Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring


{kind=link}
Abstract
1. Introduction
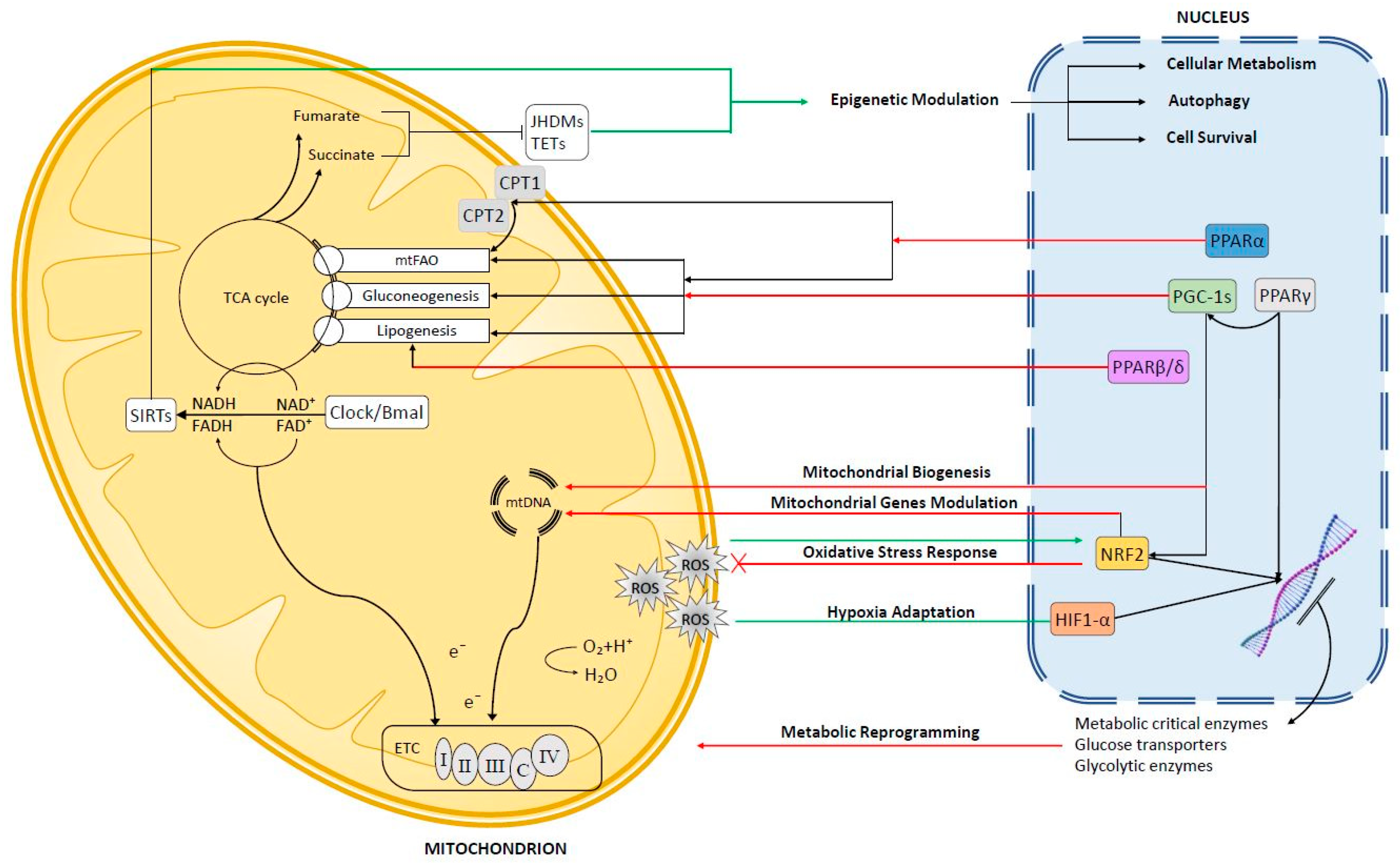
2. Anterograde Signaling
2.1. Peroxisome Proliferator-Activated Receptors (PPARs)
2.1.1. PPARα
2.1.2. PPARβ/δ
2.1.3. PPARγ
2.2. Peroxisome Proliferator Activated Receptor Co-Activators (PGC-1s)
3. Mitochondrial Retrograde Signaling in Hepatocellular Carcinoma (HCC)
3.1. Reactive Oxygen Species (ROS)-Dependent Retrograde Signaling
3.1.1. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)
3.1.2. Hypoxia-Inducible Transcription Factor (Hif1-α)
3.2. NAD+-Dependent Retrograde Signaling
3.3. Mitochondrial Metabolism and Epigenetic Regulation in HCC
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| OXPHOS | Oxidative phosphorylation system |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| HCC | Hepatocellular carcinoma |
| mTOR | Mammalian target of rapamycin |
| PI3K | Phosphoinositide 3-kinase |
| AKT | Protein kinase B |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| SREBP-1 | Sterol regulatory element-binding protein 1 |
| PPARs | Peroxisome Proliferator Activated Receptor |
| PGC-1s | Peroxisome proliferator-activated receptor gamma coactivator 1 |
| TZD | Thiazolidinedione |
| FA | Fatty acid |
| FAO | Fatty acid oxidation |
| CPT-1 | Carnitine Palmitoyltransferase 1 |
| CPT-2 | Carnitine Palmitoyltransferase 2 |
| TCA | Tricarboxylic acid cycle |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| UCP | Uncoupling Protein |
| DEN | Diethylnitrosamine |
| NF-kB | Nuclear Factor Kappa B |
| HADHA | Hydroxyacyl-CoA Dehydrogenase Trifunctional Multienzyme Complex Subunit Alpha |
| MLYCD | Malonyl-CoA Decarboxylase |
| ACC1 | Acetyl-CoA carboxylase 1 |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| CTNNB1 | Catenin Beta 1 |
| ACAC | Acetyl-CoA Carboxylase |
| GLUT2 | Glucose transporter 2 |
| GK | Glycerol Kinase |
| FASN | Fatty acid synthase |
| ACC2 | Acetyl-CoA carboxylase 2 |
| SCD1 | Stearoyl-CoA Desaturase 1 |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| FASN | Fatty Acid Synthase |
| HNF-4 | Hepatocyte Nuclear Factor 4 Alpha |
| PPP | Pentose-Phosphate-Pathway |
| GLS1 | Glutaminase |
| PTEN | Phosphatase and Tensin Homolog |
| HK | Hexokinase |
| PKM2 | Pyruvate Kinase M2 |
| MEK | Mitogen-activated Protein Kinase Kinase |
| ERK | Extracellular signal–regulated Kinases |
| PFKFB4 | 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 4 |
| MCAD | Medium-chain acyl-CoA dehydrogenase |
| ERR | Estrogen Related Receptors |
| NRF | Nuclear Respiratory Factor |
| GR | Glucocorticoid receptor |
| ER | Estrogen Receptor |
| TFB1M | Transcription Factor B1, Mitochondrial |
| TFB2M | Transcription Factor B2, Mitochondrial |
| TFAM | Transcription Factor A, Mitochondrial |
| HNFα | Hepatocyte Nuclear Factor Alpha |
| FOXO1 | Forkhead Box O1 |
| CREB | CAMP Responsive Element Binding Protein |
| PEPCK | Phosphoenolpyruvate Carboxykinase |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| LXRα | Liver X Receptor Alpha |
| SIRT1 | Sirtuin 1 |
| YAP | Yes-associated protein 1 |
| JNK | c-Jun N-terminal Kinases |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| CaMKII | Ca2+/calmodulin-dependent protein kinases II |
| NRF2 | Nuclear factor erythroid 2 related factor 2 |
| AREs | Antioxidant response elements |
| KEAP1 | Kelchlike ECH-associated protein 1 |
| CUL3 | Cullin 3 |
| RBX1 | Ring-Box 1 |
| PKCδ | Protein kinase C delta |
| sMAF | small Maf proteins |
| NQO-1 | NADPH quinone oxidoreductase |
| GSTs | Glutathione S-transferases |
| HMOX1 | Heme oxygenase-1 |
| HK II | Hexokinase 2 |
| CS | Citrate Synthase |
| TRAP1 | TNF Receptor Associated Protein 1 |
| HIF-1α | Hypoxia Inducible Factor 1 Subunit Alpha |
| HIFs | Hypoxia Inducible Factors |
| HIF-1β | Hypoxia Inducible Factor 1 Subunit Beta |
| PHD2 | α-ketoglutarate-dependent prolyl hydroxylase 2 PHD2 |
| VHL | von Hippel-Lindau ubiquitin ligase |
| HREs | Hypoxia Response Elements |
| HDAC | NAD+-dependent histone deacetylases |
| LDH | Lactate dehydrogenase |
| FXR | Farnesoid X Receptor |
| CLOCK | Clock Circadian Regulator |
| BMAL | Brain and Muscle ARNT-Like |
| PER2 | Period Circadian Regulator 2 |
| GCN5 | General control of amino acid synthesis 5 |
| MFN-1 | Mitofusin 1 |
| MFN-2 | Mitofusin 2 |
| FAK | Focal Adhesion Kinase |
| Drp1 | Dynamin-1-like Protein |
| GSK-5b | Glycogen Synthase Kinase 5 Beta |
| SOD2 | Superoxide Dismutase 2 |
| MMP2 | Matrix Metallopeptidase 2 |
| GSTP1 | Glutathione S-transferase pi 1 |
| LCAD | Long-chain acyl-coenzyme A (acyl-CoA) dehydrogenase |
| LIHC | Liver Hepatocellular Carcinoma |
| RASSF1 | Ras Association Domain Family Member 1 |
| GATA4 | GATA Binding Protein 4 |
| CDKL2 | Cyclin Dependent Kinase Like 2 |
| FH | Fumarate dehydrogenase |
| SDH | Succinate dehydrogenase |
| JHDMs | Jumonji-C histone demethylases |
| TET | Ten-eleven translocation methylcytosine dioxygenase |
References
