Sh3bp2 Gain-Of-Function Mutation Ameliorates Lupus Phenotypes in B6.MRL-Faslpr Mice

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Animal Study: Analysis of the Double-Mutant Mice
2.3. Western Blot Analysis
2.4. Urine Protein Assessment
2.5. Histopathologic Assessment
2.6. Serum Anti-dsDNA Antibody Measurement
2.7. Serum Immunoglobulin Measurement
2.8. Flow Cytometry
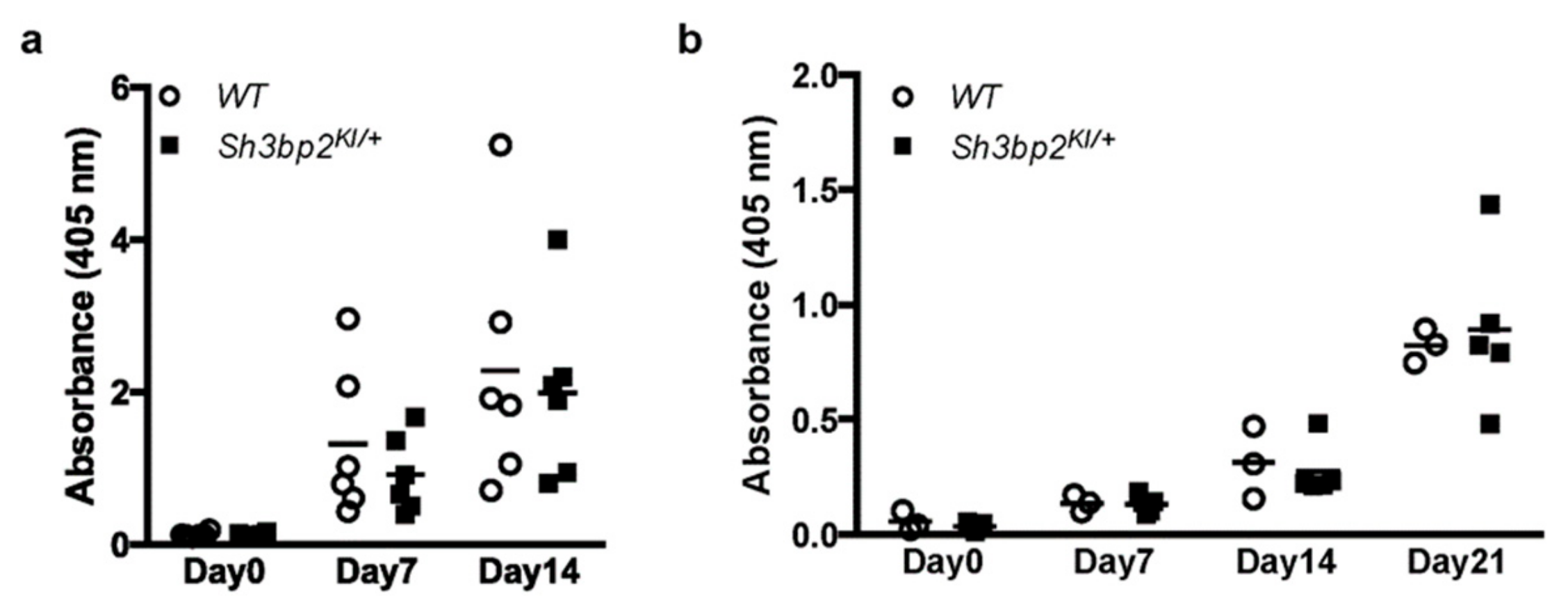
2.9. In Vivo Immunization and Measurement of Serum Immunoglobulins Levels
2.10. Real-Time Quantitative PCR (qPCR) Analysis
2.11. Bone-Marrow-Derived Dendritic Cell (BMDC) Culture
2.12. TNF Measurement by ELISA
2.13. Bone Marrow-Derived Macrophage (BMM) Culture
2.14. Phagocytosis Assay
2.14.1. Preparation of Apoptotic Cells
2.14.2. Measurement of Phagocytic Activity
2.15. Statistical Analysis
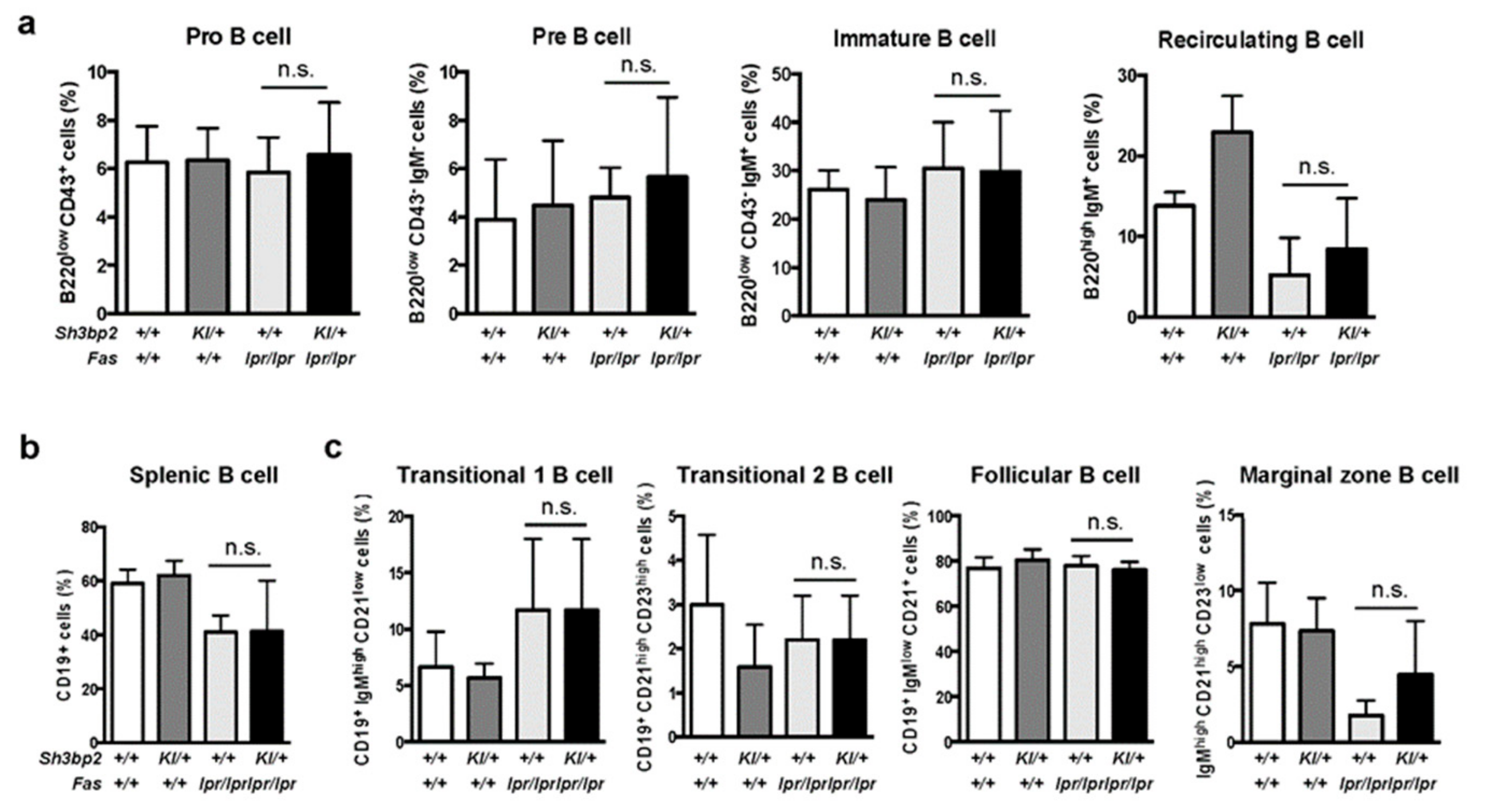
3. Results
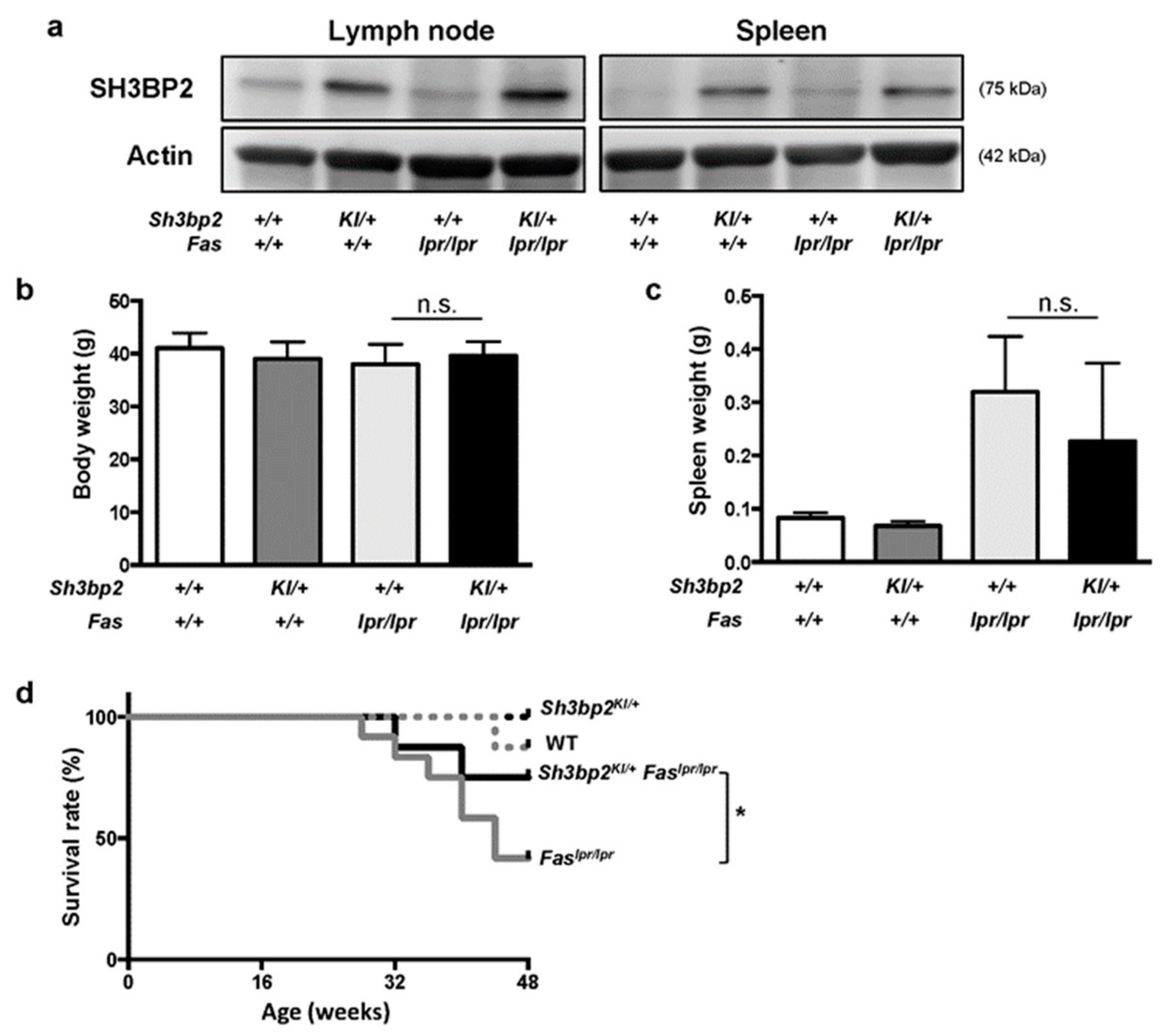
3.1. Sh3bp2 Gain-Of-Function Mutation Improves the Survival Rate of Lupus-Prone Mice
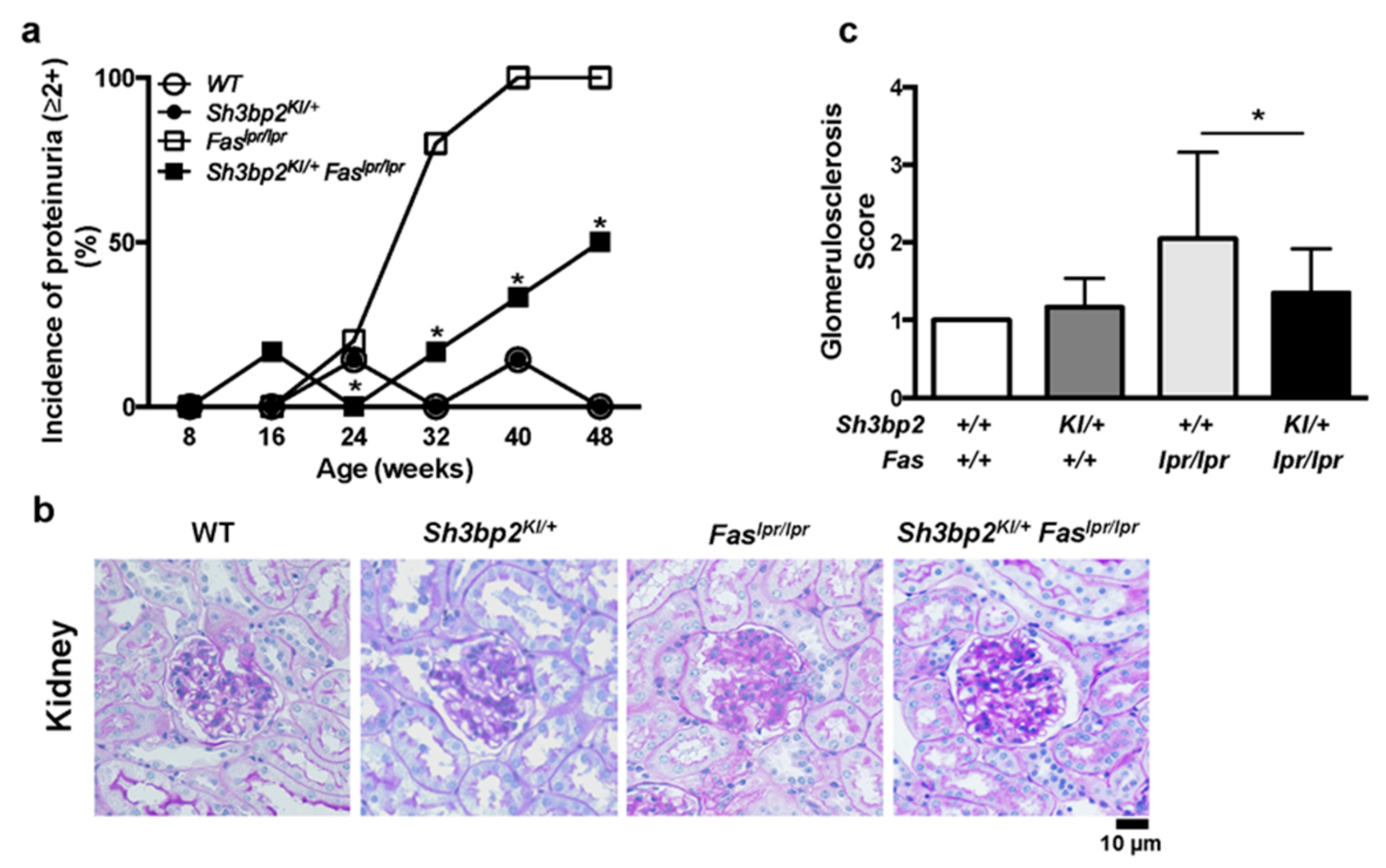
3.2. Sh3bp2 Gain-Of-Function Mutation Improves Renal Involvement in Lupus-Prone Mice
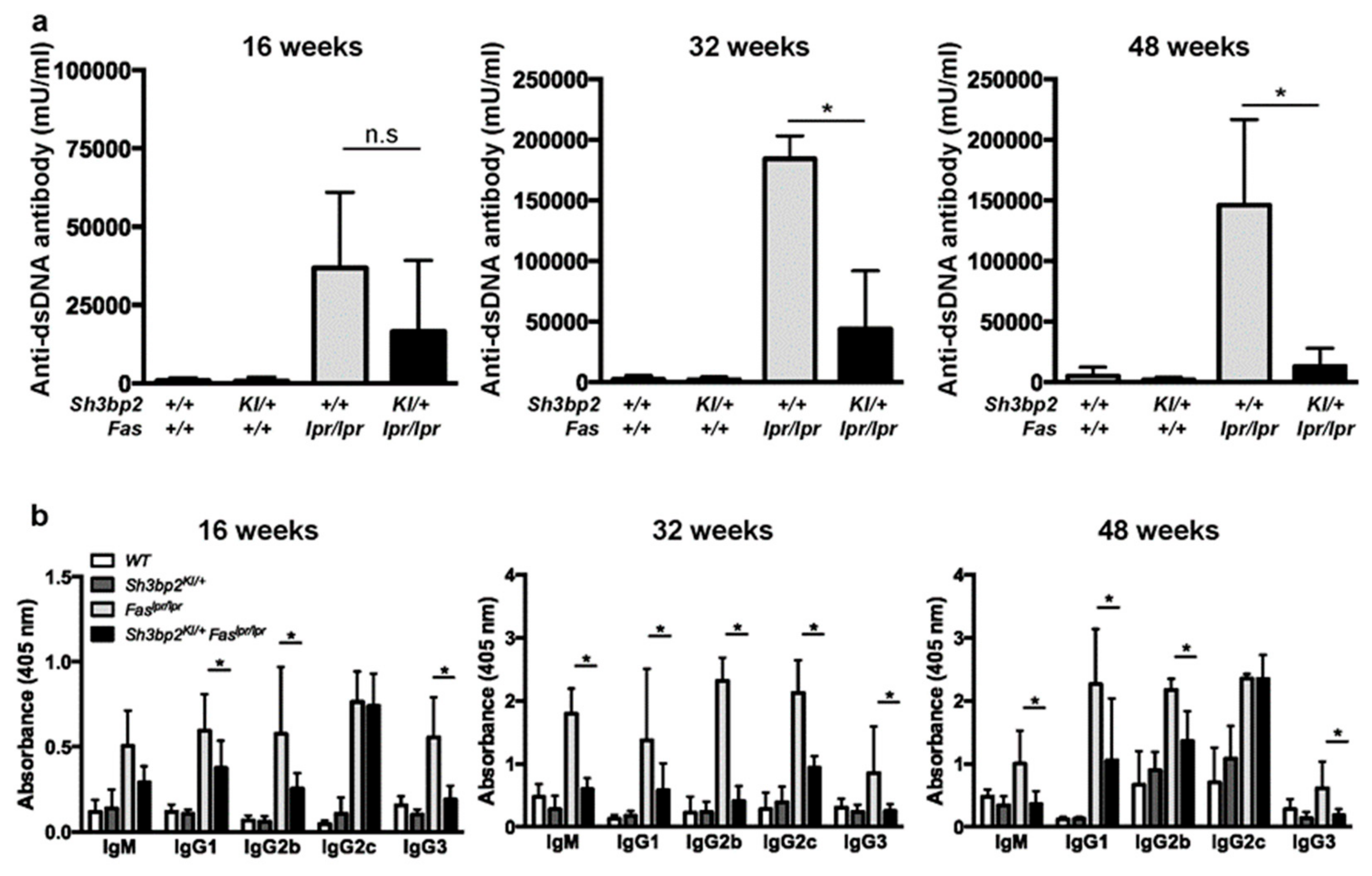
3.3. Sh3bp2 Gain-Of-Function Mutation Reduces the Production of Serum Anti-dsDNA Antibody
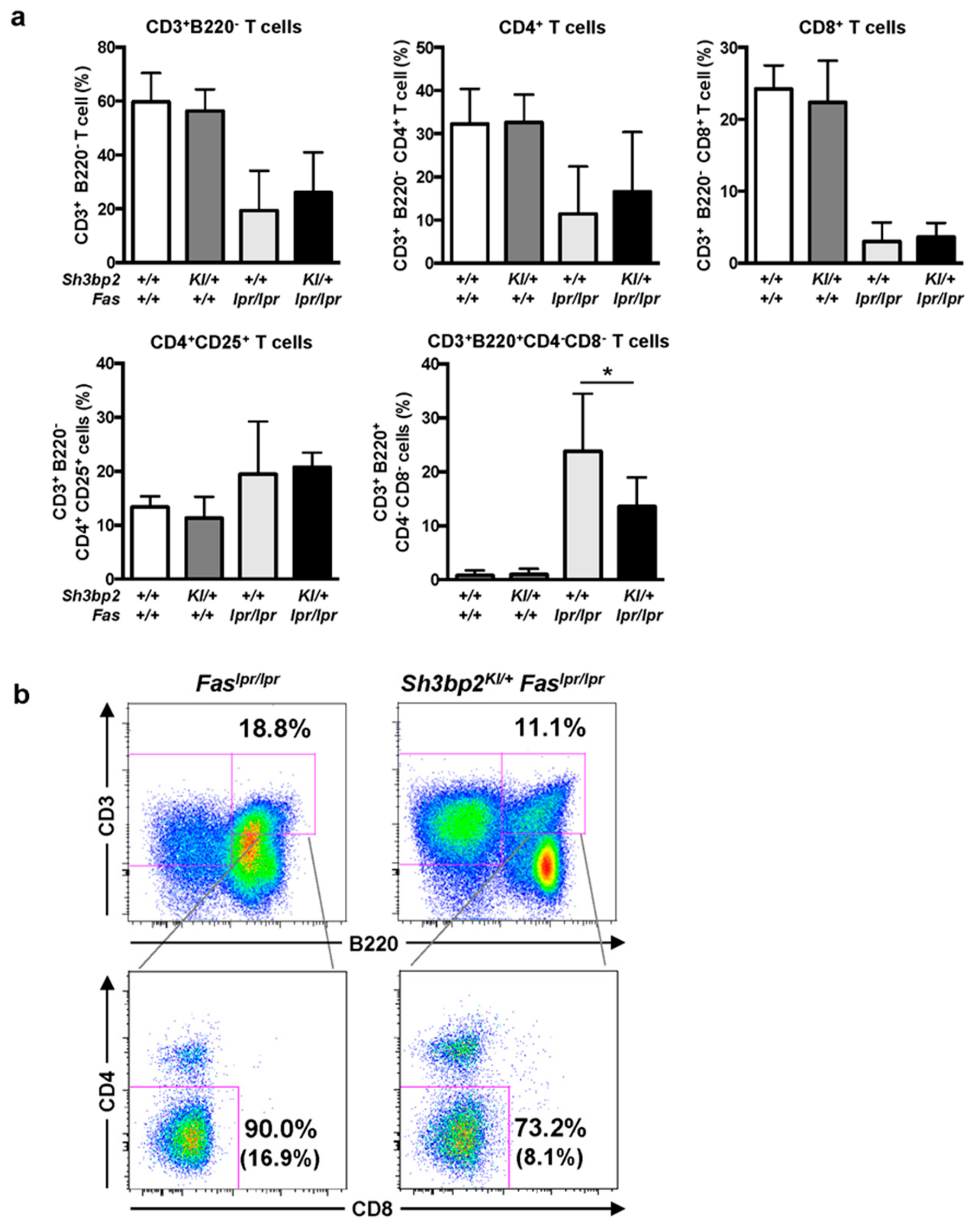
3.4. Aberrant Accumulation of the B220+CD4−CD8− T cell Population is Ameliorated in Sh3bp2KI/+Faslpr/lpr Mice
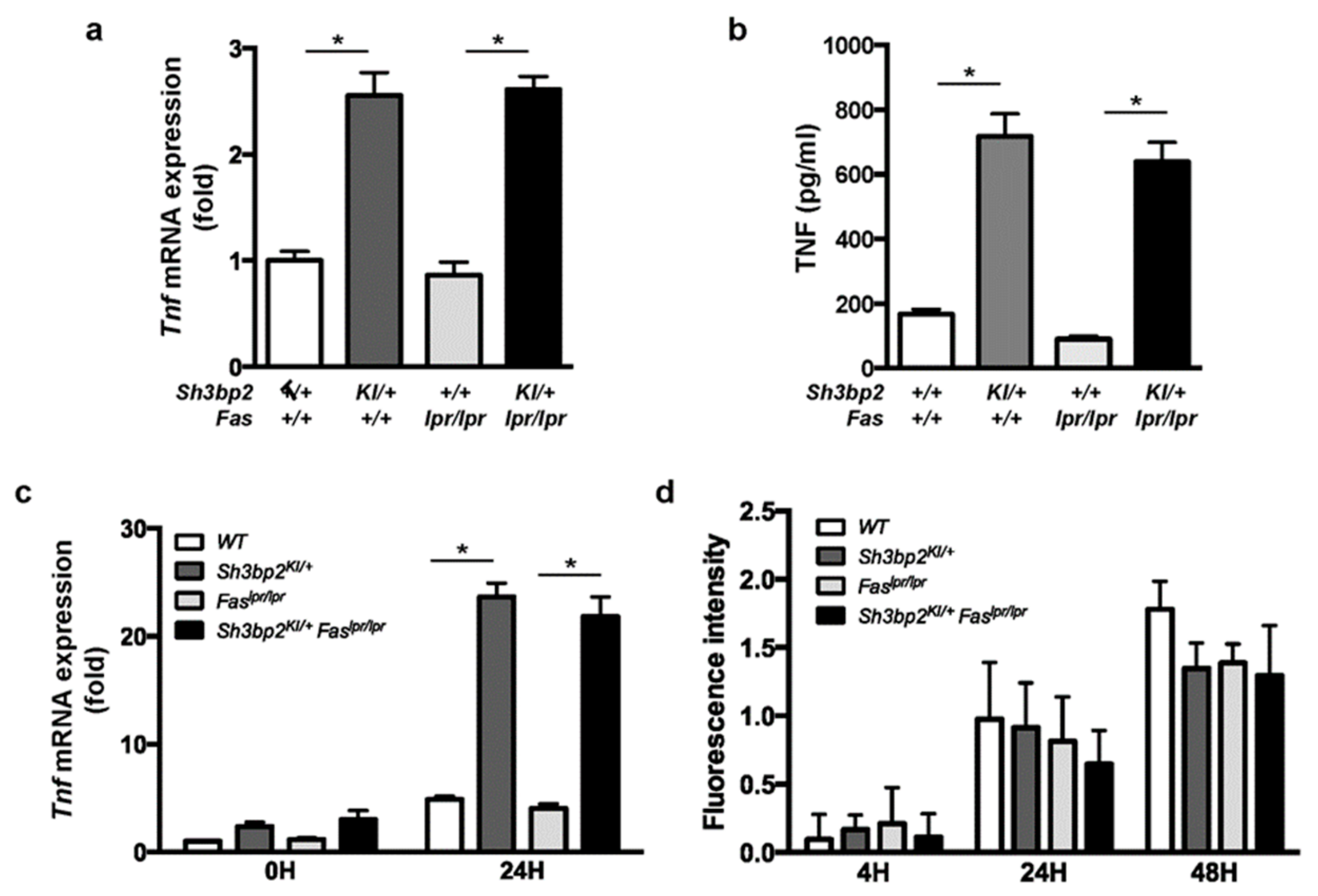
3.5. Sh3bp2KI/+Faslpr/lpr Mice Exhibit Increased Activation of Apoptosis-Inducing Cascades in the Lymph Nodes
3.6. Phagocytic Activity Was Unaltered in Sh3bp2KI/+Faslpr/lpr Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Zharkova, O.; Celhar, T.; Cravens, P.D.; Satterthwaite, A.B.; Fairhurst, A.M.; Davis, L.S. Pathways leading to an immunological disease: Systemic lupus erythematosus. Rheumatology 2017, 56, i55–i66. [Google Scholar] [CrossRef] [PubMed]
- Moulton, V.R.; Suarez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Yoshimasu, T. Animal models of spontaneous and drug-induced cutaneous lupus erythematosus. Autoimmun. Rev. 2005, 4, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Titov, A.A.; Morel, L. An update on lupus animal models. Curr. Opin. Rheumatol. 2017, 29, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.G.; Carrera, A.C.; Marshakrothstein, A.; Martineza, C.; Abbas, A.K. Apoptosis, FAS and Systemic Autoimmunity—The Mrl-Ipr/Ipr Model. Curr. Opin. Immunol. 1994, 6, 913–920. [Google Scholar] [CrossRef]
