Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Insulin Preparation and Glycation
2.3. Fluorescence Measurements
2.4. Cell Cultures and Treatments
2.5. MTT Assay
2.6. Cell Cycle Analysis
2.7. Detection of Intracellular ROS
2.8. Confocal Laser-Scanning Microscopy
2.9. Cellular Nuclear Extraction
2.10. Immunoblotting
2.11. Caspase Assay
2.12. Statistical Analysis
3. Results
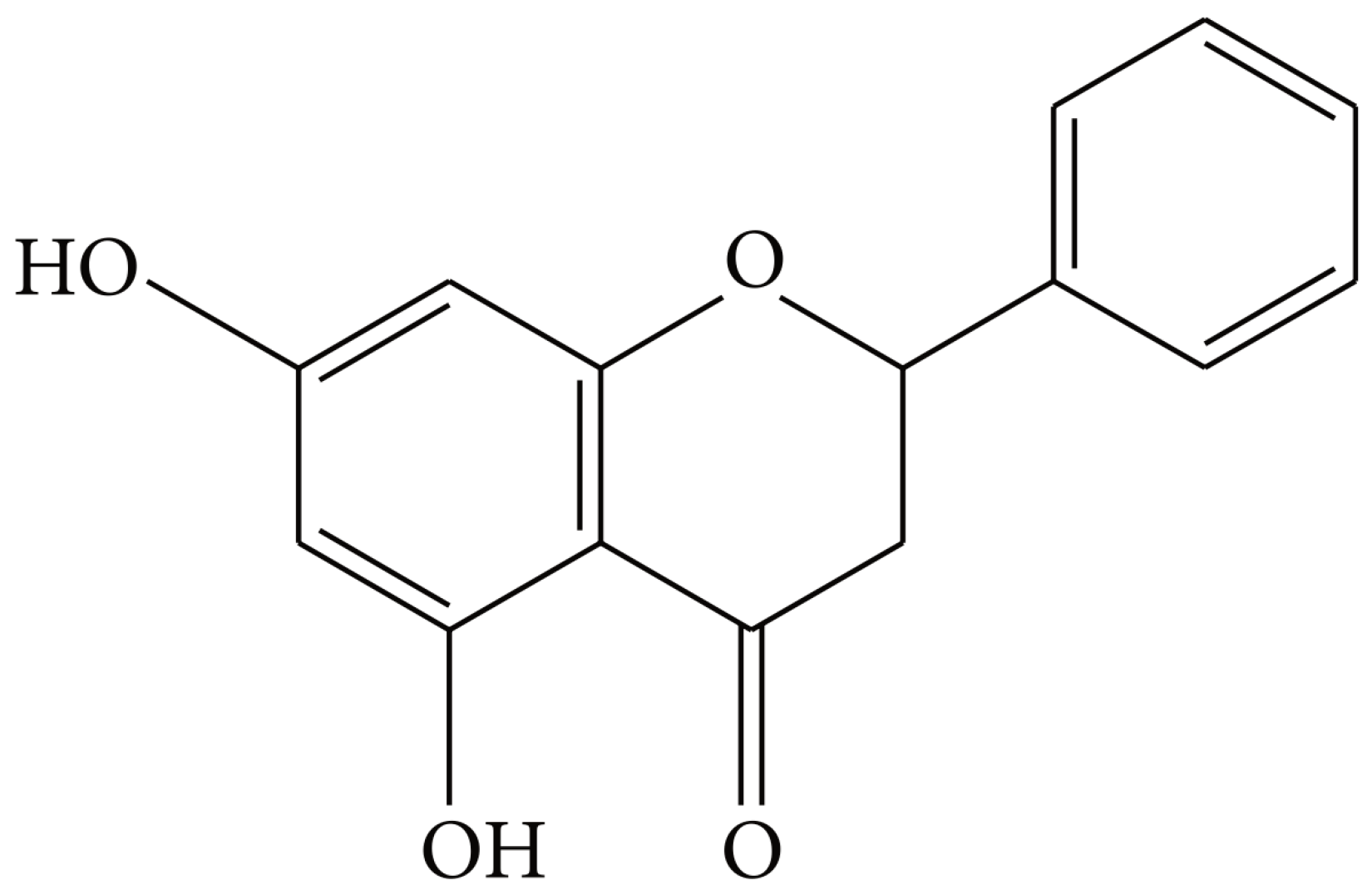
