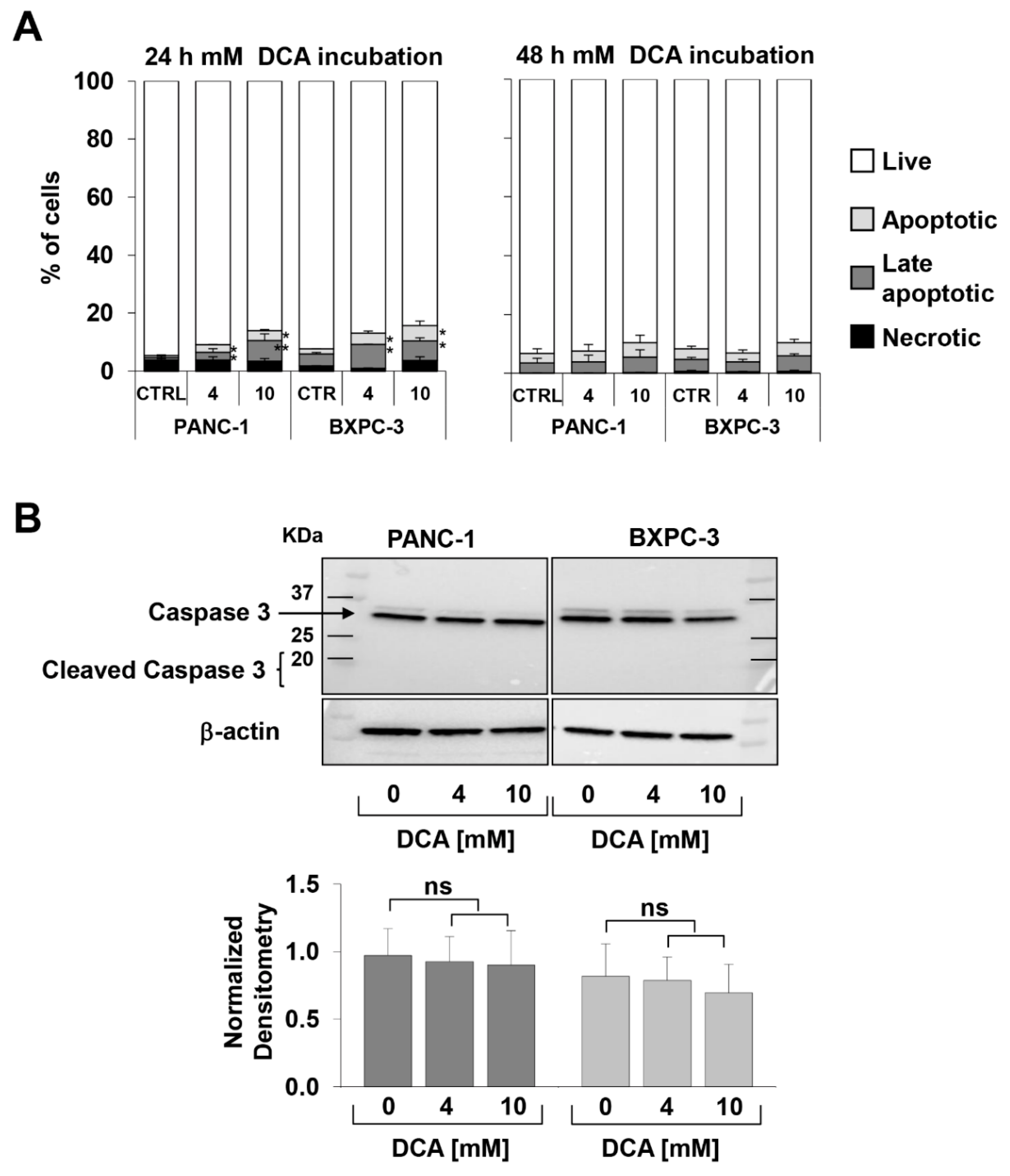
3.1. DCA Negatively Affects Cell Proliferation, Survival, and Migration in PANC-1 and BXPC-3 Cell Lines
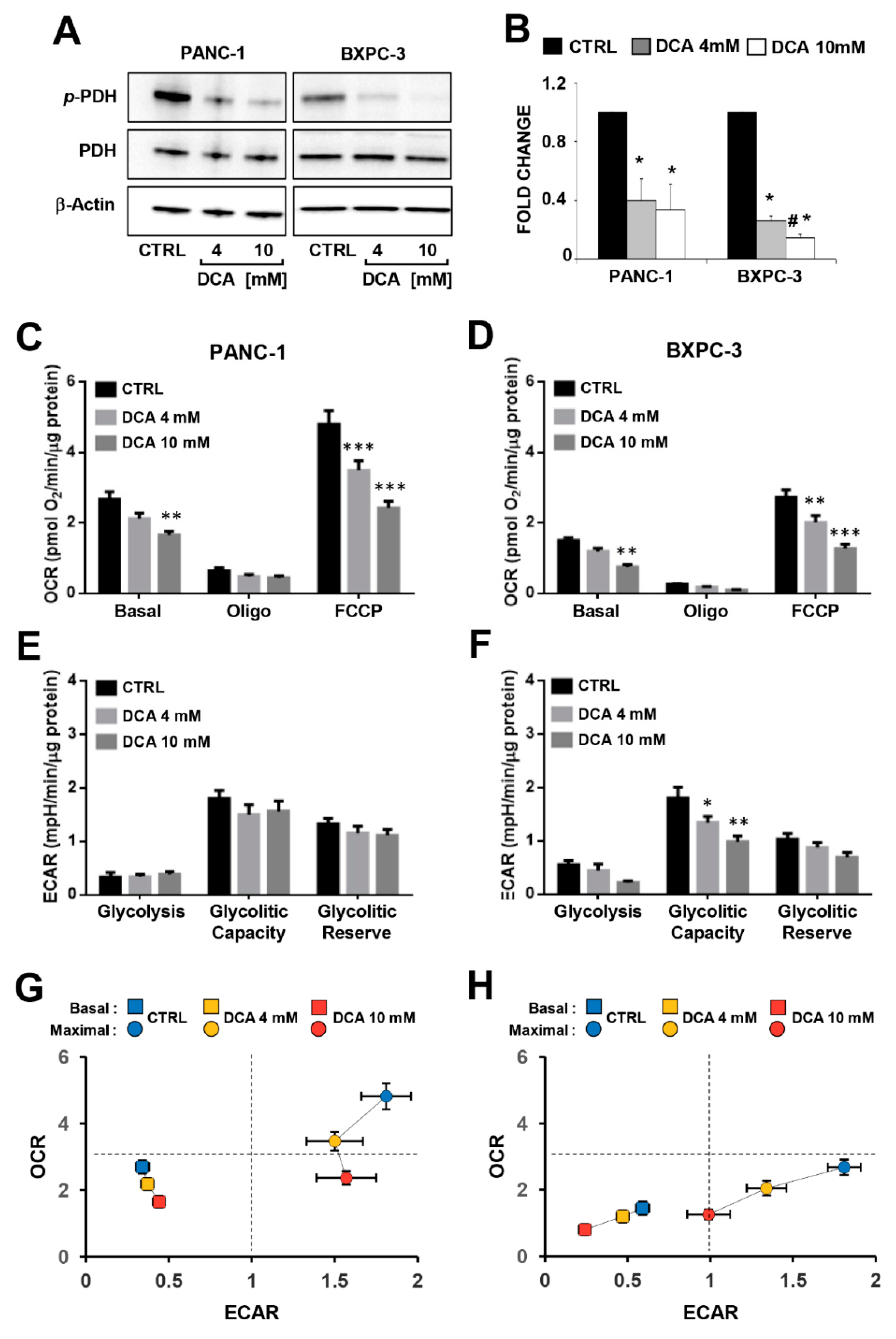
3.2. DCA Alters the Energetic Cell Metabolism in PDAC Cell Lines
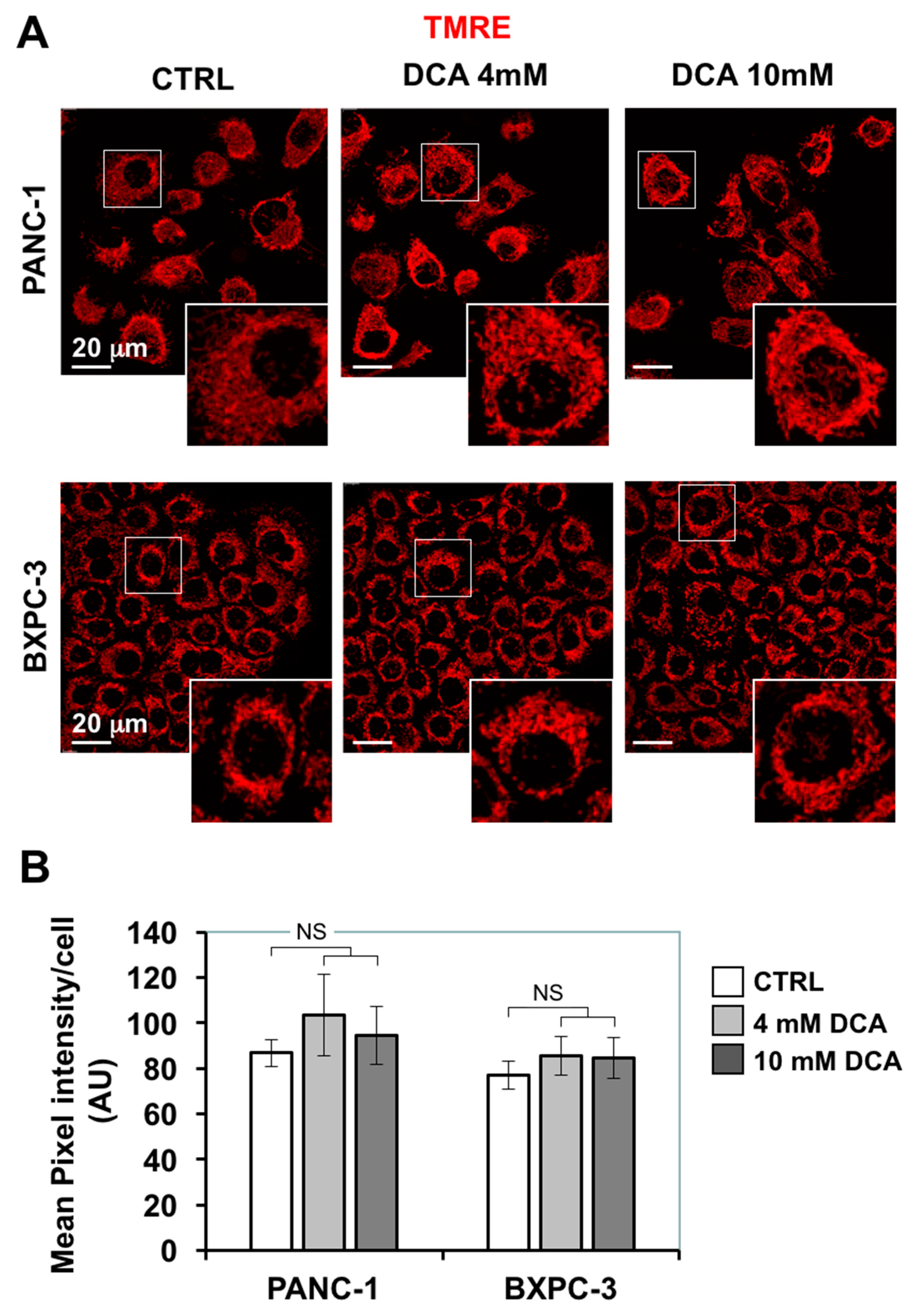
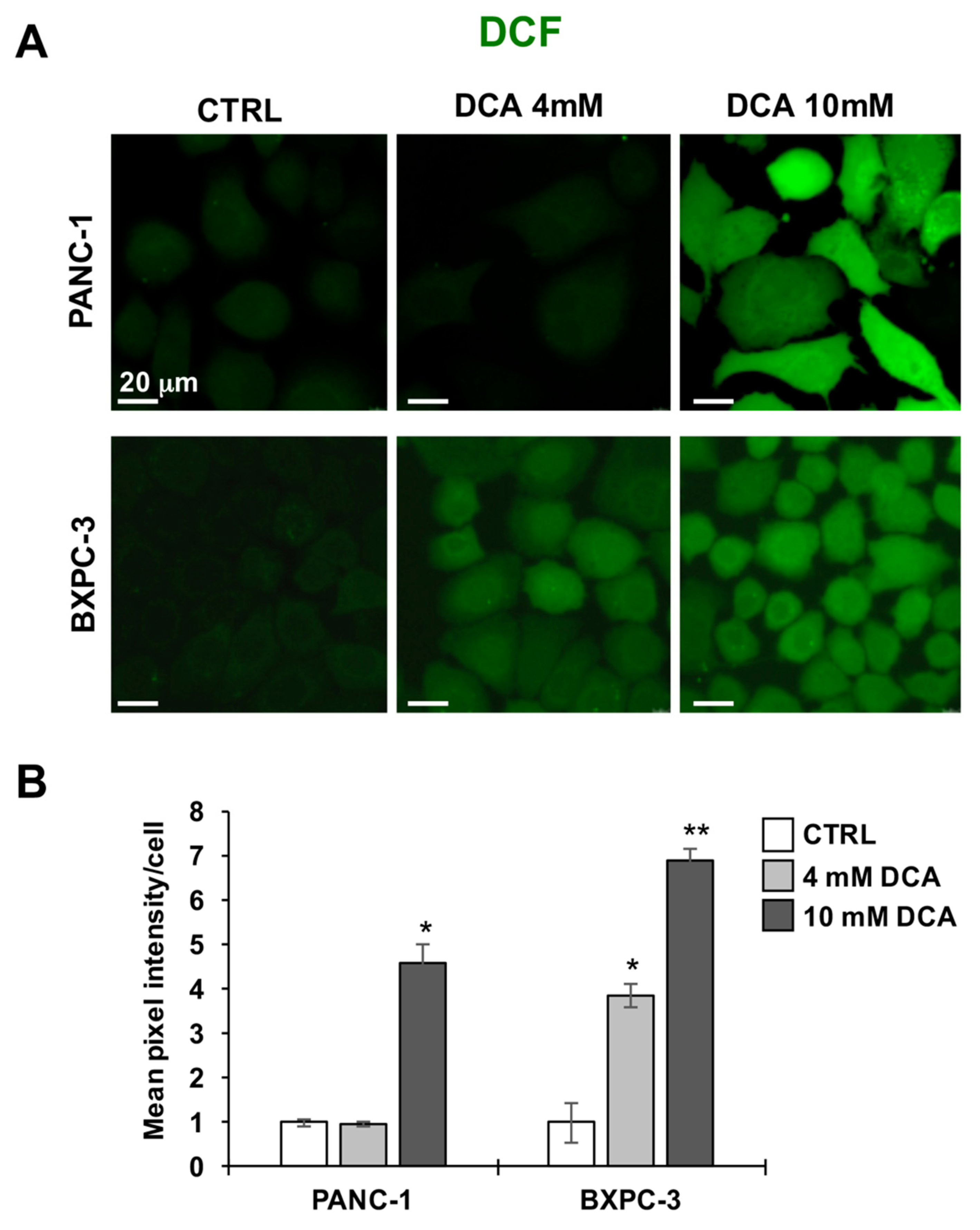
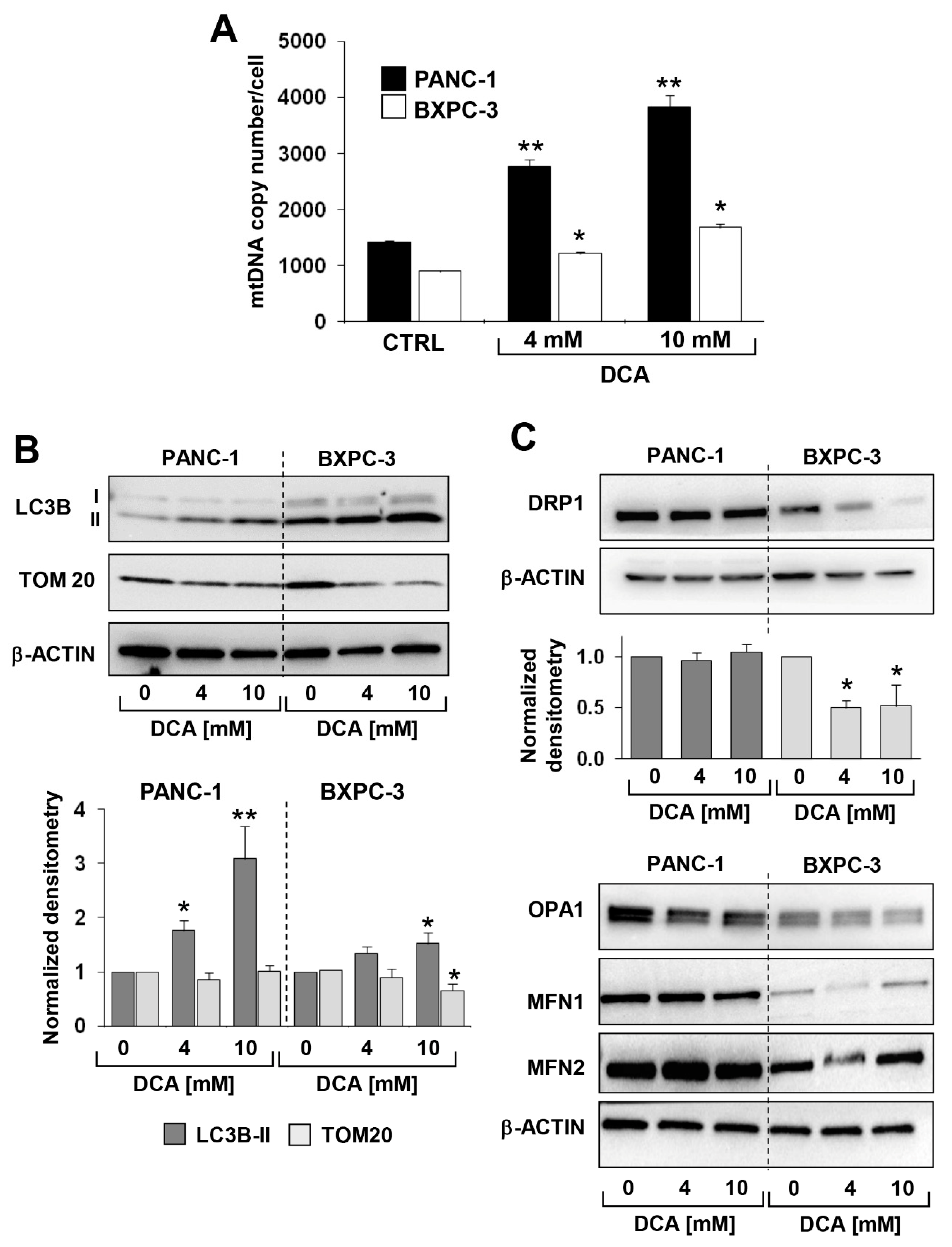
3.3. DCA Induces ROS Production in PDAC Cell Lines and Differentially Affects Mitochondrial Biogenesis and Dynamics
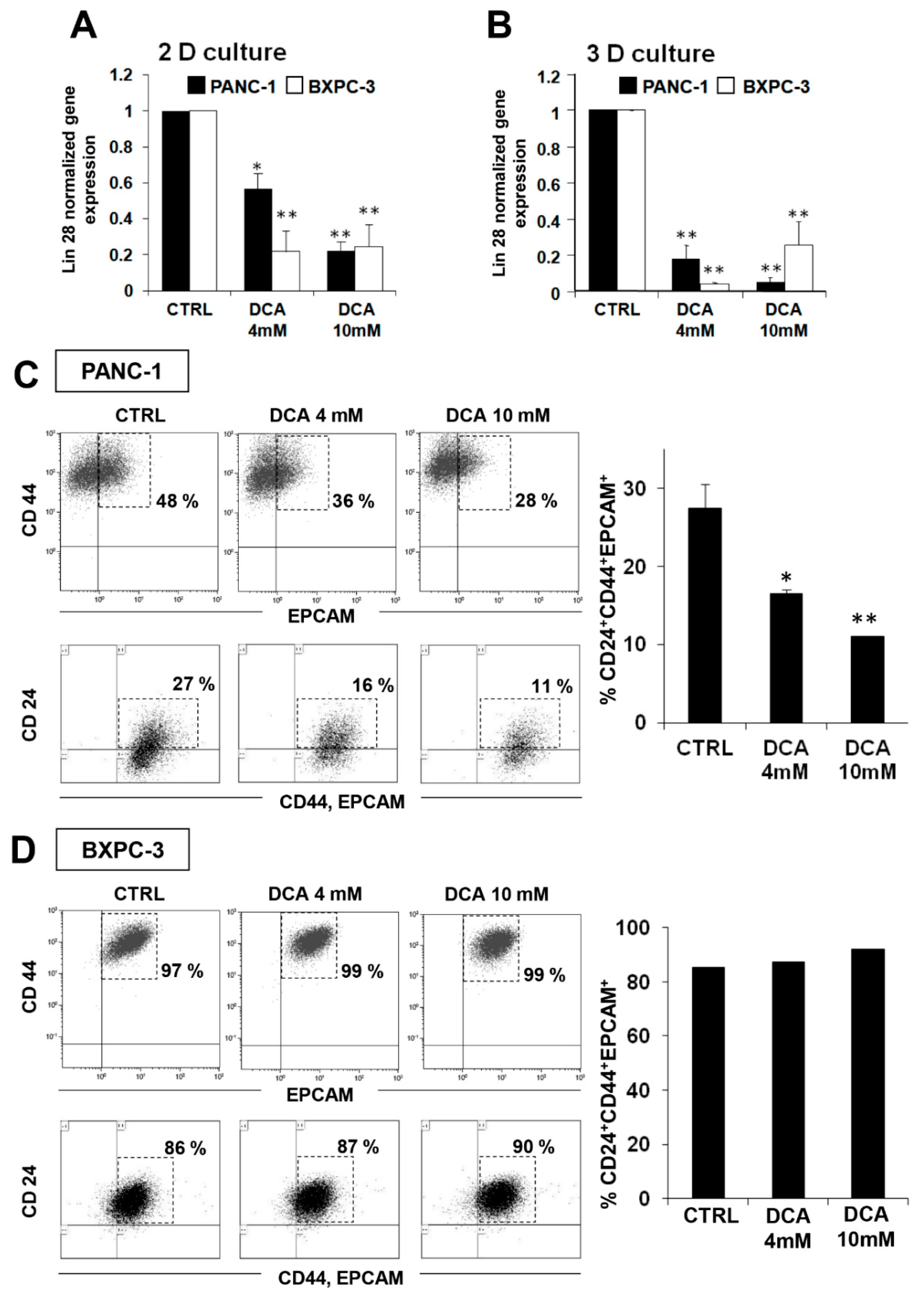
3.4. DCA Differentially Affects the Cancer Stem Cell Compartment in PDAC Cell Lines
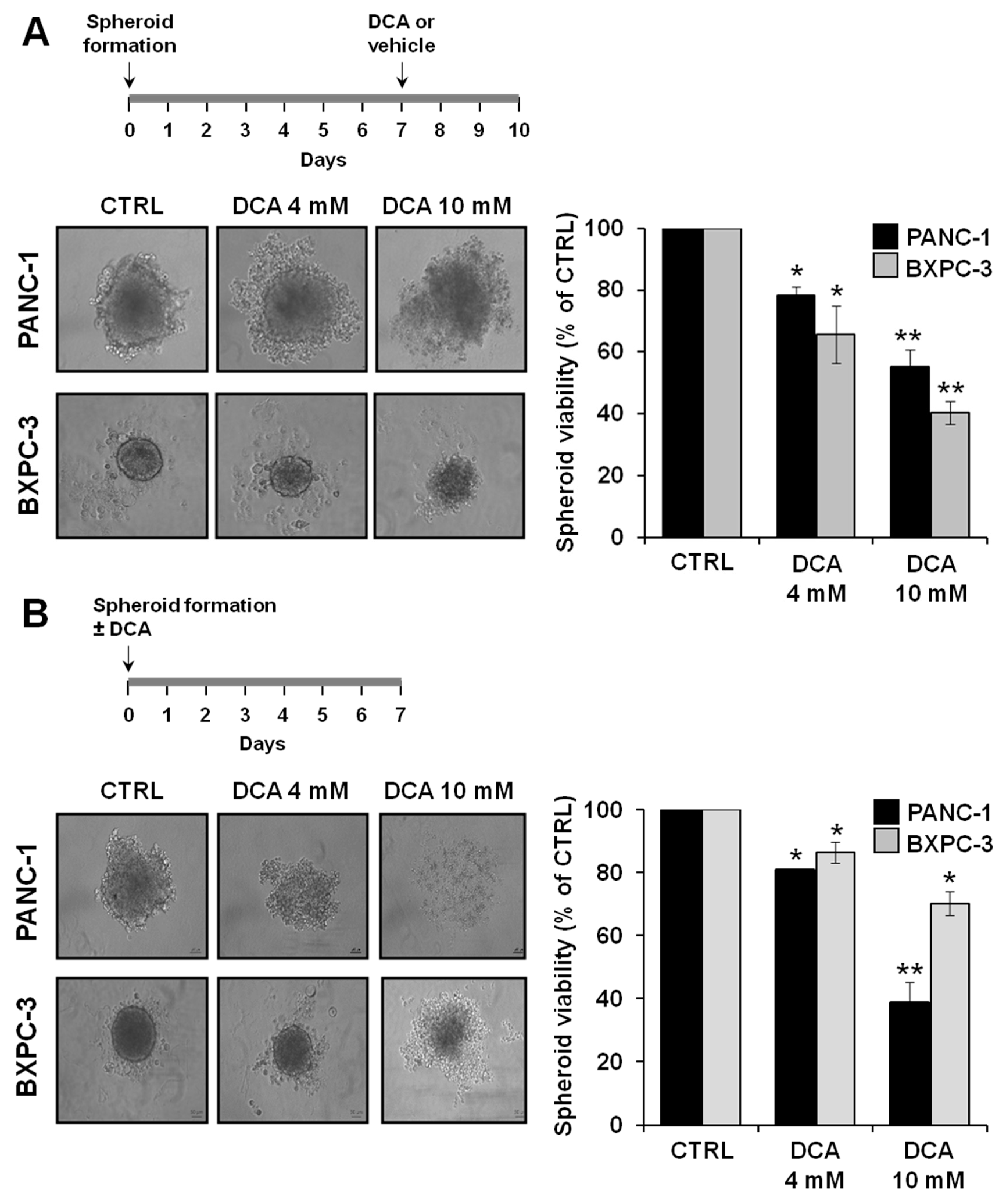
3.5. Effect of DCA on 3D Cultures
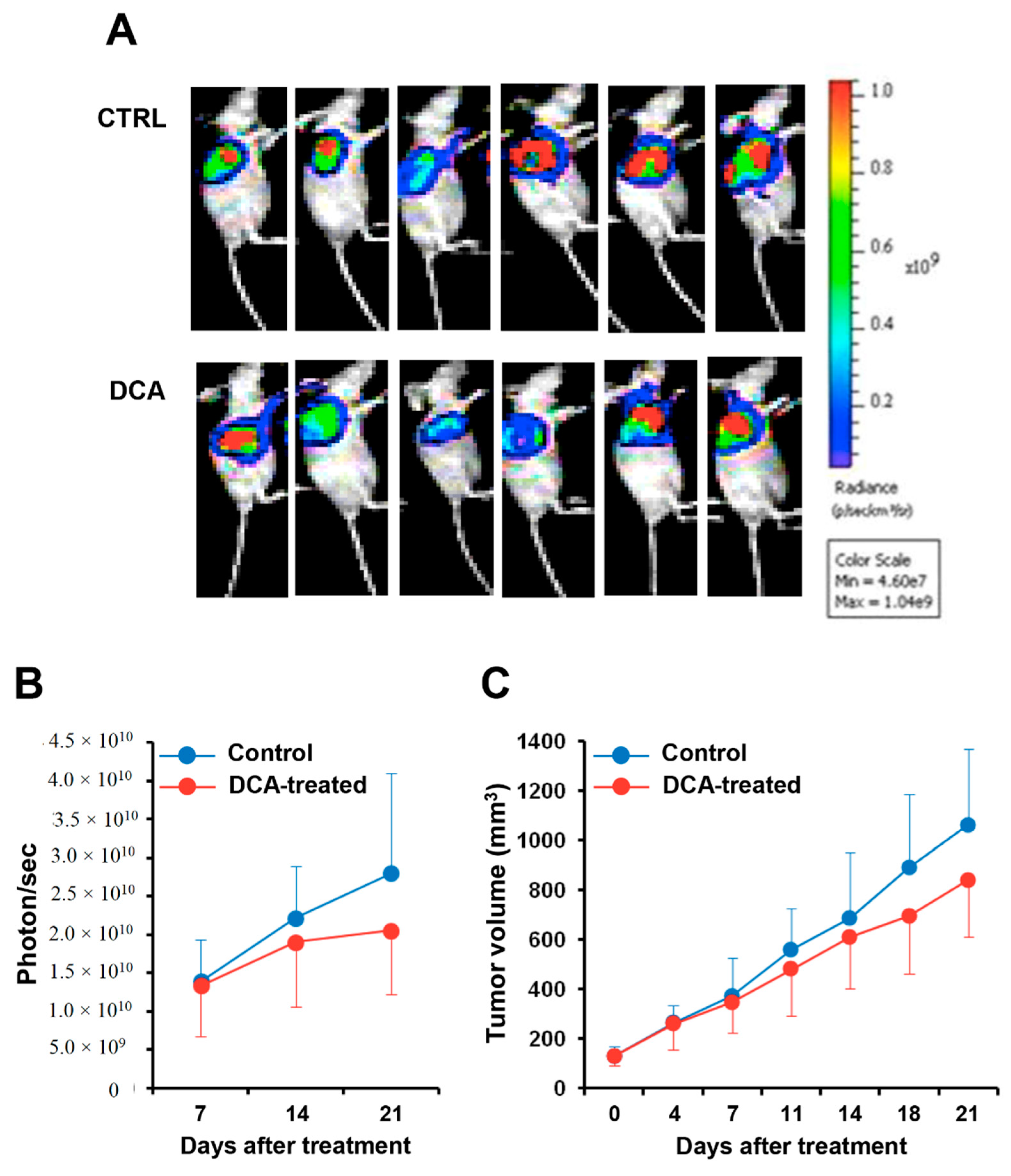
3.6. DCA Mitigates Cancer Progression in A PC Xenograft Mouse Model
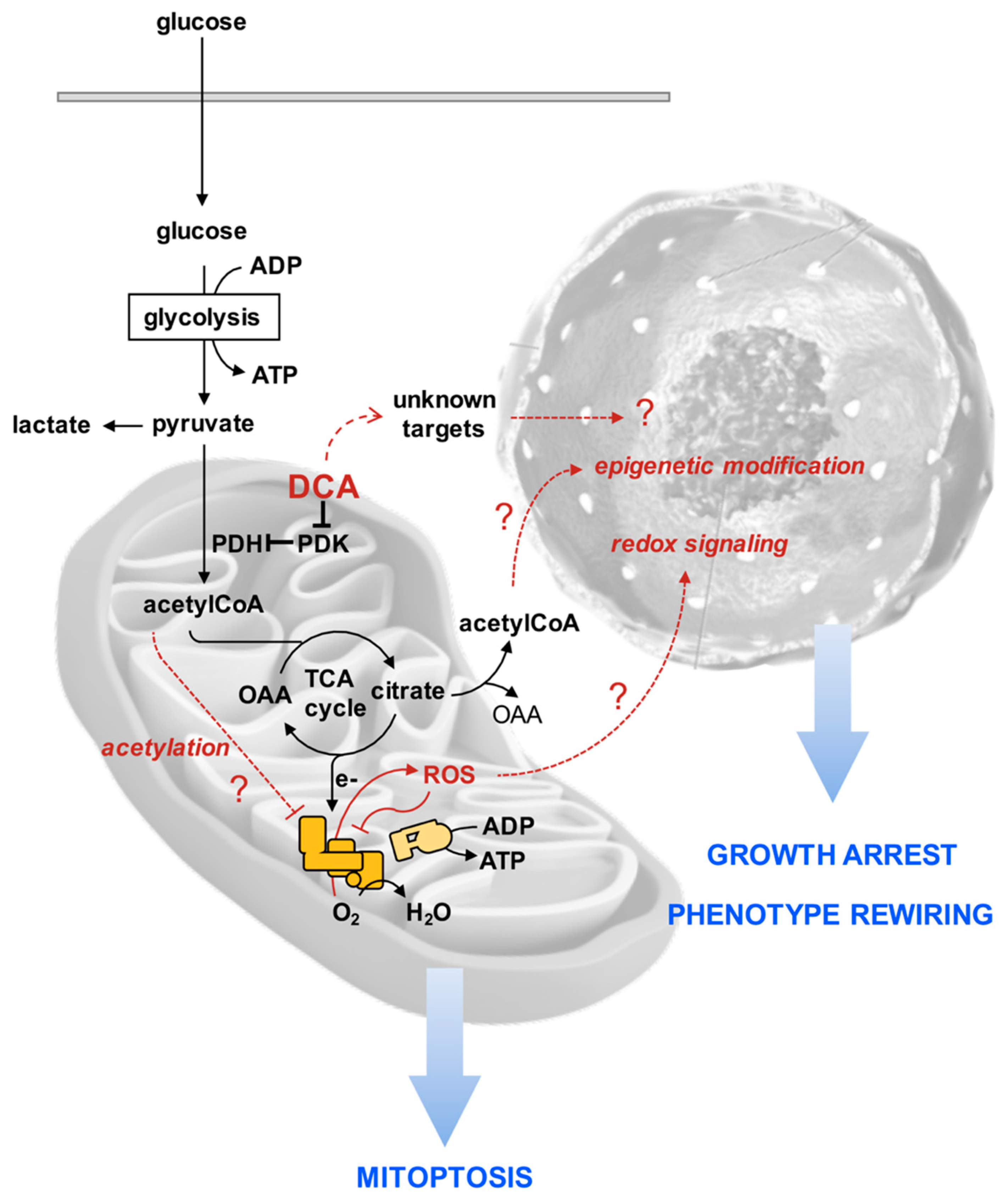
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Camelo, F.; Le, A. The Intricate Metabolism of Pancreatic Cancers. Adv. Exp. Med. Biol. 2018, 1063, 73–81. [Google Scholar] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, E1338. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Tataranni, T.; Agriesti, F.; Ruggieri, V.; Mazzoccoli, C.; Simeon, V.; Laurenzana, I.; Scrima, R.; Pazienza, V.; Capitanio, N.; Piccoli, C. Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cell lines with diverse metabolic profiles. Oncotarget 2017, 8, 41265–41281. [Google Scholar] [CrossRef]