- Singer, G.G.; Abbas, A.K. The FAS Antigen Is Involved in Peripheral but Not Thymic Deletion of T-Lymphocytes in T-Cell Receptor Transgenic Mice. Immunity 1994, 1, 365–371. [Google Scholar] [CrossRef]
- Mogil, R.J.; Radvanyi, L.; Gonzalezquintial, R.; Miller, R.; Mills, G.; Theofilopoulos, A.N.; Green, D.R. FAS (CD95) Participates in Peripheral T-Cell Deletion and Associated Apoptosis In-Vivo. Int. Immunol. 1995, 7, 1451–1458. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Kudo, Y.; Ishimaru, N. Dual Role of Fas/FasL-Mediated Signal in Peripheral Immune Tolerance. Front. Immunol. 2017, 8, 403. [Google Scholar] [CrossRef]
- Hasegawa, H.; Matsumoto, T. Mechanisms of Tolerance Induction by Dendritic Cells in Vivo. Front. Immunol. 2018, 9, 350. [Google Scholar] [CrossRef]
- Izawa, T.; Kondo, T.; Kurosawa, M.; Oura, R.; Matsumoto, K.; Tanaka, E.; Yamada, A.; Arakaki, R.; Kudo, Y.; Hayashi, Y.; et al. Fas-independent T-cell apoptosis by dendritic cells controls autoimmune arthritis in MRL/lpr mice. PLoS ONE 2012, 7, e48798. [Google Scholar] [CrossRef] [PubMed]
- Sytwu, H.K.; Liblau, R.S.; McDevitt, H.O. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity 1996, 5, 17–30. [Google Scholar] [CrossRef]
- Zhou, T.; Edwards, C.K.; Yang, P.A.; Wang, Z.; Bluethmann, H.; Mountz, J.D. Greatly accelerated lymphadenopathy and autoimmune disease in lpr mice lacking tumor necrosis factor receptor I. J. Immunol. 1996, 156, 2661–2665. [Google Scholar] [PubMed]
- Ueki, Y.; Lin, C.Y.; Senoo, M.; Ebihara, T.; Agata, N.; Onji, M.; Saheki, Y.; Kawai, T.; Mukherjee, P.M.; Reichenberger, E.; et al. Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 “cherubism” mice. Cell 2007, 128, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Yoshitaka, T.; Mukai, T.; Kittaka, M.; Alford, L.M.; Masrani, S.; Ishida, S.; Yamaguchi, K.; Yamada, M.; Mizuno, N.; Olsen, B.R.; et al. Enhanced TLR-MYD88 signaling stimulates autoinflammation in SH3BP2 cherubism mice and defines the etiology of cherubism. Cell Rep. 2014, 8, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.A.; Kumar, L.; Lu, B.; Geha, R.S. 3BP2 deficiency impairs the response of B cells, but not T cells, to antigen receptor ligation. Mol. Cell. Biol. 2006, 26, 5214–5225. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Dimitriou, I.D.; La Rose, J.; Ilangumaran, S.; Yeh, W.C.; Doody, G.; Turner, M.; Gommerman, J.; Rottapel, R. The 3BP2 adapter protein is required for optimal B-cell activation and thymus-independent type 2 humoral response. Mol. Cell. Biol. 2007, 27, 3109–3122. [Google Scholar] [CrossRef]