- WARBURG, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- WARBURG, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadel, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–786. [Google Scholar] [CrossRef]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany. NY) 2016, 8, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621. [Google Scholar] [CrossRef]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 1–15. [Google Scholar] [CrossRef]
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Zender, L.; Villanueva, A.; Tovar, V.; Sia, D.; Chiang, D.Y.; Llovet, J.M. Cancer gene discovery in hepatocellular carcinoma. J. Hepatol. 2010, 52, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef]
- Watanabe, S.; Horie, Y.; Suzuki, A. Hepatocyte-specific Pten-deficient mice as a novel model for nonalcoholic steatohepatitis and hepatocellular carcinoma. Hepatol. Res. 2005, 33, 161–166. [Google Scholar] [CrossRef]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Li, L.; Che, L.; Tharp, K.M.; Park, H.M.; Pilo, M.G.; Cao, D.; Cigliano, A.; Latte, G.; Xu, Z.; Ribback, S.; et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 2016, 63, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef]
- Xu, Z.; Hu, J.; Cao, H.; Pilo, M.G.; Cigliano, A.; Shao, Z.; Xu, M.; Ribback, S.; Dombrowski, F.; Calvisi, D.F.; et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp. Mol. Med. 2018, 50, e417. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef]
- Lee, H.C.; Wei, Y.H. Mitochondrial DNA instability and metabolic shift in human cancers. Int. J. Mol. Sci. 2009, 10, 674–701. [Google Scholar] [CrossRef]
- Nomoto, S.; Yamashita, K.; Koshikawa, K.; Nakao, A.; Sidransky, D. Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin. Cancer Res. 2002, 8, 481–487. [Google Scholar]
- Lee, H.C.; Li, S.H.; Lin, J.C.; Wu, C.C.; Yeh, D.C.; Wei, Y.H. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat. Res. 2004, 547, 71–78. [Google Scholar] [CrossRef]
- Tamori, A.; Nishiguchi, S.; Nishikawa, M.; Kubo, S.; Koh, N.; Hirohashi, K.; Shiomi, S.; Inoue, M. Correlation between clinical characteristics and mitochondrial D-loop DNA mutations in hepatocellular carcinoma. J. Gastroenterol. 2004, 39, 1063–1068. [Google Scholar] [CrossRef]
- Okochi, O.; Hibi, K.; Uemura, T.; Inoue, S.; Takeda, S.; Kaneko, T.; Nakao, A. Detection of mitochondrial DNA alterations in the serum of hepatocellular carcinoma patients. Clin. Cancer Res. 2002, 8, 2875–2878. [Google Scholar]
- Yin, P.H.; Lee, H.C.; Chau, G.Y.; Wu, Y.T.; Li, S.H.; Lui, W.Y.; Wei, Y.H.; Liu, T.Y.; Chi, C.W. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br. J. Cancer 2004, 90, 2390–2396. [Google Scholar] [CrossRef]
- Guo, Z.S.; Jin, C.L.; Yao, Z.J.; Wang, Y.M.; Xu, B.T. Analysis of the Mitochondrial 4977 Bp Deletion in Patients with Hepatocellular Carcinoma. Balkan. J. Med. Genet. 2017, 20, 81–86. [Google Scholar] [CrossRef]
- Qiao, L.; Ru, G.; Mao, Z.; Wang, C.; Nie, Z.; Li, Q.; Huang-Yang, Y.; Zhu, L.; Liang, X.; Yu, J.; et al. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017, 8, 84373–84383. [Google Scholar] [CrossRef]
- Ke, S.; Chen, S.; Dong, Z.; Hong, C.S.; Zhang, Q.; Tang, L.; Yang, P.; Zhai, J.; Yan, H.; Shen, F.; et al. Erythrocytosis in hepatocellular carcinoma portends poor prognosis by respiratory dysfunction secondary to mitochondrial DNA mutations. Hepatology 2017, 65, 134–151. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Hou, Y.; Wang, H.; Wang, K.; Xiang, H.; Wan, X.; Xia, Y.; Li, J.; Wei, W.; Xu, S.; et al. Aspartate beta-hydroxylase disrupts mitochondrial DNA stability and function in hepatocellular carcinoma. Oncogenesis 2017, 6, e362. [Google Scholar] [CrossRef]
- Yu, C.; Wang, X.; Huang, L.; Tong, Y.; Chen, L.; Wu, H.; Xia, Q.; Kong, X. Deciphering the Spectrum of Mitochondrial DNA Mutations in Hepatocellular Carcinoma Using High-Throughput Sequencing. Gene Expr. 2018, 18, 125–134. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, E.; Marin, J.J.; Perez, M.J. The expression of genes involved in hepatocellular carcinoma chemoresistance is affected by mitochondrial genome depletion. Mol. Pharm. 2014, 11, 1856–1868. [Google Scholar] [CrossRef]
- Marin, J.J.; Hernandez, A.; Revuelta, I.E.; Gonzalez-Sanchez, E.; Gonzalez-Buitrago, J.M.; Perez, M.J. Mitochondrial genome depletion in human liver cells abolishes bile acid-induced apoptosis: Role of the Akt/mTOR survival pathway and Bcl-2 family proteins. Free Radic. Biol. Med. 2013, 61, 218–228. [Google Scholar] [CrossRef]
- Li, X.; Guo, X.; Li, D.; Du, X.; Yin, C.; Chen, C.; Fang, W.; Bian, Z.; Zhang, J.; Li, B.; et al. Multi-regional sequencing reveals intratumor heterogeneity and positive selection of somatic mtDNA mutations in hepatocellular carcinoma and colorectal cancer. Int. J. Cancer 2018, 143, 1143–1152. [Google Scholar] [CrossRef]
- Gwak, G.Y.; Lee, D.H.; Moon, T.G.; Choi, M.S.; Lee, J.H.; Koh, K.C.; Paik, S.W.; Joh, J.W.; Yoo, B.C. The correlation of hepatitis B virus pre-S mutation with mitochondrial D-loop mutations and common deletions in hepatocellular carcinoma. Hepatogastroenterology 2011, 58, 522–528. [Google Scholar]