- Ruggieri, V.; Agriesti, F.; Scrima, R.; Laurenzana, I.; Perrone, D.; Tataranni, T.; Mazzoccoli, C.; Lo Muzio, L.; Capitanio, N.; Piccoli, C. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: A metabolic perspective of treatment. Oncotarget 2015, 6, 1217–1230. [Google Scholar] [CrossRef]
- Anderson, K.M.; Jajeh, J.; Guinan, P.; Rubenstein, M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res. 2009, 29, 4579–4588. [Google Scholar]
- Chen, Y.; Cairns, R.; Papandreou, I.; Koong, A.; Denko, N.C. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS ONE 2009, 4, e7033. [Google Scholar] [CrossRef]
- Lu, X.; Zhou, D.; Hou, B.; Liu, Q.X.; Chen, Q.; Deng, X.F.; Yu, Z.B.; Dai, J.G.; Zheng, H. Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer. Cancer Manag. Res. 2018, 10, 1231–1241. [Google Scholar] [CrossRef]
- Yang, C.; Wu, T.; Qin, Y.; Qi, Y.; Sun, Y.; Kong, M.; Jiang, X.; Qin, X.; Shen, Y.; Zhang, Z. A facile doxorubicin-dichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery. Int. J. Nanomed. 2018, 13, 1281–1293. [Google Scholar] [CrossRef]
- Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; De Oliveira, E.; Kamphorst, J.J.; Rabinowitz, J.D.; Tejero, H.; Al-Shahrour, F.; Hidalgo, M.; Maitra, A.; et al. Treatment of Pancreatic Cancer Patient-Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin. Cancer Res. 2017, 23, 5639–5647. [Google Scholar] [CrossRef]
- Khan, A.; Marier, D.; Marsden, E.; Andrews, D.; Eliaz, I. A novel form of dichloroacetate therapy for patients with advanced cancer: A report of 3 cases. Altern. Ther. Health Med. 2014, 20 (Suppl. 2), 21–28. [Google Scholar]