- Dimitriou, I.D.; Lee, K.; Akpan, I.; Lind, E.F.; Barr, V.A.; Ohashi, P.S.; Samelson, L.E.; Rottapel, R. Timed Regulation of 3BP2 Induction Is Critical for Sustaining CD8(+) T Cell Expansion and Differentiation. Cell Rep. 2018, 24, 1123–1135. [Google Scholar] [CrossRef]
- Deckert, M.; Tartare-Deckert, S.; Hernandez, J.; Rottapel, R.; Altman, A. Adaptor function for the Syk kinases-interacting protein 3BP2 in IL-2 gene activation. Immunity 1998, 9, 595–605. [Google Scholar] [CrossRef]
- Chihara, K.; Kato, Y.; Yoshiki, H.; Takeuchi, K.; Fujieda, S.; Sada, K. Syk-dependent tyrosine phosphorylation of 3BP2 is required for optimal FcRgamma-mediated phagocytosis and chemokine expression in U937 cells. Sci. Rep. 2017, 7, 11480. [Google Scholar] [CrossRef]
- Jevremovic, D.; Billadeau, D.D.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. Regulation of NK cell-mediated cytotoxicity by the adaptor protein 3BP2. J. Immunol. 2001, 166, 7219–7228. [Google Scholar] [CrossRef] [PubMed]
- Chihara, K.; Kimura, Y.; Honjoh, C.; Yamauchi, S.; Takeuchi, K.; Sada, K. Tyrosine phosphorylation of 3BP2 is indispensable for the interaction with VAV3 in chicken DT40 cells. Exp. Cell Res. 2014, 322, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Foucault, I.; Le Bras, S.; Charvet, C.; Moon, C.; Altman, A.; Deckert, M. The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood 2005, 105, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Levaot, N.; Simoncic, P.D.; Dimitriou, I.D.; Scotter, A.; La Rose, J.; Ng, A.H.; Willett, T.L.; Wang, C.J.; Janmohamed, S.; Grynpas, M.; et al. 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J. Clin. Investig. 2011, 121, 3244–3257. [Google Scholar] [CrossRef]
- GuezGuez, A.; Prod’homme, V.; Mouska, X.; Baudot, A.; Blin-Wakkach, C.; Rottapel, R.; Deckert, M. 3BP2 adapter protein is required for receptor activator of NFkappaB ligand (RANKL)-induced osteoclast differentiation of RAW264.7 cells. J. Biol. Chem. 2010, 285, 20952–20963. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Tiziani, V.; Santanna, C.; Fukai, N.; Maulik, C.; Garfinkle, J.; Ninomiya, C.; doAmaral, C.; Peters, H.; Habal, M.; et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat. Genet. 2001, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Gallant, R.; Ishida, S.; Yoshitaka, T.; Kittaka, M.; Nishida, K.; Fox, D.A.; Morita, Y.; Ueki, Y. SH3BP2 gain-of-function mutation exacerbates inflammation and bone loss in a murine collagen-induced arthritis model. PLoS ONE 2014, 9, e105518. [Google Scholar] [CrossRef]