- Vivekanandan, P.; Daniel, H.; Yeh, M.M.; Torbenson, M. Mitochondrial mutations in hepatocellular carcinomas and fibrolamellar carcinomas. Mod. Pathol. 2010, 23, 790–798. [Google Scholar] [CrossRef][Green Version]
- Szalowska, E.; Tesfay, H.A.; van Hijum, S.A.; Kersten, S. Transcriptomic signatures of peroxisome proliferator-activated receptor alpha (PPARalpha) in different mouse liver models identify novel aspects of its biology. BMC. Genom. 2014, 15, 1106. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR. Res. 2010, 2010. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef]
- Villarroya, F.; Iglesias, R.; Giralt, M. PPARs in the Control of Uncoupling Proteins Gene Expression. PPAR. Res. 2007, 2007. [Google Scholar] [CrossRef]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Mello, T.; Materozzi, M.; Galli, A. PPARs and Mitochondrial Metabolism: From NAFLD to HCC. PPAR. Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef]
- Zhang, N.; Chu, E.S.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.; et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-kappaB signaling pathway. Oncotarget 2014, 5, 8330–8340. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Holness, M.J. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch. Physiol Biochem. 2006, 112, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Reiss, K. Peroxisome proliferator activated receptor alpha ligands as anticancer drugs targeting mitochondrial metabolism. Curr. Pharm. Biotechnol. 2013, 14, 342–356. [Google Scholar] [CrossRef]
- Xu, P.; Oosterveer, M.H.; Stein, S.; Demagny, H.; Ryu, D.; Moullan, N.; Wang, X.; Can, E.; Zamboni, N.; Comment, A.; et al. LRH-1-dependent programming of mitochondrial glutamine processing drives liver cancer. Genes Dev. 2016, 30, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Oppliger, W.; Albert, V.; Robitaille, A.M.; Trapani, F.; Quagliata, L.; Fuhrer, T.; Sauer, U.; Terracciano, L.; Hall, M.N. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc. Natl. Acad. Sci. USA 2014, 111, 11592–11599. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Becker, J.R.; Hanse, E.A.; Mashek, D.G.; Mashek, M.T.; Hendrickson, A.M.; Mullany, L.K.; Albrecht, J.H. Cyclin D1 represses peroxisome proliferator-activated receptor alpha and inhibits fatty acid oxidation. Oncotarget 2016, 7, 47674–47686. [Google Scholar] [CrossRef]
- Tanaka, M.; Masaki, Y.; Tanaka, K.; Miyazaki, M.; Kato, M.; Sugimoto, R.; Nakamura, K.; Aishima, S.; Shirabe, K.; Nakamuta, M.; et al. Reduction of fatty acid oxidation and responses to hypoxia correlate with the progression of de-differentiation in HCC. Mol. Med. Rep. 2013, 7, 365–370. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kim, N.H.; Zhao, Z.S.; Cha, B.S.; Kim, Y.S. Peroxisomal-proliferator-activated receptor alpha activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: A key regulation of malonyl-CoA level. Biochem. J. 2004, 378, 983–990. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182. [Google Scholar] [CrossRef]
- Senni, N.; Savall, M.; Cabrerizo, G.D.; ves-Guerra, M.C.; Sartor, C.; Lagoutte, I.; Gougelet, A.; Terris, B.; Gilgenkrantz, H.; Perret, C.; et al. beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut 2019, 68, 322–334. [Google Scholar] [CrossRef]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Wu, J.Y.; Li, H.; Yan, J.P.; Guo, M.Y.; Wo, Y.B.; Lou, Y.J. PPAR-beta facilitating maturation of hepatic-like tissue derived from mouse embryonic stem cells accompanied by mitochondriogenesis and membrane potential retention. J. Cell Biochem. 2010, 109, 498–508. [Google Scholar]
- Liu, H.X.; Fang, Y.; Hu, Y.; Gonzalez, F.J.; Fang, J.; Wan, Y.J. PPARbeta Regulates Liver Regeneration by Modulating Akt and E2f Signaling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Farra, R.; Grassi, G.; Tonon, F.; Abrami, M.; Grassi, M.; Pozzato, G.; Fiotti, N.; Forte, G.; Dapas, B. The Role of the Transcription Factor E2F1 in Hepatocellular Carcinoma. Curr. Drug Deliv. 2017, 14, 272–281. [Google Scholar]
- Ladu, S.; Calvisi, D.F.; Conner, E.A.; Farina, M.; Factor, V.M.; Thorgeirsson, S.S. E2F1 inhibits c-Myc-driven apoptosis via PIK3CA/Akt/mTOR and COX-2 in a mouse model of human liver cancer. Gastroenterology 2008, 135, 1322–1332. [Google Scholar] [CrossRef]
- Blanchet, E.; Annicotte, J.S.; Lagarrigue, S.; Aguilar, V.; Clape, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef]
- Vacca, M.; D’Amore, S.; Graziano, G.; D’Orazio, A.; Cariello, M.; Massafra, V.; Salvatore, L.; Martelli, N.; Murzilli, S.; Lo, S.G.; et al. Clustering nuclear receptors in liver regeneration identifies candidate modulators of hepatocyte proliferation and hepatocarcinoma. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Kim, M.J.; Choi, Y.K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.G.; Jeon, H.J.; Kim, J.H.; Kim, J.G.; et al. PPARdelta Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Ceni, E.; Crabb, D.W.; Mello, T.; Salzano, R.; Grappone, C.; Milani, S.; Surrenti, E.; Surrenti, C.; Casini, A. Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic cancer cells via PPARgamma independent mechanisms. Gut 2004, 53, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Ceni, E.; Mello, T.; Polvani, S.; Tarocchi, M.; Buccoliero, F.; Lisi, F.; Cioni, L.; Ottanelli, B.; Foresta, V.; et al. Thiazolidinediones inhibit hepatocarcinogenesis in hepatitis B virus-transgenic mice by peroxisome proliferator-activated receptor gamma-independent regulation of nucleophosmin. Hepatology 2010, 52, 493–505. [Google Scholar] [CrossRef]