- Hanberry, B.S.; Berger, R.; Zastre, J.A. High-dose vitamin B1 reduces proliferation in cancer cell lines analogous to dichloroacetate. Cancer Chemother. Pharm. 2014, 73, 585–594. [Google Scholar] [CrossRef]
- Lowe, A.W.; Olsen, M.; Hao, Y.; Lee, S.P.; Taek Lee, K.; Chen, X.; van de Rijn, M.; Brown, P.O. Gene expression patterns in pancreatic tumors, cells and tissues. PLoS ONE 2007, 2, e323. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef]
- Mazzoccoli, C.; Ruggieri, V.; Tataranni, T.; Agriesti, F.; Laurenzana, I.; Fratello, A.; Capitanio, N.; Piccoli, C. N-acetylaspartate (NAA) induces neuronal differentiation of SH-SY5Y neuroblastoma cell line and sensitizes it to chemotherapeutic agents. Oncotarget 2016, 7, 26235–26246. [Google Scholar] [CrossRef]
- Scrima, R.; Menga, M.; Pacelli, C.; Agriesti, F.; Cela, O.; Piccoli, C.; Cotoia, A.; De Gregorio, A.; Gefter, J.V.; Cinnella, G.; et al. Para-hydroxyphenylpyruvate inhibits the pro-inflammatory stimulation of macrophage preventing LPS-mediated nitro-oxidative unbalance and immunometabolic shift. PLoS ONE 2017, 12, e0188683. [Google Scholar] [CrossRef]
- Fredebohm, J.; Boettcher, M.; Eisen, C.; Gaida, M.M.; Heller, A.; Keleg, S.; Tost, J.; Greulich-Bode, K.M.; Hotz-Wagenblatt, A.; Lathrop, M.; et al. Establishment and characterization of a highly tumourigenic and cancer stem cell enriched pancreatic cancer cell line as a well defined model system. PLoS ONE 2012, 7, e48503. [Google Scholar] [CrossRef]
- Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD™ lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar]