- Kittaka, M.; Mayahara, K.; Mukai, T.; Yoshimoto, T.; Yoshitaka, T.; Gorski, J.P.; Ueki, Y. Cherubism Mice Also Deficient in c-Fos Exhibit Inflammatory Bone Destruction Executed by Macrophages That Express MMP14 Despite the Absence of TRAP+ Osteoclasts. J. Bone Miner. Res. 2017. [Google Scholar] [CrossRef]
- Prod’Homme, V.; Boyer, L.; Dubois, N.; Mallavialle, A.; Munro, P.; Mouska, X.; Coste, I.; Rottapel, R.; Tartare-Deckert, S.; Deckert, M. Cherubism allele heterozygosity amplifies microbe-induced inflammatory responses in murine macrophages. J. Clin. Investig. 2015, 125, 1396–1400. [Google Scholar] [CrossRef]
- Mukai, T.; Ishida, S.; Ishikawa, R.; Yoshitaka, T.; Kittaka, M.; Gallant, R.; Lin, Y.L.; Rottapel, R.; Brotto, M.; Reichenberger, E.J.; et al. SH3BP2 cherubism mutation potentiates TNF-alpha-induced osteoclastogenesis via NFATc1 and TNF-alpha-mediated inflammatory bone loss. J. Bone Miner. Res. 2014, 29, 2618–2635. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Mukai, T.; Mito, T.; Kodama, S.; Nagasu, A.; Kittaka, M.; Sone, T.; Ueki, Y.; Morita, Y. Pharmacological inhibition of tankyrase induces bone loss in mice by increasing osteoclastogenesis. Bone 2018, 106, 156–166. [Google Scholar] [CrossRef]
- Sogawa, Y.; Nagasu, H.; Itano, S.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Kadoya, H.; Satoh, M.; Sasaki, T.; Kashihara, N. The eNOS-NO pathway attenuates kidney dysfunction via suppression of inflammasome activation in aldosterone-induced renal injury model mice. PLoS ONE 2018, 13, e0203823. [Google Scholar] [CrossRef]
- Nagasu, H.; Satoh, M.; Kuwabara, A.; Yorimitsu, D.; Sakuta, T.; Tomita, N.; Kashihara, N. Renal denervation reduces glomerular injury by suppressing NAD(P)H oxidase activity in Dahl salt-sensitive rats. Nephrol. Dial. Transplant. 2010, 25, 2889–2898. [Google Scholar] [CrossRef]
- Iseki, M.; Kubo, C.; Kwon, S.M.; Yamaguchi, A.; Kataoka, Y.; Yoshida, N.; Takatsu, K.; Takaki, S. Increased numbers of B-1 cells and enhanced responses against TI-2 antigen in mice lacking APS, an adaptor molecule containing PH and SH2 domains. Mol. Cell. Biol. 2004, 24, 2243–2250. [Google Scholar] [CrossRef]