- Gao, M.; Liu, D. CRISPR/Cas9-based Pten knock-out and Sleeping Beauty Transposon-mediated Nras knock-in induces hepatocellular carcinoma and hepatic lipid accumulation in mice. Cancer Biol. Ther. 2017, 18, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef]
- Shu, Y.; Lu, Y.; Pang, X.; Zheng, W.; Huang, Y.; Li, J.; Ji, J.; Zhang, C.; Shen, P. Phosphorylation of PPARgamma at Ser84 promotes glycolysis and cell proliferation in hepatocellular carcinoma by targeting PFKFB4. Oncotarget 2016, 7, 76984–76994. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Q.; Qi, J.; Wang, W.; Zhang, D.; Li, Z.; Qin, C. lncRNA Ftx promotes aerobic glycolysis and tumor progression through the PPARgamma pathway in hepatocellular carcinoma. Int. J. Oncol. 2018, 53, 551–566. [Google Scholar] [PubMed]
- Liu, Z.; Wang, Y.; Dou, C.; Sun, L.; Li, Q.; Wang, L.; Xu, Q.; Yang, W.; Liu, Q.; Tu, K. MicroRNA-1468 promotes tumor progression by activating PPAR-gamma-mediated AKT signaling in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37. [Google Scholar] [CrossRef]
- Chang, J.S.; Ha, K. A truncated PPAR gamma 2 localizes to mitochondria and regulates mitochondrial respiration in brown adipocytes. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARgamma for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Leveille, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-gamma Coactivator 1alpha to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef]
- Galmes-Pascual, B.M.; Nadal-Casellas, A.; Bauza-Thorbrugge, M.; Sbert-Roig, M.; Garcia-Palmer, F.J.; Proenza, A.M.; Gianotti, M.; Llado, I. 17beta-estradiol improves hepatic mitochondrial biogenesis and function through PGC1B. J. Endocrinol. 2017, 232, 297–308. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef]
- Stiles, A.R.; Simon, M.T.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jones, A.W.; Fassone, E.; Sweeney, M.G.; Lebiedzinska, M.; Suski, J.M.; Wieckowski, M.R.; Tajeddine, N.; Hargreaves, I.P.; Yasukawa, T.; et al. PGC-1beta mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene 2013, 32, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, J.; Lu, N.; Fang, F.; Liu, J.; Wong, C.W. Mitogen-activated protein kinase kinases promote mitochondrial biogenesis in part through inducing peroxisome proliferator-activated receptor gamma coactivator-1beta expression. Biochim. Biophys. Acta 2011, 1813, 1239–1244. [Google Scholar] [CrossRef][Green Version]
- Lin, J.; Tarr, P.T.; Yang, R.; Rhee, J.; Puigserver, P.; Newgard, C.B.; Spiegelman, B.M. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Biol. Chem. 2003, 278, 30843–30848. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef]
- Lin, J.; Yang, R.; Tarr, P.T.; Wu, P.H.; Handschin, C.; Li, S.; Yang, W.; Pei, L.; Uldry, M.; Tontonoz, P.; et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 2005, 120, 261–273. [Google Scholar] [CrossRef]
- Li, S.; Lin, J.D. Transcriptional control of circadian metabolic rhythms in the liver. Diabetes Obes. Metab. 2015, 17 (Suppl. 1), 33–38. [Google Scholar] [CrossRef]
- Sonoda, J.; Mehl, I.R.; Chong, L.W.; Nofsinger, R.R.; Evans, R.M. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2007, 104, 5223–5228. [Google Scholar] [CrossRef]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef]
- Gravel, S.P. Deciphering the Dichotomous Effects of PGC-1alpha on Tumorigenesis and Metastasis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Mastropasqua, F.; Girolimetti, G.; Shoshan, M. PGC1alpha: Friend or Foe in Cancer? Genes 2018, 9, 48. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; McGill, M.R.; Mansouri, A.; Asselah, T.; Farhood, A.; Woolbright, B.L.; Ding, W.X.; Jaeschke, H. Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem. Toxicol. 2017, 108, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; van der Windt, D.J.; Huang, H.; Simmons, R.L.; Shiva, S.; Tai, S.; Tsung, A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017, 66, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Li, J.; Zheng, L.; Feng, M.; Wang, X.; Han, K.; Pi, H.; Li, M.; Huang, X.; et al. SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1alpha-mediated mitochondrial biogenesis. Oncotarget 2016, 7, 29255–29274. [Google Scholar]