- Yin, T.; Wei, H.; Gou, S.; Shi, P.; Yang, Z.; Zhao, G.; Wang, C. Cancer stem-like cells enriched in Panc-1 spheres possess increased migration ability and resistance to gemcitabine. Int. J. Mol. Sci. 2011, 12, 1595–1604. [Google Scholar] [CrossRef]
- Jun-Hao, E.T.; Gupta, R.R.; Shyh-Chang, N. Lin28 and let-7 in the Metabolic Physiology of Aging. Trends Endocrinol. Metab. 2016, 27, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ratanasirintrawoot, S.; Chandrasekaran, S.; Wu, Z.; Ficarro, S.B.; Yu, C.; Ross, C.A.; Cacchiarelli, D.; Xia, Q.; Seligson, M.; et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell 2016, 19, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Weng, M.; Jin, Y.; Yang, W.; Wu, D.; Wang, T.; Li, X. Beyond an oncogene, Lin28 is a master regulator of cancer progression. Histol. Histopathol. 2018, 33, 327–334. [Google Scholar] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Ohara, Y.; Oda, T.; Sugano, M.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Akashi, Y.; Miyamoto, R.; Kobayashi, A.; Fukunaga, K.; et al. Histological and prognostic importance of CD44(+)/CD24(+)/EpCAM(+) expression in clinical pancreatic cancer. Cancer Sci. 2013, 104, 1127–1134. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Author Correction: Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2018, 8, 4276. [Google Scholar] [CrossRef] [PubMed]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef]
- Vivanco, I. Targeting molecular addictions in cancer. Br. J. Cancer 2014, 111, 2033–2038. [Google Scholar] [CrossRef]