- Mori, T.; Iwasaki, Y.; Seki, Y.; Iseki, M.; Katayama, H.; Yamamoto, K.; Takatsu, K.; Takaki, S. Lnk/Sh2b3 controls the production and function of dendritic cells and regulates the induction of IFN-gamma-producing T cells. J. Immunol. 2014, 193, 1728–1736. [Google Scholar] [CrossRef]
- Mukai, T.; Gallant, R.; Ishida, S.; Kittaka, M.; Yoshitaka, T.; Fox, D.A.; Morita, Y.; Nishida, K.; Rottapel, R.; Ueki, Y. Loss of SH3 domain-binding protein 2 function suppresses bone destruction in tumor necrosis factor-driven and collagen-induced arthritis in mice. Arthritis Rheumatol. 2015, 67, 656–667. [Google Scholar] [CrossRef]
- Fossati-Jimack, L.; Ling, G.S.; Cortini, A.; Szajna, M.; Malik, T.H.; McDonald, J.U.; Pickering, M.C.; Cook, H.T.; Taylor, P.R.; Botto, M. Phagocytosis is the main CR3-mediated function affected by the lupus-associated variant of CD11b in human myeloid cells. PLoS ONE 2013, 8, e57082. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Fujita, S.; Morita, Y. Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2. Cells 2019, 8, 195. [Google Scholar] [CrossRef]
- Levaot, N.; Voytyuk, O.; Dimitriou, I.; Sircoulomb, F.; Chandrakumar, A.; Deckert, M.; Krzyzanowski, P.M.; Scotter, A.; Gu, S.; Janmohamed, S.; et al. Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 2011, 147, 1324–1339. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Eisenberg, R.A. Lpr and gld: Single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 1991, 9, 243–269. [Google Scholar] [CrossRef]
- Watanabe-Fukunaga, R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Nagata, S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992, 356, 314–317. [Google Scholar] [CrossRef]
- Martina, M.N.; Noel, S.; Saxena, A.; Rabb, H.; Hamad, A.R. Double negative (DN) alphabeta T cells: Misperception and overdue recognition. Immunol. Cell Biol. 2015, 93, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.L.; Ramos, P.; Rosendorff, A.; Nikolic-Zugic, J.; Lacy, E.; Matsuzawa, A.; Elkon, K.B. Massive upregulation of the Fas ligand in lpr and gld mice: Implications for Fas regulation and the graft-versus-host disease-like wasting syndrome. J. Exp. Med. 1995, 181, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Licht, R.; Dieker, J.W.; Jacobs, C.W.; Tax, W.J.; Berden, J.H. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J. Autoimmun. 2004, 22, 139–145. [Google Scholar] [CrossRef]