- Shalaby, R.E.; Iram, S.; Oropeza, C.E.; McLachlan, A. Peroxisome proliferator-activated receptor gamma coactivator family members competitively regulate hepatitis b virus biosynthesis. Virology 2019, 526, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, Y.; Huang, C.; Ying, L.; Xue, J.; Wu, H.; Chen, Z.; Yang, Z. Resveratrol enhances HBV replication through activating Sirt1-PGC-1alpha-PPARalpha pathway. Sci. Rep. 2016, 6, 24744. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Chou, S.F.; Lee, J.W.; Chen, H.L.; Chen, C.M.; Tao, M.H.; Shih, C. MicroRNA-130a can inhibit hepatitis B virus replication via targeting PGC1alpha and PPARgamma. RNA 2015, 21, 385–400. [Google Scholar] [CrossRef]
- Curtil, C.; Enache, L.S.; Radreau, P.; Dron, A.G.; Scholtes, C.; Deloire, A.; Roche, D.; Lotteau, V.; Andre, P.; Ramiere, C. The metabolic sensors FXRalpha, PGC-1alpha, and SIRT1 cooperatively regulate hepatitis B virus transcription. FASEB J. 2014, 28, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhao, F.; Cheng, Z.; Zhou, M.; Zhi, X.; Li, J.; Hu, K. GCN5 acetyltransferase inhibits PGC1alpha-induced hepatitis B virus biosynthesis. Virol. Sin. 2013, 28, 216–222. [Google Scholar] [CrossRef]
- Deng, J.J.; Kong, K.E.; Gao, W.W.; Tang, H.V.; Chaudhary, V.; Cheng, Y.; Zhou, J.; Chan, C.P.; Wong, D.K.; Yuen, M.F.; et al. Interplay between SIRT1 and hepatitis B virus X protein in the activation of viral transcription. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 491–501. [Google Scholar]
- Lee, H.J.; Su, Y.; Yin, P.H.; Lee, H.C.; Chi, C.W. PPAR(gamma)/PGC-1(alpha) pathway in E-cadherin expression and motility of HepG2 cells. Anticancer Res. 2009, 29, 5057–5063. [Google Scholar] [PubMed]
- Liu, R.; Zhang, H.; Zhang, Y.; Li, S.; Wang, X.; Wang, X.; Wang, C.; Liu, B.; Zen, K.; Zhang, C.Y.; et al. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha acts as a tumor suppressor in hepatocellular carcinoma. Tumour. Biol. 2017, 39. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.H.; Frankel, W.; Ghoshal, K.; Jacob, S.T. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 2012, 56, 186–197. [Google Scholar] [CrossRef]
- Mohamed, A.A.; li-Eldin, Z.A.; Elbedewy, T.A.; El-Serafy, M.; li-Eldin, F.A.; bdelAziz, H. MicroRNAs and clinical implications in hepatocellular carcinoma. World J. Hepatol. 2017, 9, 1001–1007. [Google Scholar] [CrossRef]
- Du, J.; Hang, P.; Pan, Y.; Feng, B.; Zheng, Y.; Chen, T.; Zhao, L.; Du, Z. Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1alpha/Drp1 pathway. Toxicol. Appl. Pharmacol. 2019, 369, 73–81. [Google Scholar] [CrossRef]
- Sun, L.Y.; Wang, N.; Ban, T.; Sun, Y.H.; Han, Y.; Sun, L.L.; Yan, Y.; Kang, X.H.; Chen, S.; Sun, L.H.; et al. MicroRNA-23a mediates mitochondrial compromise in estrogen deficiency-induced concentric remodeling via targeting PGC-1alpha. J. Mol. Cell Cardiol. 2014, 75, 1–11. [Google Scholar] [CrossRef]
- Shang, J.; Yang, F.; Wang, Y.; Wang, Y.; Xue, G.; Mei, Q.; Wang, F.; Sun, S. MicroRNA-23a antisense enhances 5-fluorouracil chemosensitivity through APAF-1/caspase-9 apoptotic pathway in colorectal cancer cells. J. Cell Biochem. 2014, 115, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, K.; Yuan, D.H.; Meng, C.Y. Overexpression of NAD(P)H: Quinone Oxidoreductase 1 Inhibits Hepatocellular Carcinoma Cell Proliferation and Induced Apoptosis by Activating AMPK/PGC-1alpha Pathway. DNA Cell Biol. 2017, 36, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Kim, G.Y.; Mansfield, B.C.; Chou, J.Y. Sirtuin signaling controls mitochondrial function in glycogen storage disease type Ia. J. Inherit. Metab. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shin, D.J.; Pan, H.; Lin, Z.; Dreyfuss, J.M.; Camargo, F.D.; Miao, J.; Biddinger, S.B. YAP suppresses gluconeogenic gene expression through PGC1alpha. Hepatology 2017, 66, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Cai, Y.; Li, Y.; Li, Y.; Hu, N.; Ma, S.; Hu, S.; Zhu, P.; Wang, W.; Zhou, H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox. Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, E.; Peres, C.; Bellafante, E.; Ducheix, S.; Pinto, C.; Villani, G.; Moschetta, A. Hepatic peroxisome proliferator-activated receptor gamma coactivator 1beta drives mitochondrial and anabolic signatures that contribute to hepatocellular carcinoma progression in mice. Hepatology 2018, 67, 884–898. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.E.; Lahiri, S.; Chow, J.D.; Byrne, F.L.; Hargett, S.R.; Breen, D.S.; Olzomer, E.M.; Wu, L.E.; Cooney, G.J.; Turner, N.; et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat. Commun. 2017, 8, 14689. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Mello, T.; Zanieri, F.; Ceni, E.; Galli, A. Oxidative Stress in the Healthy and Wounded Hepatocyte: A Cellular Organelles Perspective. Oxid. Med. Cell Longev. 2016, 2016, 8327410. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Hayes, J.D.; nkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Baird, L.; Lleres, D.; Swift, S.; nkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox. Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Lu, Y.F.; Chen, H.; Shen, Z.Y.; Liu, J. Liver expression of Nrf2-related genes in different liver diseases. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 485–491. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; nkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox. Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chung, F.L. Oxidative stress and hepatocarcinogenesis. Hepatoma. Res. 2018, 4. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxid. Med. Cell Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef]