- Belizário, J.E.; Sangiuliano, B.A.; Perez-Sosa, M.; Neyra, J.M.; Moreira, D.F. Using Pharmacogenomic Databases for Discovering Patient-Target Genes and Small Molecule Candidates to Cancer Therapy. Front. Pharmacol. 2016, 7, 312. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer:Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125. [Google Scholar] [CrossRef]
- Fedorchuk, A.G.; Pyaskovskaya, O.N.; Gorbik, G.V.; Prokhorova, I.V.; Kolesnik, D.L.; Solyanik, G.I. Effectiveness of sodium dichloroacetate against glioma C6 depends on administration schedule and dosage. Exp. Oncol. 2016, 38, 80–83. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, X.; Wang, K.; Liu, J.; Huang, G. Dichloroacetic acid (DCA) synergizes with the SIRT2 inhibitor Sirtinol and AGK2 to enhance anti-tumor efficacy in non-small cell lung cancer. Cancer Biol. Ther. 2018, 19, 835–846. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef]
- Papandreou, I.; Goliasova, T.; Denko, N.C. Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int. J. Cancer 2011, 128, 1001–1008. [Google Scholar] [CrossRef]
- Yan, C.; Li, T.S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: Its implication in cancer therapeutics. Cell Mol. Life Sci. 2018. [Google Scholar] [CrossRef]
- Dubuis, S.; Ortmayr, K.; Zampieri, M. A framework for large-scale metabolome drug profiling links coenzyme A metabolism to the toxicity of anti-cancer drug dichloroacetate. Commun. Biol. 2018, 1, 101. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604–617. [Google Scholar] [CrossRef]