- Bijl, M.; Reefman, E.; Horst, G.; Limburg, P.C.; Kallenberg, C.G.M. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: Correlates with decreased serum levels of complement. Ann. Rheum. Dis. 2006, 65, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Harte, M.T.; Kluck, R.M.; Wolf, B.B.; Casiano, C.A.; Newmeyer, D.D.; Wang, H.G.; Reed, J.C.; Nicholson, D.W.; Alnemri, E.S.; et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 1999, 144, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Noman, M.; Van Assche, G.; Baert, F.; Van Steen, K.; Esters, N.; Joossens, S.; Bossuyt, X.; Rutgeerts, P. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn’s disease: A prospective cohort study. Gastroenterology 2003, 125, 32–39. [Google Scholar] [CrossRef]
- Vaglio, A.; Grayson, P.C.; Fenaroli, P.; Gianfreda, D.; Boccaletti, V.; Ghiggeri, G.M.; Moroni, G. Drug-induced lupus: Traditional and new concepts. Autoimmun. Rev. 2018, 17, 912–918. [Google Scholar] [CrossRef]
- D’Auria, F.; Rovere-Querini, P.; Giazzon, M.; Ajello, P.; Baldissera, E.; Manfredi, A.A.; Sabbadini, M.G. Accumulation of plasma nucleosomes upon treatment with anti-tumour necrosis factor-alpha antibodies. J. Intern. Med. 2004, 255, 409–418. [Google Scholar] [CrossRef]
- Ferraccioli, G.; Mecchia, F.; Di Poi, E.; Fabris, M. Anticardiolipin antibodies in rheumatoid patients treated with etanercept or conventional combination therapy: Direct and indirect evidence for a possible association with infections. Ann. Rheum. Dis. 2002, 61, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Steiner, G.; Smolen, J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res. 2002, 4 (Suppl. S2), S1–S5. [Google Scholar] [CrossRef]
- Gordon, C.; Ranges, G.E.; Greenspan, J.S.; Wofsy, D. Chronic therapy with recombinant tumor necrosis factor-alpha in autoimmune NZB/NZW F1 mice. Clin. Immunol. Immunopathol. 1989, 52, 421–434. [Google Scholar] [CrossRef]
- Xu, Y.; Zhuang, H.; Han, S.; Liu, C.; Wang, H.; Mathews, C.E.; Massini, J.; Yang, L.; Reeves, W.H. Mechanisms of tumor necrosis factor alpha antagonist-induced lupus in a murine model. Arthritis Rheumatol. 2015, 67, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Korner, H.; Cretney, E.; Wilhelm, P.; Kelly, J.M.; Rollinghoff, M.; Sedgwick, J.D.; Smyth, M.J. Tumor necrosis factor sustains the generalized lymphoproliferative disorder (gld) phenotype. J. Exp. Med. 2000, 191, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Segal, R.; Dayan, M.; Zinger, H.; Mozes, E. Suppression of experimental systemic lupus erythematosus (SLE) in mice via TNF inhibition by an anti-TNFalpha monoclonal antibody and by pentoxiphylline. Lupus 2001, 10, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kollias, G.; Kontoyiannis, D. Role of TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002, 13, 315–321. [Google Scholar] [CrossRef]