- Kurzawski, M.; Dziedziejko, V.; Urasinska, E.; Post, M.; Wojcicki, M.; Mietkiewski, J.; Drozdzik, M. Nuclear factor erythroid 2-like 2 (Nrf2) expression in end-stage liver disease. Environ. Toxicol. Pharm. 2012, 34, 87–95. [Google Scholar] [CrossRef]
- Chen, J.; Yu, Y.; Ji, T.; Ma, R.; Chen, M.; Li, G.; Li, F.; Ding, Q.; Kang, Q.; Huang, D.; et al. Clinical implication of Keap1 and phosphorylated Nrf2 expression in hepatocellular carcinoma. Cancer Med. 2016, 5, 2678–2687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC. Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Zhu, L.; Pan, H.; Li, W.; Zhou, Y.; Cong, Z.; Yan, F.; Chen, S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Int. J. Cancer 2014, 135, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; cquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Lin, D.; Wu, J. Hypoxia inducible factor in hepatocellular carcinoma: A therapeutic target. World J. Gastroenterol. 2015, 21, 12171–12178. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Cavadas, M.A.S.; Cheong, A.; Taylor, C.T. The regulation of transcriptional repression in hypoxia. Exp. Cell Res. 2017, 356, 173–181. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Liu, L.P.; Jiang, J.X.; Xiong, Z.F.; He, Q.J.; Wu, C. The correlation of expression levels of HIF-1alpha and HIF-2alpha in hepatocellular carcinoma with capsular invasion, portal vein tumor thrombi and patients’ clinical outcome. Jpn. J. Clin. Oncol. 2014, 44, 159–167. [Google Scholar] [CrossRef]
- Bangoura, G.; Liu, Z.S.; Qian, Q.; Jiang, C.Q.; Yang, G.F.; Jing, S. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 3176–3182. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.X.; Gao, Q.; Qiu, S.J.; Ju, M.J.; Cai, M.Y.; Xu, Y.F.; Zhou, J.; Zhang, B.H.; Fan, J. Hypoxia-inducible factor-1 alpha, in association with inflammation, angiogenesis and MYC, is a critical prognostic factor in patients with HCC after surgery. BMC Cancer 2009, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Iizuka, N.; Tsunedomi, R.; Hamamoto, Y.; Miyamoto, T.; Iida, M.; Tokuhisa, Y.; Sakamoto, K.; Takashima, M.; Tamesa, T.; et al. Glycolysis module activated by hypoxia-inducible factor 1alpha is related to the aggressive phenotype of hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 725–731. [Google Scholar]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed]
- Gwak, G.Y.; Yoon, J.H.; Kim, K.M.; Lee, H.S.; Chung, J.W.; Gores, G.J. Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 2005, 42, 358–364. [Google Scholar] [CrossRef]
- Piret, J.P.; Mottet, D.; Raes, M.; Michiels, C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar] [CrossRef]
- Matteucci, E.; Modora, S.; Simone, M.; Desiderio, M.A. Hepatocyte growth factor induces apoptosis through the extrinsic pathway in hepatoma cells: Favouring role of hypoxia-inducible factor-1 deficiency. Oncogene 2003, 22, 4062–4073. [Google Scholar] [CrossRef]
- Piret, J.P.; Lecocq, C.; Toffoli, S.; Ninane, N.; Raes, M.; Michiels, C. Hypoxia and CoCl2 protect HepG2 cells against serum deprivation- and t-BHP-induced apoptosis: A possible anti-apoptotic role for HIF-1. Exp. Cell Res. 2004, 295, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.P.; Minet, E.; Cosse, J.P.; Ninane, N.; Debacq, C.; Raes, M.; Michiels, C. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J. Biol. Chem. 2005, 280, 9336–9344. [Google Scholar] [CrossRef] [PubMed]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction in cancer chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Zhang, N.Y.; Hu, X.; Chen, J.L.; Rao, M.J.; Wu, L.W.; Li, Q.Y.; Zhang, B.; Yan, W.; Zhang, C. Evodiamine induces apoptosis and promotes hepatocellular carcinoma cell death induced by vorinostat via downregulating HIF-1alpha under hypoxia. Biochem. Biophys. Res. Commun. 2018, 498, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Li, Q.G.; Xing, T.Y.; Zhang, M.; Zhang, J.J.; Xia, Q. HIF1 regulates WSB-1 expression to promote hypoxia-induced chemoresistance in hepatocellular carcinoma cells. FEBS Lett. 2013, 587, 2530–2535. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2010, 106, 526–535. [Google Scholar] [CrossRef]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef]
- Cash, T.P.; Pan, Y.; Simon, M.C. Reactive oxygen species and cellular oxygen sensing. Free Radic. Biol. Med. 2007, 43, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, R.A.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol. Med. 2017, 23, 320–331. [Google Scholar]