- El Sayed, S.M.; Baghdadi, H.; Ahmed, N.S.; Almaramhy, H.H.; Mahmoud, A.A.; El-Sawy, S.A.; Ayat, M.; Elshazley, M.; Abdel-Aziz, W.; Abdel-Latif, H.M.; et al. Dichloroacetate is an antimetabolite that antagonizes acetate and deprives cancer cells from its benefits: A novel evidence-based medical hypothesis. Med. Hypotheses 2019, 122, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Dixit, V.; Liu, H.; Shroads, A.L.; Henderson, G.N.; James, M.O.; Stacpoole, P.W. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab. Dispos. 2006, 34, 36–42. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, X.; Zhou, W.; Tam, K.Y. Liquid Chromatography-Tandem Mass Spectrometry Method Revealed that Lung Cancer Cells Exhibited Distinct Metabolite Profiles upon the Treatment with Different Pyruvate Dehydrogenase Kinase Inhibitors. J. Proteome Res. 2018, 17, 3012–3021. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Wang, A.X.; Dong, B.; Pu, K.F.; Yuan, L.H.; Zhu, Y.M. A new prospect in cancer therapy: Targeting cancer stem cells to eradicate cancer. Chin. J. Cancer 2012, 31, 564–572. [Google Scholar] [CrossRef]
- Ma, X.; Li, C.; Sun, L.; Huang, D.; Li, T.; He, X.; Wu, G.; Yang, Z.; Zhong, X.; Song, L.; et al. Lin28/let-7 axis regulates aerobic glycolysis and cancer progression via PDK1. Nat. Commun. 2014, 5, 5212. [Google Scholar] [CrossRef]
- Zhou, J.; Ng, S.B.; Chng, W.J. LIN28/LIN28B: An emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol. 2013, 45, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Guo, S.; Ouyang, Y.; Yin, L.; Liu, S.; Zhao, Z.; Yang, J.; Huang, W.; Qin, H.; et al. Lin28B facilitates the progression and metastasis of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 60414–60428. [Google Scholar] [CrossRef] [PubMed]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef]