- Hatani, T.; Sada, K. Adaptor protein 3BP2 and cherubism. Curr. Med. Chem. 2008, 15, 549–554. [Google Scholar]
- Alexopoulou, L.; Pasparakis, M.; Kollias, G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur. J. Immunol. 1997, 27, 2588–2592. [Google Scholar] [CrossRef]
- Lipsky, P.E.; van der Heijde, D.M.; St Clair, E.W.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; Feldmann, M.; et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef]
- Aringer, M.; Smolen, J.S. Therapeutic blockade of TNF in patients with SLE-promising or crazy? Autoimmun. Rev. 2012, 11, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, P.Y.; Reeves, W.H. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch. Immunol. Ther. Exp. 2010, 58, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.W.; Quartier, P.; Botto, M.; Fossati-Jimack, L. Macrophages from patients with SLE and rheumatoid arthritis have defective adhesion in vitro, while only SLE macrophages have impaired uptake of apoptotic cells. Ann. Rheum. Dis. 2006, 65, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Mak, A.; Lee, B.; Lee, H.Y.; Poidinger, M.; Connolly, J.E.; Fairhurst, A.M. Monocyte Siglec-14 expression is upregulated in patients with systemic lupus erythematosus and correlates with lupus disease activity. Rheumatology 2017, 56, 1025–1030. [Google Scholar] [CrossRef][Green Version]
- Mak, A.; Thornhill, S.I.; Lee, H.Y.; Lee, B.; Poidinger, M.; Connolly, J.E.; Fairhurst, A.M. Brief report: Decreased expression of CD244 (SLAMF4) on monocytes and platelets in patients with systemic lupus erythematosus. Clin. Rheumatol. 2018, 37, 811–816. [Google Scholar] [CrossRef]
- Oliveira, J.J.; Karrar, S.; Rainbow, D.B.; Pinder, C.L.; Clarke, P.; Rubio Garcia, A.; Al-Assar, O.; Burling, K.; Morris, S.; Stratton, R.; et al. The plasma biomarker soluble SIGLEC-1 is associated with the type I interferon transcriptional signature, ethnic background and renal disease in systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 152. [Google Scholar] [CrossRef]