- George, J.; Ahmad, N. Mitochondrial Sirtuins in Cancer: Emerging Roles and Therapeutic Potential. Cancer Res. 2016, 76, 2500–2506. [Google Scholar] [CrossRef]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Ohashi, K.; Kawai, S.; Murata, K. Identification and characterization of a human mitochondrial NAD kinase. Nat. Commun. 2012, 3, 1248. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Meng, X.; Huang, C.; Li, J. Emerging role of silent information regulator 1 (SIRT1) in hepatocellular carcinoma: A potential therapeutic target. Tumour. Biol. 2015, 36, 4063–4074. [Google Scholar] [CrossRef]
- Lerin, C.; Rodgers, J.T.; Kalume, D.E.; Kim, S.H.; Pandey, A.; Puigserver, P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006, 3, 429–438. [Google Scholar] [CrossRef]
- Kelly, T.J.; Lerin, C.; Haas, W.; Gygi, S.P.; Puigserver, P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 2009, 284, 19945–19952. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, B.; Wong, N.; Lo, A.W.; To, K.F.; Chan, A.W.; Ng, M.H.; Ho, C.Y.; Cheng, S.H.; Lai, P.B.; et al. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res. 2011, 71, 4138–4149. [Google Scholar] [CrossRef]
- Choi, H.N.; Bae, J.S.; Jamiyandorj, U.; Noh, S.J.; Park, H.S.; Jang, K.Y.; Chung, M.J.; Kang, M.J.; Lee, D.G.; Moon, W.S. Expression and role of SIRT1 in hepatocellular carcinoma. Oncol. Rep. 2011, 26, 503–510. [Google Scholar] [PubMed]
- Zhang, Z.Y.; Hong, D.; Nam, S.H.; Kim, J.M.; Paik, Y.H.; Joh, J.W.; Kwon, C.H.; Park, J.B.; Choi, G.S.; Jang, K.Y.; et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J. Hepatol. 2015, 62, 121–130. [Google Scholar] [CrossRef]
- Biel, T.G.; Lee, S.; Flores-Toro, J.A.; Dean, J.W.; Go, K.L.; Lee, M.H.; Law, B.K.; Law, M.E.; Dunn, W.A., Jr.; Zendejas, I.; et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 2016, 23, 279–290. [Google Scholar] [CrossRef]
- Chun, S.K.; Go, K.; Yang, M.J.; Zendejas, I.; Behrns, K.E.; Kim, J.S. Autophagy in Ischemic Livers: A Critical Role of Sirtuin 1/Mitofusin 2 Axis in Autophagy Induction. Toxicol. Res. 2016, 32, 35–46. [Google Scholar] [CrossRef]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lu, J.; Zhu, F.; Wei, J.; Jia, C.; Zhang, Y.; Zhou, L.; Xie, H.; Zheng, S. Pro-apoptotic and anti-proliferative effects of mitofusin-2 via Bax signaling in hepatocellular carcinoma cells. Med. Oncol. 2012, 29, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, D.; Xu, X.; Zhao, X.; Huang, P.; Zhou, X.; Song, W.; Guo, H.; Wang, W.; Zheng, S. Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma. World J. Surg. Oncol. 2016, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Sun, J.; Gong, W.; Sun, P.; Kong, X.; Yang, M.; Zhang, W. Mitofusin-2 acts as biomarker for predicting poor prognosis in hepatitis B virus related hepatocellular carcinoma. Infect. Agent. Cancer 2018, 13, 36. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Sun, X.; Cao, H.; Zhan, L.; Yin, C.; Wang, G.; Liang, P.; Li, J.; Wang, Z.; Liu, B.; Huang, Q.; et al. Mitochondrial fission promotes cell migration by Ca(2+) /CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver Int. 2018, 38, 1263–1272. [Google Scholar] [CrossRef]
- Dashzeveg, N.; Yoshida, K. Cell death decision by p53 via control of the mitochondrial membrane. Cancer Lett. 2015, 367, 108–112. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.L.; Cheng, W.; Yin, X.M.; Jiang, B. The expression of SIRT3 in primary hepatocellular carcinoma and the mechanism of its tumor suppressing effects. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 978–998. [Google Scholar]
- Song, C.L.; Tang, H.; Ran, L.K.; Ko, B.C.; Zhang, Z.Z.; Chen, X.; Ren, J.H.; Tao, N.N.; Li, W.Y.; Huang, A.L.; et al. Sirtuin 3 inhibits hepatocellular carcinoma growth through the glycogen synthase kinase-3beta/BCL2-associated X protein-dependent apoptotic pathway. Oncogene 2016, 35, 631–641. [Google Scholar] [CrossRef]
- Wang, J.X.; Yi, Y.; Li, Y.W.; Cai, X.Y.; He, H.W.; Ni, X.C.; Zhou, J.; Cheng, Y.F.; Jin, J.J.; Fan, J.; et al. Down-regulation of sirtuin 3 is associated with poor prognosis in hepatocellular carcinoma after resection. BMC Cancer 2014, 14, 297. [Google Scholar] [CrossRef]
- Zhang, B.; Qin, L.; Zhou, C.J.; Liu, Y.L.; Qian, H.X.; He, S.B. SIRT3 expression in hepatocellular carcinoma and its impact on proliferation and invasion of hepatoma cells. Asian Pac. J. Trop. Med. 2013, 6, 649–652. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Liu, L.; Cai, M.; Pan, Y.; Fu, J.; Cao, Y.; Yun, J. Low SIRT3 expression correlates with poor differentiation and unfavorable prognosis in primary hepatocellular carcinoma. PLoS ONE 2012, 7, e51703. [Google Scholar] [CrossRef]
- Zhou, Y.; Cheng, S.; Chen, S.; Zhao, Y. Prognostic and clinicopathological value of SIRT3 expression in various cancers: A systematic review and meta-analysis. Onco. Targets. Ther. 2018, 11, 2157–2167. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef]
- Tao, N.N.; Zhou, H.Z.; Tang, H.; Cai, X.F.; Zhang, W.L.; Ren, J.H.; Zhou, L.; Chen, X.; Chen, K.; Li, W.Y.; et al. Sirtuin 3 enhanced drug sensitivity of human hepatoma cells through glutathione S-transferase pi 1/JNK signaling pathway. Oncotarget 2016, 7, 50117–50130. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Huang, J.Y.; Schwer, B.; Verdin, E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 267–277. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar]