- Ravi, M.; Ramesh, A.; Pattabhi, A. Contributions of 3D Cell Cultures for Cancer Research. J. Cell Physiol. 2017, 232, 2679–2697. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Gutierrez-Barrera, A.M.; Menter, D.G.; Abruzzese, J.L.; Reddy, S.A. Establishment of three-dimensional cultures of human pancreatic duct epithelial cells. Biochem. Biophys. Res. Commun. 2007, 358, 698–703. [Google Scholar] [CrossRef][Green Version]
- Dröse, S.; Brandt, U.; Wittig, I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim. Biophys. Acta 2014, 1844, 1344–1354. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- Chaudhari, P.; Ye, Z.; Jang, Y.Y. Roles of reactive oxygen species in the fate of stem cells. Antioxid. Redox Signal. 2014, 20, 1881–1890. [Google Scholar] [CrossRef]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Trisciuoglio, D.; Di Martile, M.; Del Bufalo, D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem Cells Int. 2018, 2018, 8908751. [Google Scholar] [CrossRef]
- Yadav, T.; Quivy, J.P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332–1336. [Google Scholar] [CrossRef]
- Abánades Lázaro, I.; Haddad, S.; Rodrigo-Muñoz, J.M.; Orellana-Tavra, C.; Del Pozo, V.; Fairen-Jimenez, D.; Forgan, R.S. Mechanistic Investigation into the Selective Anticancer Cytotoxicity and Immune System Response of Surface-Functionalized, Dichloroacetate-Loaded, UiO-66 Nanoparticles. ACS Appl. Mater Interfaces 2018, 10, 5255–5268. [Google Scholar] [CrossRef] [PubMed]