- Serrano-Candelas, E.; Ainsua-Enrich, E.; Navines-Ferrer, A.; Rodrigues, P.; Garcia-Valverde, A.; Bazzocco, S.; Macaya, I.; Arribas, J.; Serrano, C.; Sayos, J.; et al. Silencing of adaptor protein SH3BP2 reduces KIT/PDGFRA receptors expression and impairs gastrointestinal stromal tumors growth. Mol. Oncol. 2018, 12, 1383–1397. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Gene Accession Number | Sequence (5′-3′) | Amplicon Size (bp) | |
|---|---|---|---|---|
| Fas | NM_007987.2 | Forward: Reverse: | taaaccagacttctactgcgattct gggttccatgttcacacga | 73 |
| Fasl | NM_010177.4 | Forward: Reverse: | aaaaagagccgaggagtgtg attccagagggatggacctt | 66 |
| Tnf | NM_013693.2 | Forward: Reverse: | catcttctcaaaattcgagtgaca tgggagtagacaaggtacaaccc | 175 |
| Tnfr1 | NM_011609.4 | Forward: Reverse: | ggaaagtatgtccattctaagaacaa agtcactcaccaagtaggttcctt | 76 |
| Tnfr2 | NM_011610.3 | Forward: Reverse: | gaggcccaagggtttcag ggcttccgtgggaagaat | 64 |
| Trail | NM_ 009425.2 | Forward: Reverse: | ggctgcaagtctgcattggg cctgcagctgcttcatctcgt | 192 |
| Trailr2 | NM_ 020275.4 | Forward: Reverse: | ccacaacacggaacctggca tgtctgtgaggcccaactgc | 167 |
| Tweak | NM_ 011614.3 | Forward: Reverse: | caggatggagcacaagcag ggctggagctgttgattttg | 73 |
| Dr3 | NM_ 001291010.1 | Forward: Reverse: | ccctggcttatcccagact agatgccagaggagttccaa | 97 |
| Hprt | NM_013556.2 | Forward: Reverse: | tcctcctcagaccgctttt cctggttcatcatcgctaatc | 90 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagasu, A.; Mukai, T.; Iseki, M.; Kawahara, K.; Tsuji, S.; Nagasu, H.; Ueki, Y.; Ishihara, K.; Kashihara, N.; Morita, Y. Sh3bp2 Gain-Of-Function Mutation Ameliorates Lupus Phenotypes in B6.MRL-Faslpr Mice. Cells 2019, 8, 402. https://doi.org/10.3390/cells8050402
Nagasu A, Mukai T, Iseki M, Kawahara K, Tsuji S, Nagasu H, Ueki Y, Ishihara K, Kashihara N, Morita Y. Sh3bp2 Gain-Of-Function Mutation Ameliorates Lupus Phenotypes in B6.MRL-Faslpr Mice. Cells. 2019; 8(5):402. https://doi.org/10.3390/cells8050402
Chicago/Turabian StyleNagasu, Akiko, Tomoyuki Mukai, Masanori Iseki, Kyoko Kawahara, Shoko Tsuji, Hajime Nagasu, Yasuyoshi Ueki, Katsuhiko Ishihara, Naoki Kashihara, and Yoshitaka Morita. 2019. "Sh3bp2 Gain-Of-Function Mutation Ameliorates Lupus Phenotypes in B6.MRL-Faslpr Mice" Cells 8, no. 5: 402. https://doi.org/10.3390/cells8050402
APA StyleNagasu, A., Mukai, T., Iseki, M., Kawahara, K., Tsuji, S., Nagasu, H., Ueki, Y., Ishihara, K., Kashihara, N., & Morita, Y. (2019). Sh3bp2 Gain-Of-Function Mutation Ameliorates Lupus Phenotypes in B6.MRL-Faslpr Mice. Cells, 8(5), 402. https://doi.org/10.3390/cells8050402