- Wang, Y.S.; Du, L.; Liang, X.; Meng, P.; Bi, L.; Wang, Y.L.; Wang, C.; Tang, B. Sirtuin 4 Depletion Promotes Hepatocellular Carcinoma Tumorigenesis Through Regulating Adenosine-Monophosphate-Activated Protein Kinase Alpha/Mammalian Target of Rapamycin Axis in Mice. Hepatology 2018. [Google Scholar] [CrossRef]
- Jeong, S.M.; Xiao, C.; Finley, L.W.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Chang, L.; Xi, L.; Liu, Y.; Liu, R.; Wu, Z.; Jian, Z. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol. Med. Rep. 2018, 17, 342–349. [Google Scholar] [CrossRef]
- Dang, S.; Zhou, J.; Wang, Z.; Wang, K.; Dai, S.; He, S. MiR-299-3p functions as a tumor suppressor via targeting Sirtuin 5 in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 106, 966–975. [Google Scholar] [CrossRef]
- Guo, D.; Song, X.; Guo, T.; Gu, S.; Chang, X.; Su, T.; Yang, X.; Liang, B.; Huang, D. Vimentin acetylation is involved in SIRT5-mediated hepatocellular carcinoma migration. Am. J. Cancer Res. 2018, 8, 2453–2466. [Google Scholar]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Yang, W.; Nagasawa, K.; Munch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, M.P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016, 167, 985–1000. [Google Scholar] [CrossRef]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef]
- Han, T.S.; Ban, H.S.; Hur, K.; Cho, H.S. The Epigenetic Regulation of HCC Metastasis. Int. J. Mol. Sci. 2018, 19, 3978. [Google Scholar] [CrossRef]
- Wilson, C.L.; Mann, D.A.; Borthwick, L.A. Epigenetic reprogramming in liver fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 124–132. [Google Scholar] [CrossRef]
- Herceg, Z.; Paliwal, A. Epigenetic mechanisms in hepatocellular carcinoma: How environmental factors influence the epigenome. Mutat. Res. 2011, 727, 55–61. [Google Scholar] [CrossRef]
- Puszyk, W.M.; Trinh, T.L.; Chapple, S.J.; Liu, C. Linking metabolism and epigenetic regulation in development of hepatocellular carcinoma. Lab. Investig. 2013, 93, 983–990. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef]
- Keating, S.T.; El-Osta, A. Epigenetics and metabolism. Circ. Res. 2015, 116, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.L.; Wu, W.H.; Hu, T.H.; Chen, C.W.; Cheng, H.C.; Li, C.F.; Tsai, W.H.; Tsai, H.J.; Hsieh, M.C.; Chuang, J.H.; et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci. Rep. 2018, 8, 3081. [Google Scholar] [CrossRef]
- Gao, X.; Sheng, Y.; Yang, J.; Wang, C.; Zhang, R.; Zhu, Y.; Zhang, Z.; Zhang, K.; Yan, S.; Sun, H.; et al. Osteopontin alters DNA methylation through up-regulating DNMT1 and sensitizes CD133+/CD44+ cancer stem cells to 5 azacytidine in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 179. [Google Scholar] [CrossRef]
- Kanematsu, T.; Maehara, Y.; Matsumata, T.; Shirabe, K.; Akazawa, K.; Sugimachi, K. Human hepatocellular carcinoma sensitivity to antitumor drugs assayed using the succinate dehydrogenase inhibition test. Oncology 1991, 48, 34–38. [Google Scholar] [CrossRef]
- Scheffler, I.E. Molecular genetics of succinate: Quinone oxidoreductase in eukaryotes. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 267–315. [Google Scholar]
- Cervera, A.M.; Bayley, J.P.; Devilee, P.; McCreath, K.J. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol. Cancer 2009, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; bdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.H.; Janknecht, R.; Maher, L.J., III. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum. Mol. Genet. 2007, 16, 3136–3148. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, F.; Tan, L.; Kong, L.; Xiong, L.; Deng, J.; Barbera, A.J.; Zheng, L.; Zhang, H.; Huang, S.; et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell 2011, 42, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, L.; Chen, X.; Shen, J.; Shan, J.; Xu, Y.; Yang, Z.; Wu, L.; Xia, F.; Bie, P.; et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS ONE 2013, 8, e62828. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar] [CrossRef]
- Chen, K.; Ma, J.; Jia, X.; Ai, W.; Ma, Z.; Pan, Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 117–125. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mello, T.; Simeone, I.; Galli, A. Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring. Cells 2019, 8, 417. https://doi.org/10.3390/cells8050417
Mello T, Simeone I, Galli A. Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring. Cells. 2019; 8(5):417. https://doi.org/10.3390/cells8050417
Chicago/Turabian StyleMello, Tommaso, Irene Simeone, and Andrea Galli. 2019. "Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring" Cells 8, no. 5: 417. https://doi.org/10.3390/cells8050417
APA StyleMello, T., Simeone, I., & Galli, A. (2019). Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring. Cells, 8(5), 417. https://doi.org/10.3390/cells8050417