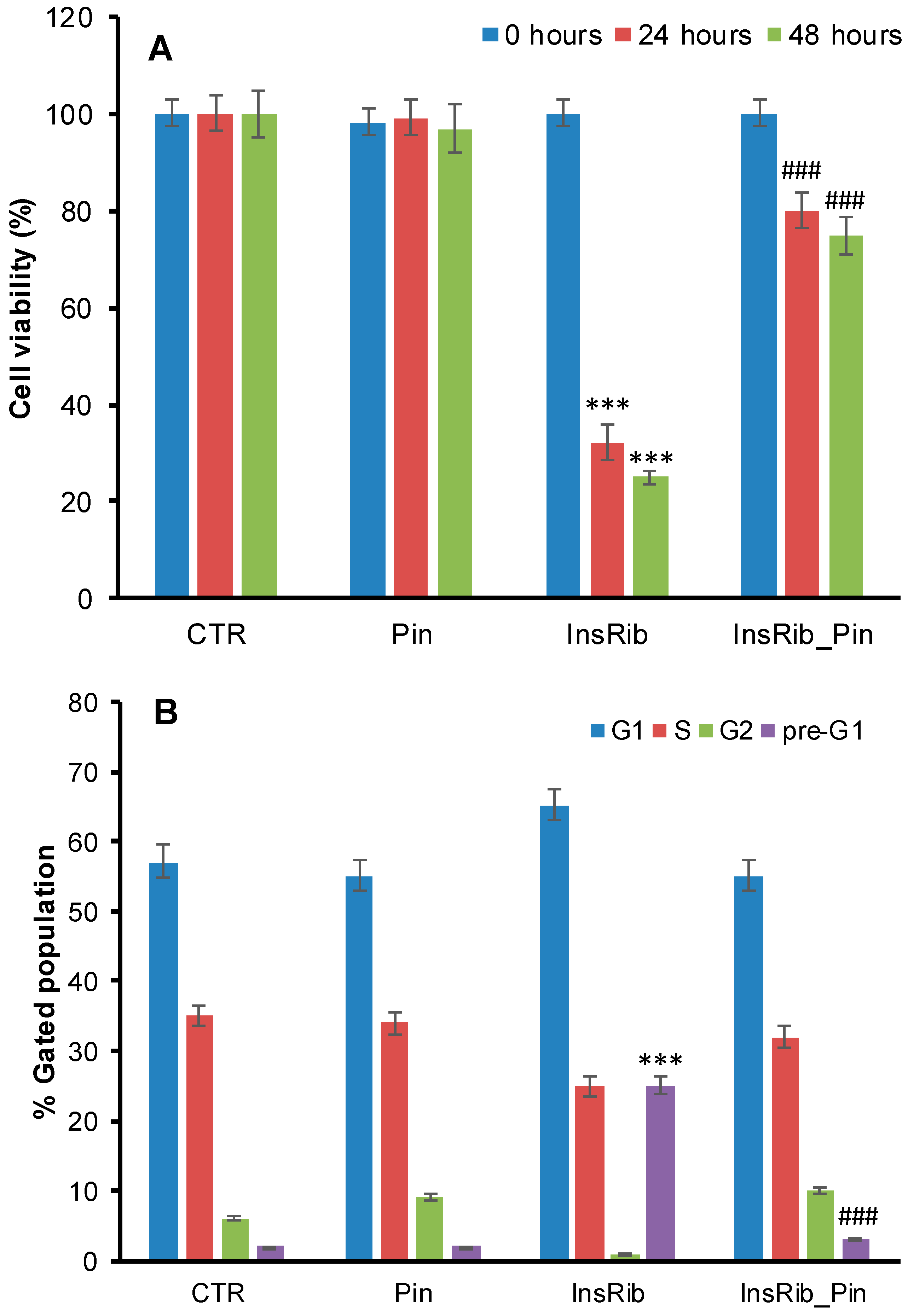
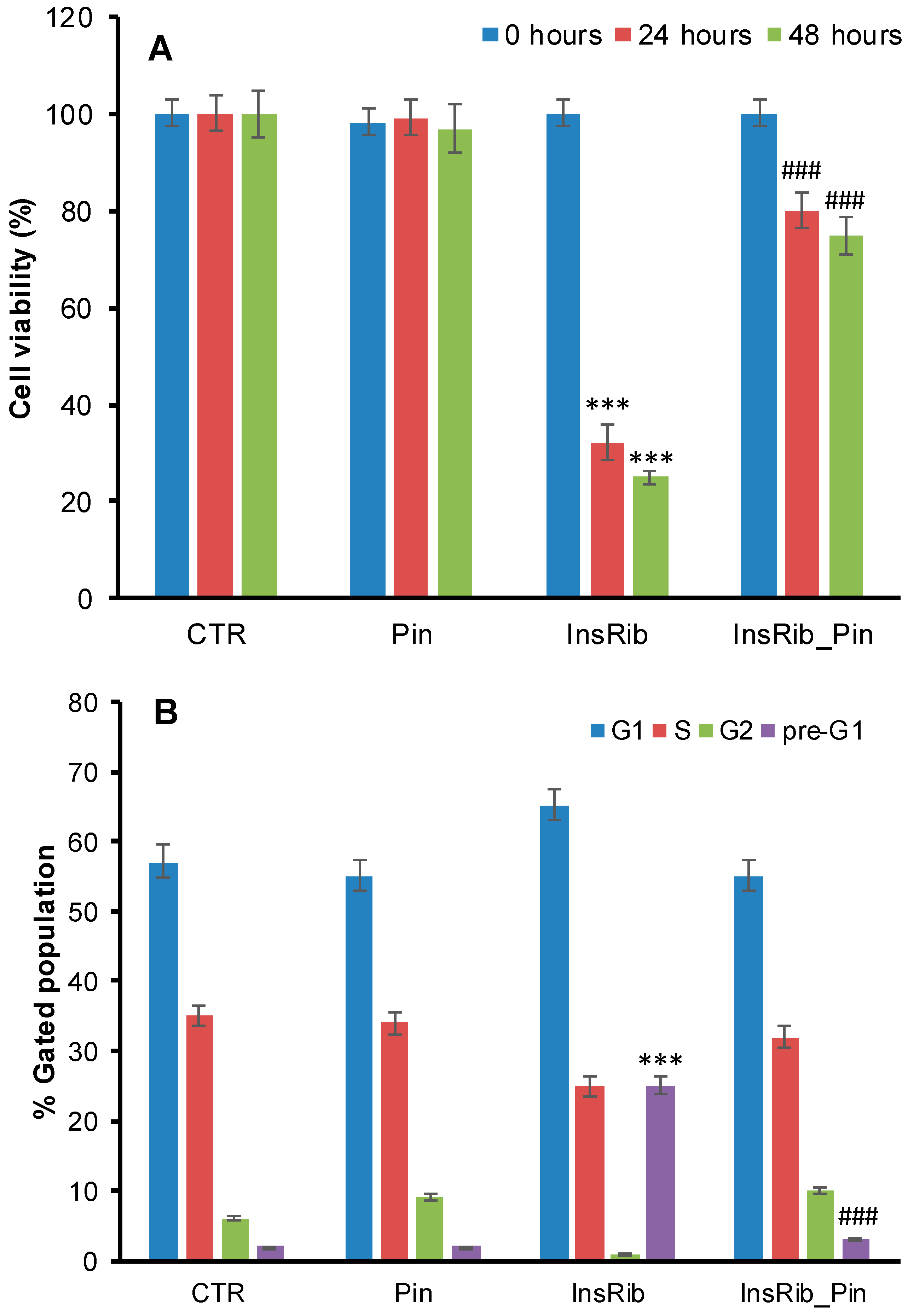
3.1. Pinocembrin Effect on AGE-Induced Toxicity
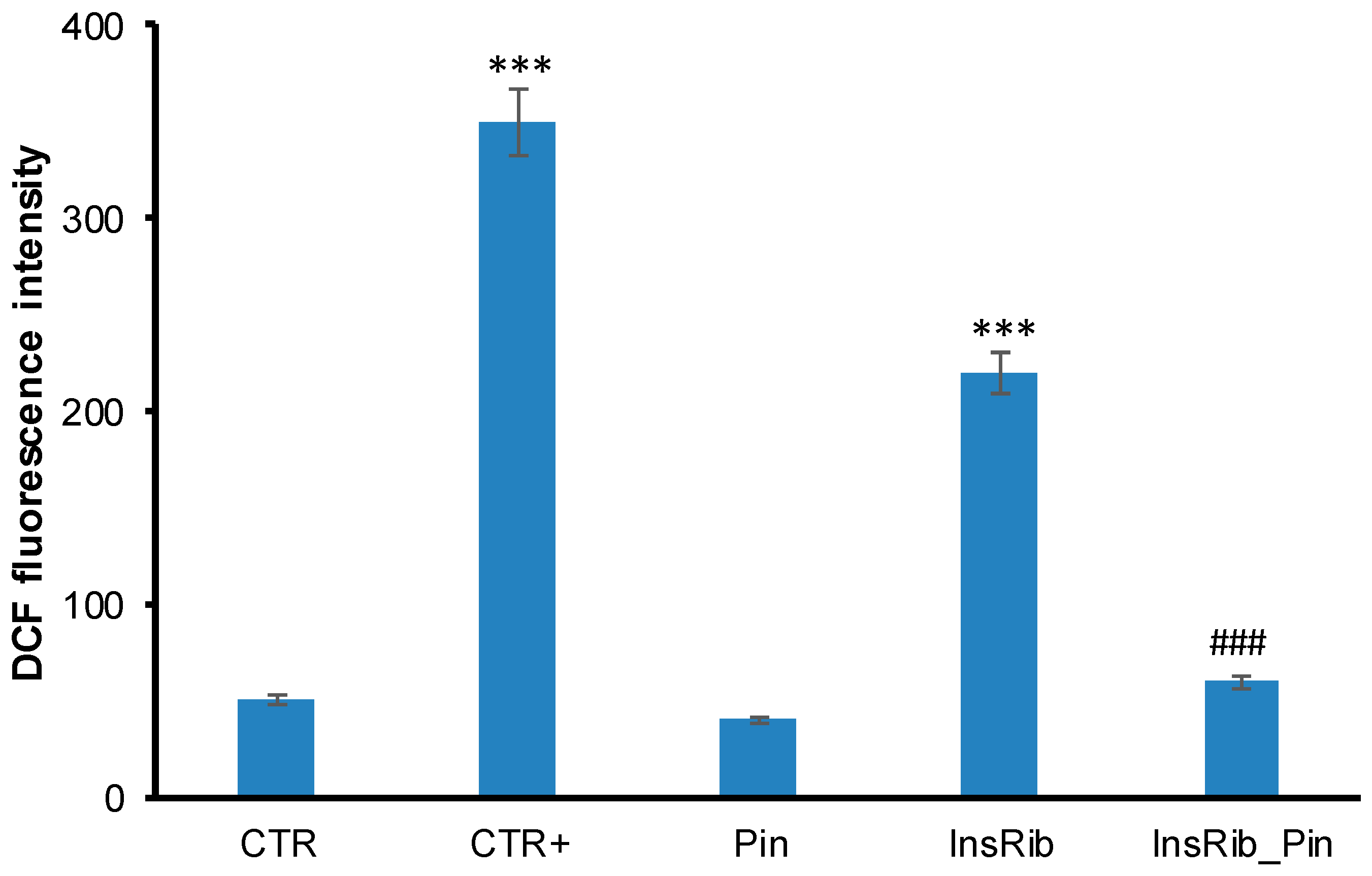
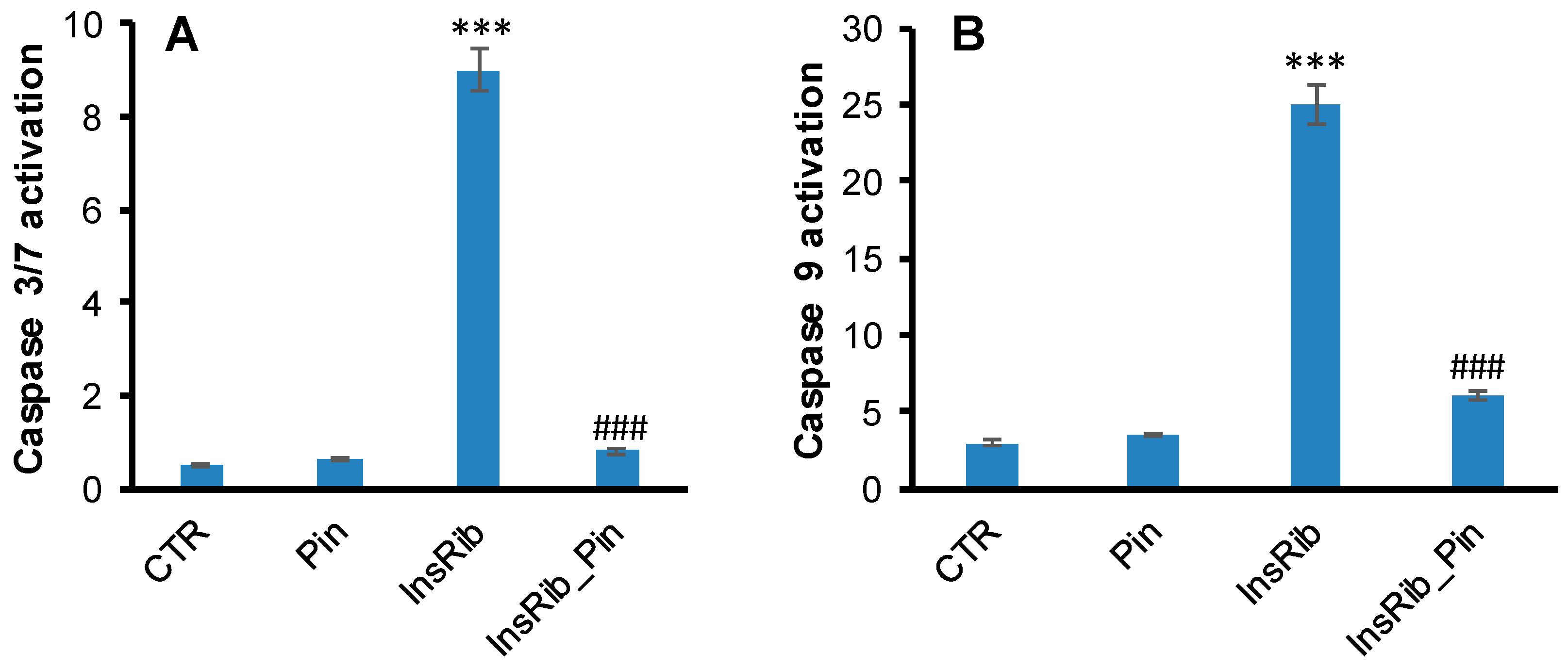
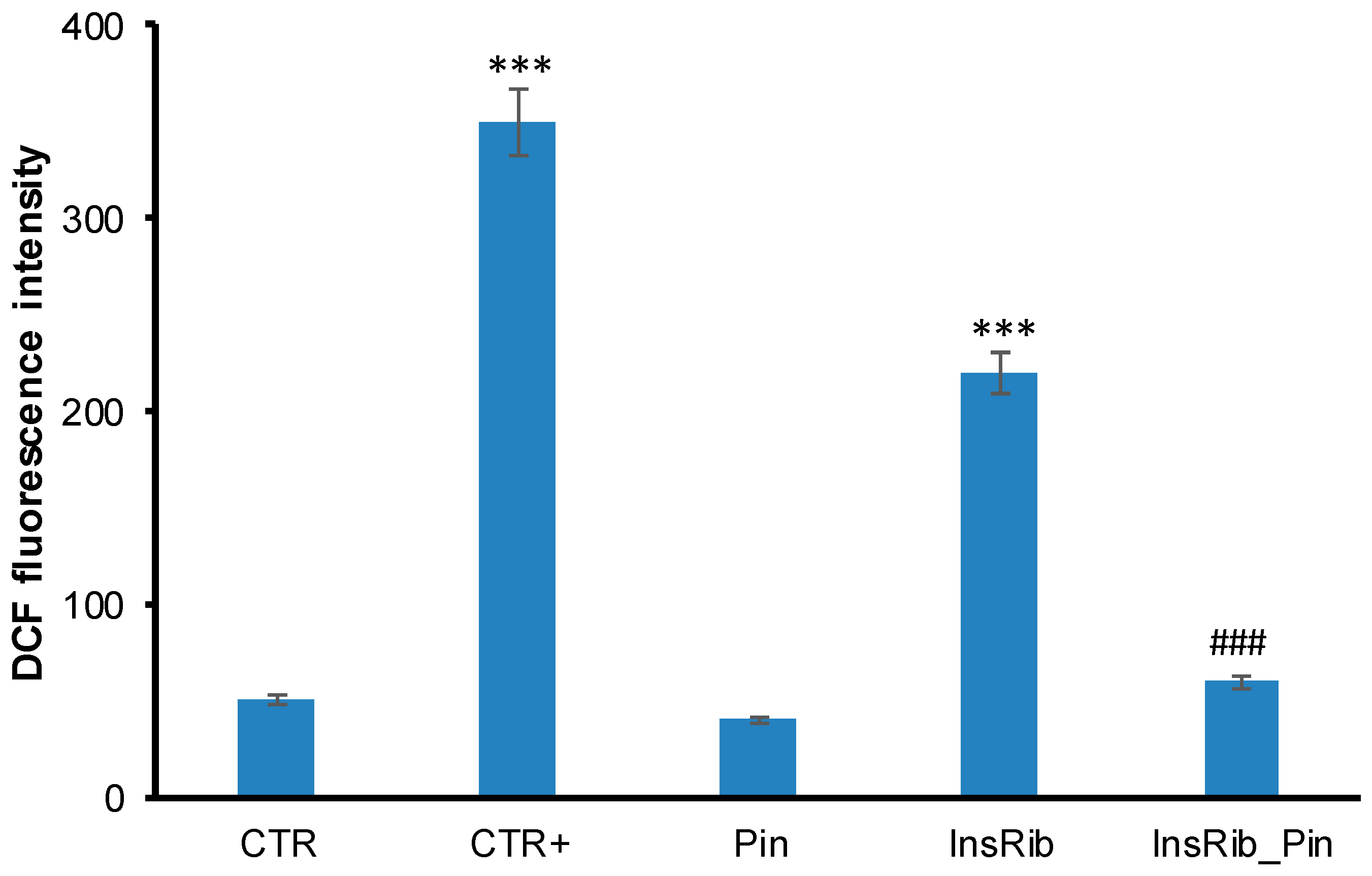
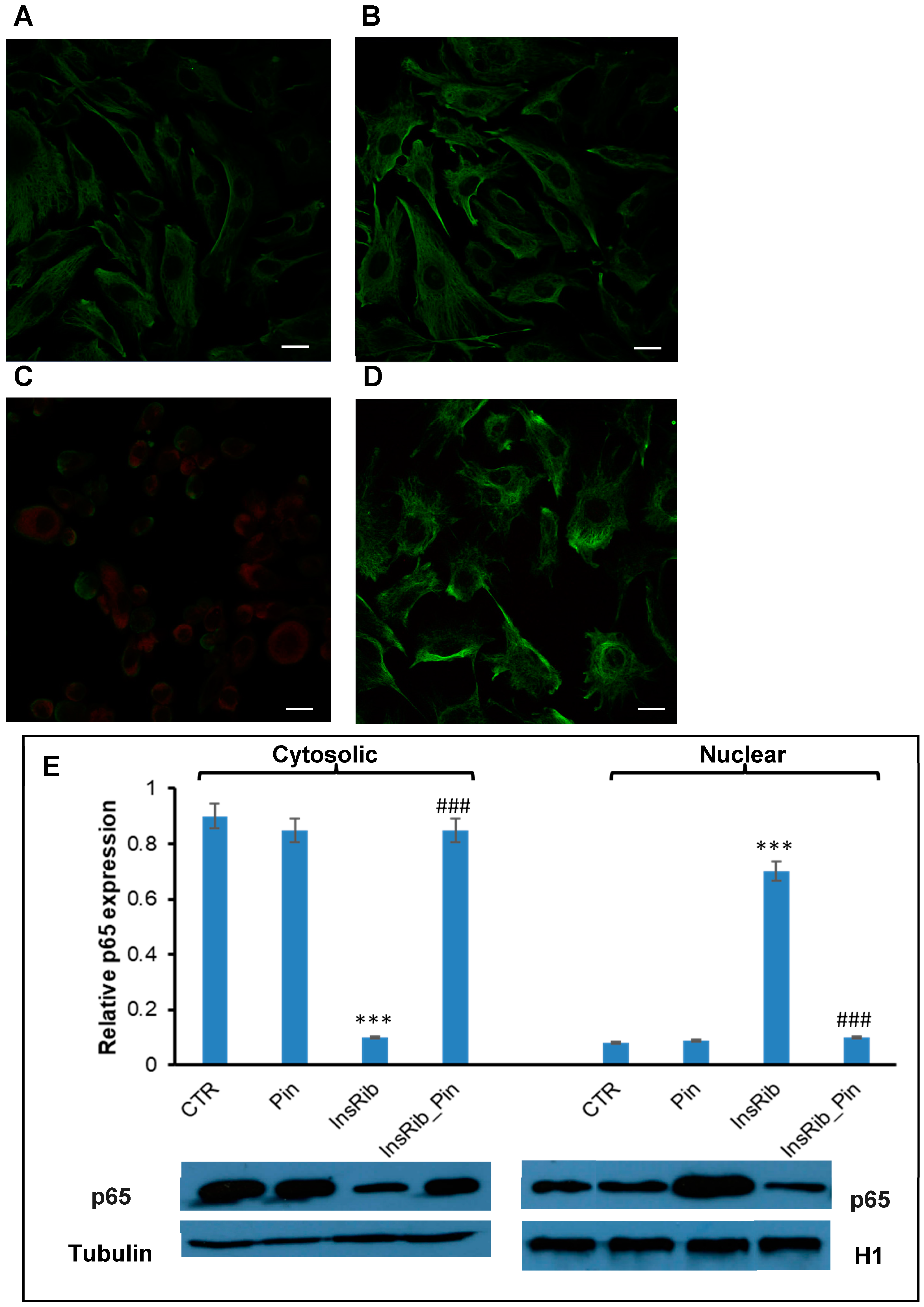
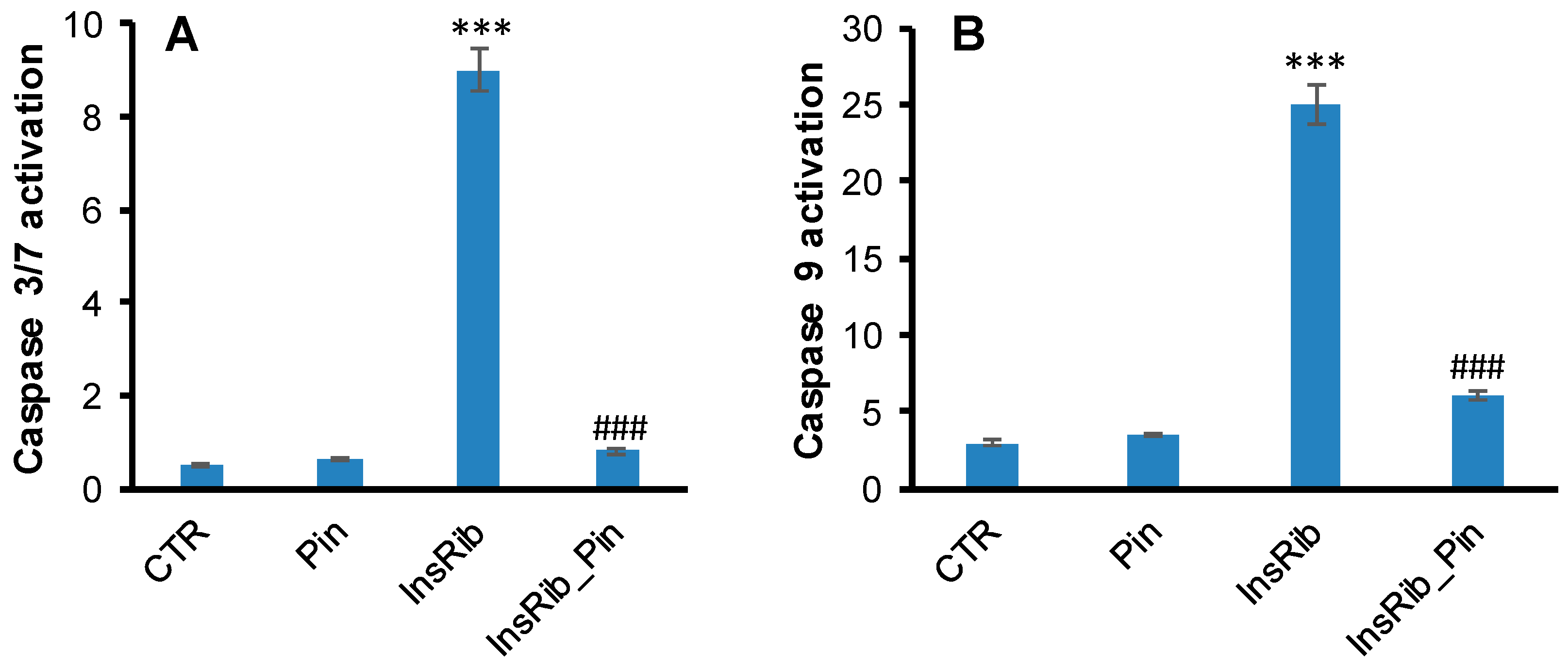
3.2. Pinocembrin Effect on AGE-Induced Oxidative Stress, NF-kB, and Caspase Activation
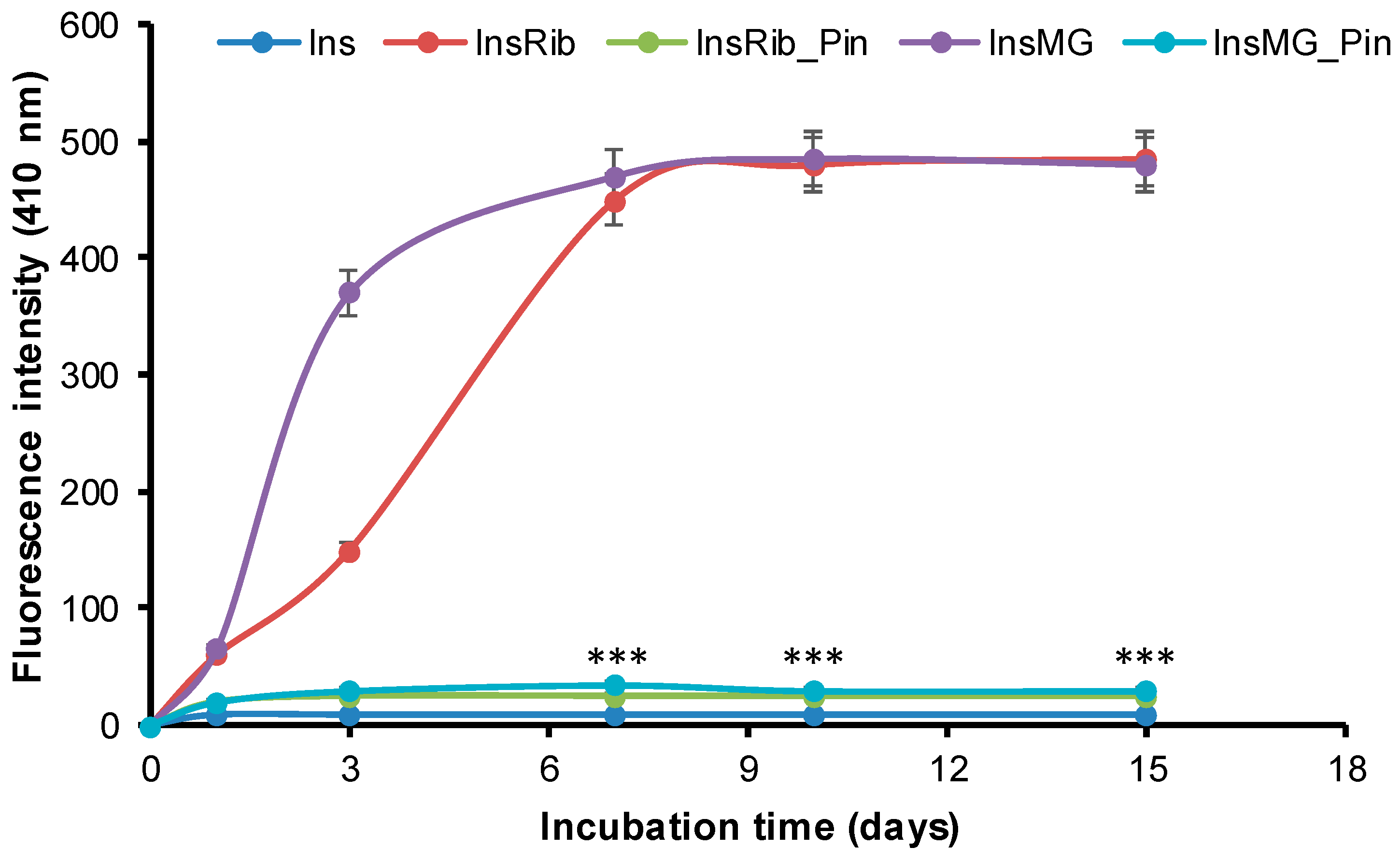