- Petruzzella, E.; Sirota, R.; Solazzo, I.; Gandin, V.; Gibson, D. Triple action Pt(iv) derivatives of cisplatin: A new class of potent anticancer agents that overcome resistance. Chem. Sci. 2018, 9, 4299–4307. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tataranni, T.; Agriesti, F.; Pacelli, C.; Ruggieri, V.; Laurenzana, I.; Mazzoccoli, C.; Della Sala, G.; Panebianco, C.; Pazienza, V.; Capitanio, N.; et al. Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells 2019, 8, 478. https://doi.org/10.3390/cells8050478
Tataranni T, Agriesti F, Pacelli C, Ruggieri V, Laurenzana I, Mazzoccoli C, Della Sala G, Panebianco C, Pazienza V, Capitanio N, et al. Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells. 2019; 8(5):478. https://doi.org/10.3390/cells8050478
Chicago/Turabian StyleTataranni, Tiziana, Francesca Agriesti, Consiglia Pacelli, Vitalba Ruggieri, Ilaria Laurenzana, Carmela Mazzoccoli, Gerardo Della Sala, Concetta Panebianco, Valerio Pazienza, Nazzareno Capitanio, and et al. 2019. "Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines" Cells 8, no. 5: 478. https://doi.org/10.3390/cells8050478
APA StyleTataranni, T., Agriesti, F., Pacelli, C., Ruggieri, V., Laurenzana, I., Mazzoccoli, C., Della Sala, G., Panebianco, C., Pazienza, V., Capitanio, N., & Piccoli, C. (2019). Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells, 8(5), 478. https://doi.org/10.3390/cells8050478