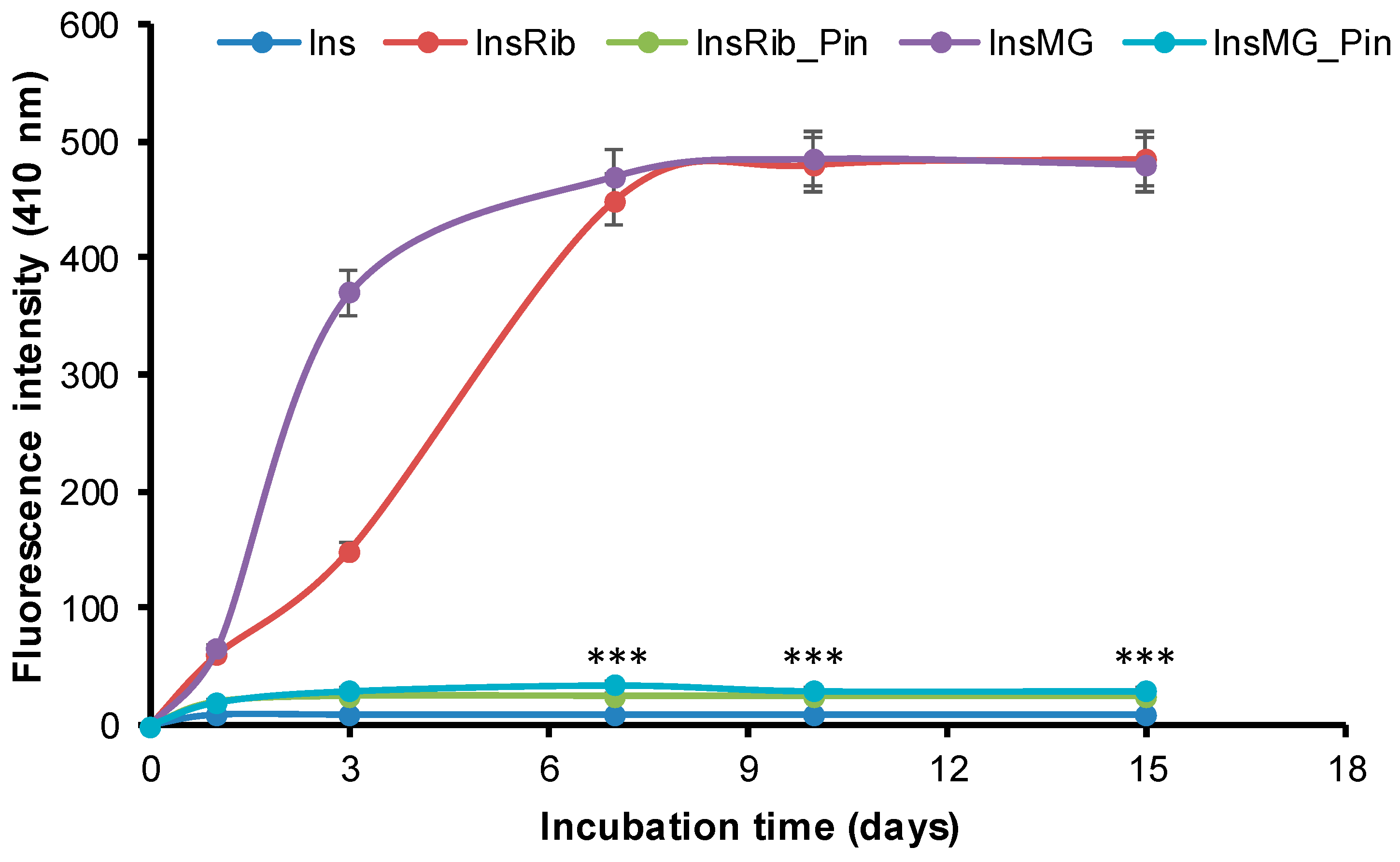
3.3. Pinocembrin Effect in Insulin Glycation Reaction
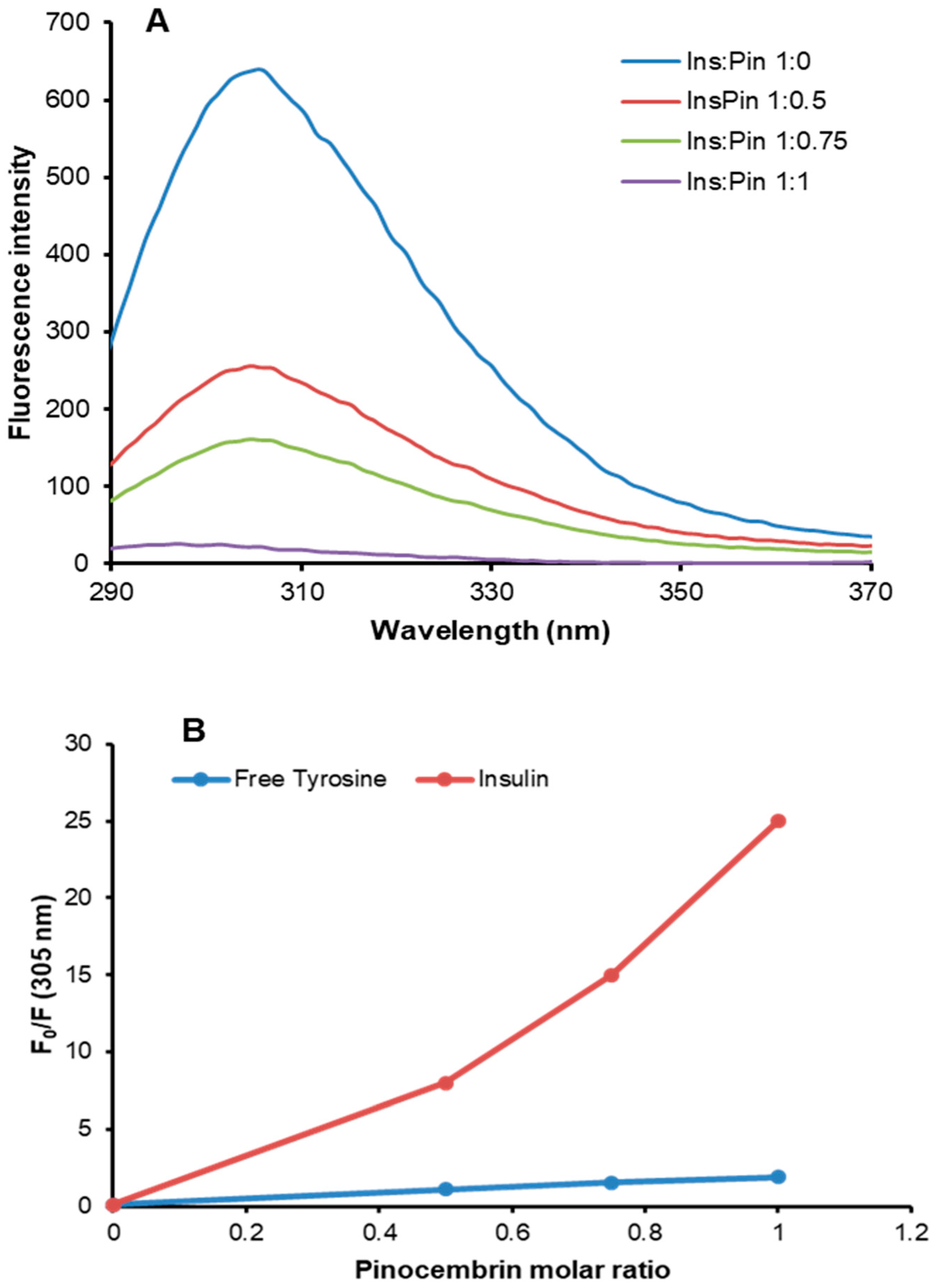
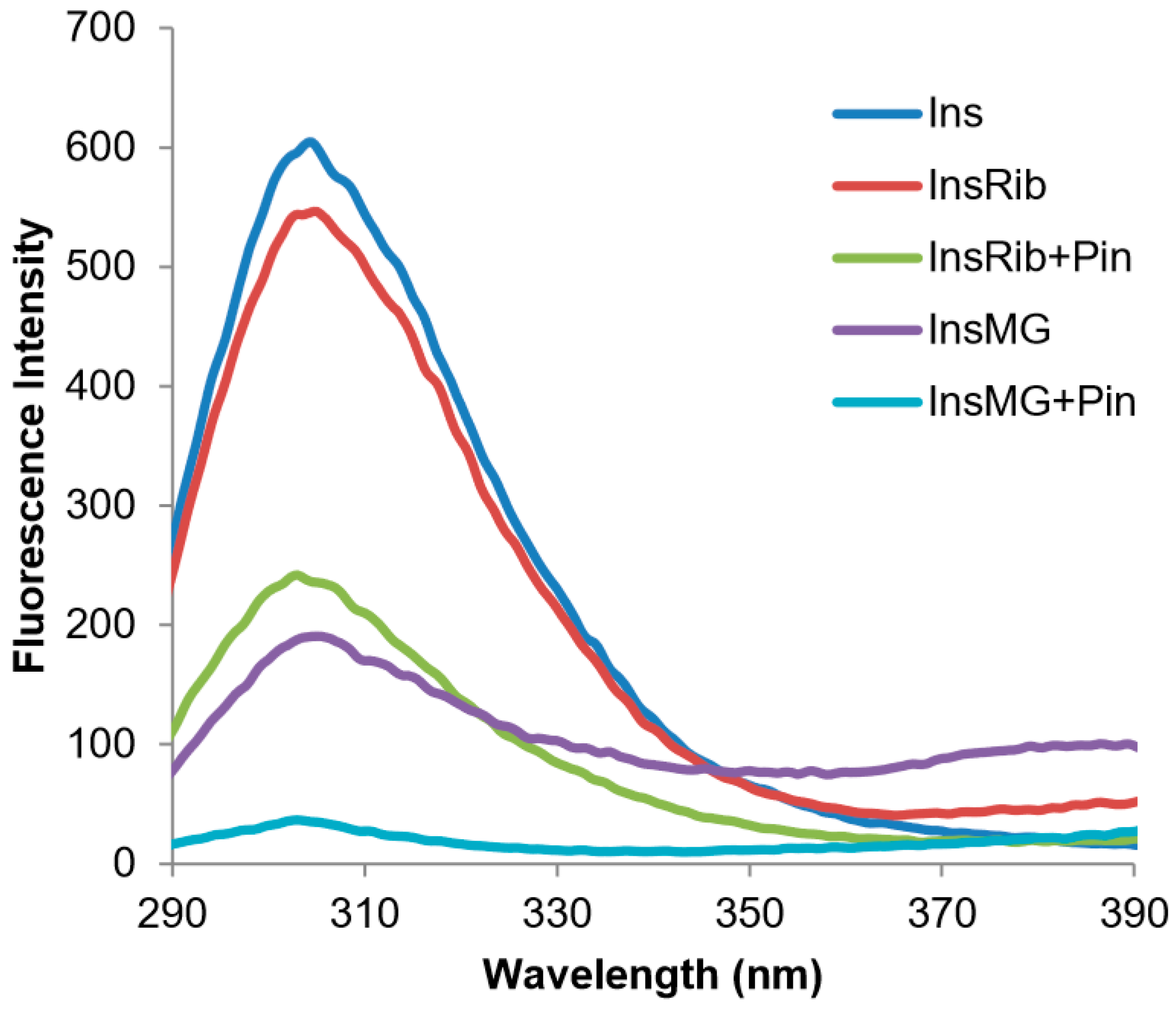
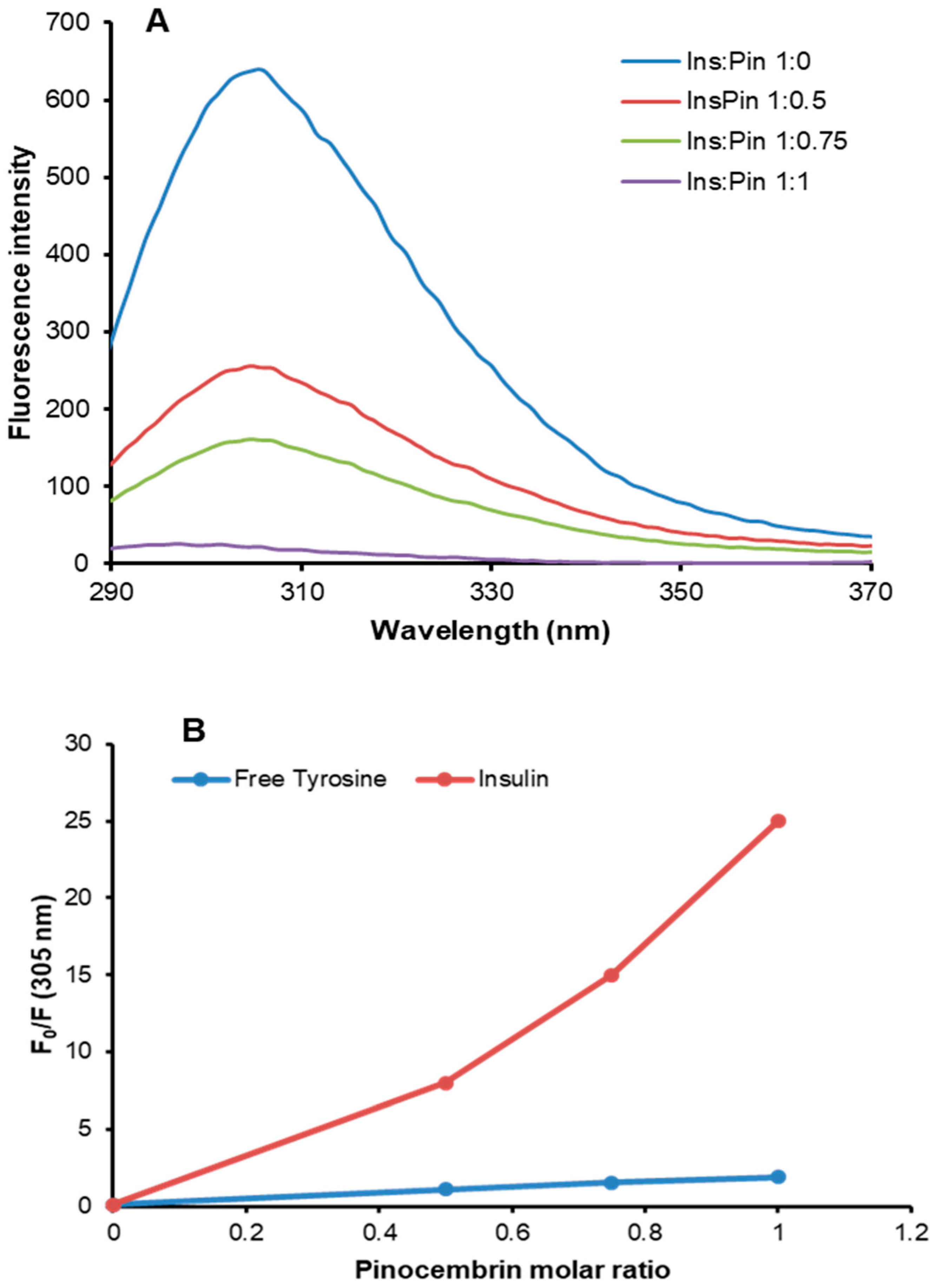
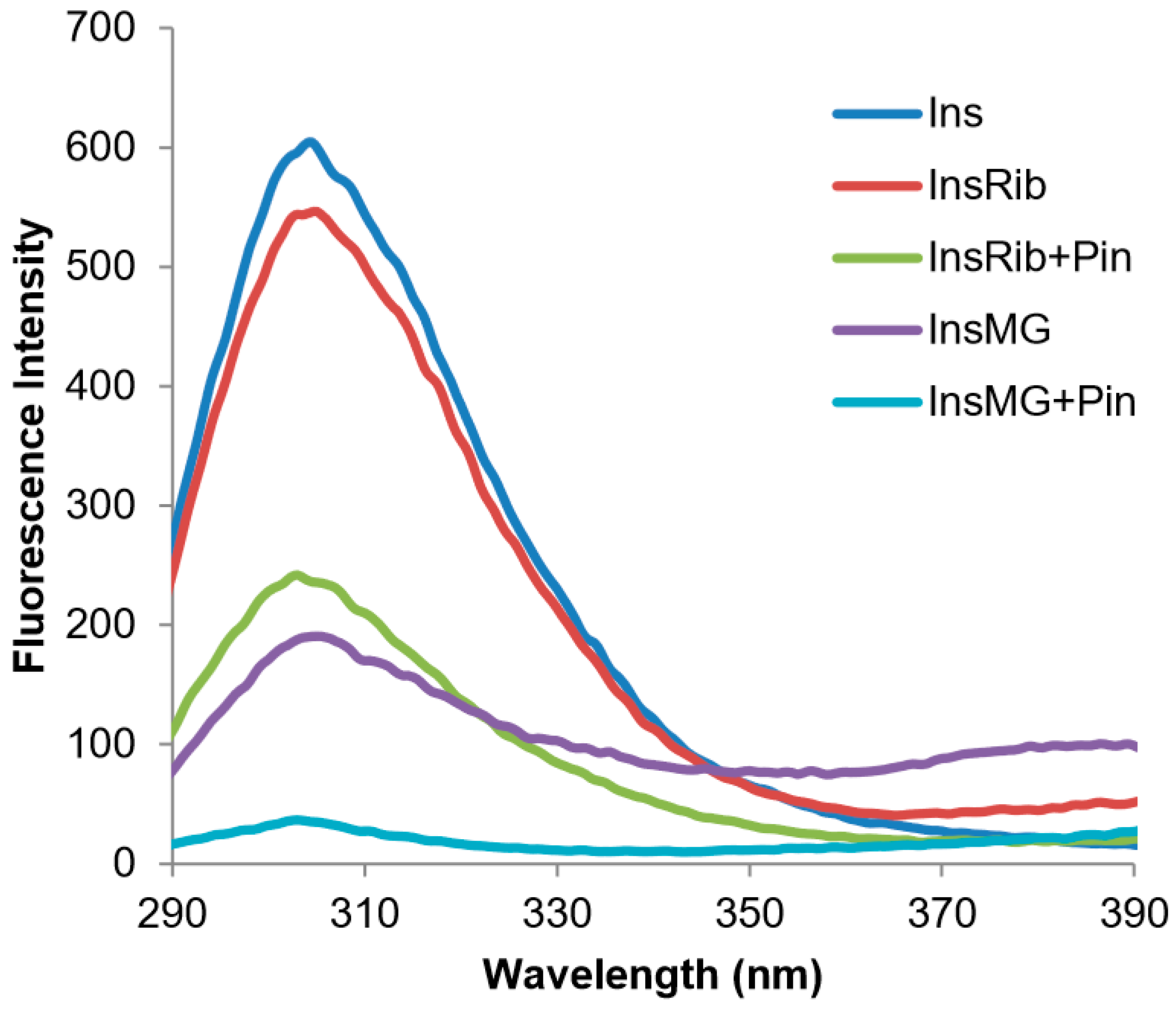
3.4. Characterization of Insulin–Pinocembrin Interaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rasul, A.; Millimouno, F.M.; Ali Eltayb, W.; Ali, M.; Li, J.; Li, X. Pinocembrin: A novel natural compound with versatile pharmacological and biological activities. Biomed. Res. Int. 2013, 2013, 379850. [Google Scholar] [CrossRef]
- Lan, X.; Wang, W.; Li, Q.; Wang, J. The Natural Flavonoid Pinocembrin: Molecular Targets and Potential Therapeutic Applications. Mol. Neurobiol. 2016, 53, 1794–1801. [Google Scholar] [CrossRef]
- Izuta, H.; Shimazawa, M.; Tazawa, S.; Araki, Y.; Mishima, S.; Hara, H. Protective effects of Chinese propolis and its component, chrysin, against neuronal cell death via inhibition of mitochondrial apoptosis pathway in SH-SY5Y cells. J. Agric. Food. Chem. 2008, 56, 8944–8953. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, W.C.; Liu, Q.S.; Hu, J.J.; Liu, G.T.; Du, G.H. Pinocembrin prevents glutamate-induced apoptosis in SH-SY5Y neuronal cells via decrease of bax/bcl-2 ratio. Eur. J. Pharmacol. 2008, 591, 73–79. [Google Scholar] [CrossRef]
- Gao, M.; Liu, R.; Zhu, S.Y.; Du, G.H. Acute neurovascular unit protective action of pinocembrin against permanent cerebral ischemia in rats. J. Asian Nat. Prod. Res. 2008, 10, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, M.; Yang, Z.H.; Du, G.H. Pinocembrin protects rat brain against oxidation and apoptosis induced by ischemia-reperfusion both in vivo and in vitro. Brain. Res. 2008, 1216, 104–115. [Google Scholar] [CrossRef]
- Shi, L.L.; Chen, B.N.; Gao, M.; Zhang, H.A.; Li, Y.J.; Wang, L.; Du, G.H. The characteristics of therapeutic effect of pinocembrin in transient global brain ischemia/reperfusion rats. Life Sci. 2011, 88, 521–528. [Google Scholar] [CrossRef]
- Wang, S.B.; Pang, X.B.; Gao, M.; Fang, L.H.; Du, G.H. Pinocembrin protects rats against cerebral ischemic damage through soluble epoxide hydrolase and epoxyeicosatrienoic acids. Chin. J. Nat. Med. 2013, 11, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, L.; Kong, L.; Zhu, Z.; Zhang, W.; Song, J.; Chang, J.; Du, G. Pinocembrin Protects Blood-Brain Barrier Function and Expands the Therapeutic Time Window for Tissue-Type Plasminogen Activator Treatment in a Rat Thromboembolic Stroke Model. Biomed. Res. Int. 2018, 8943210. [Google Scholar] [CrossRef] [PubMed]
- Sayre, C.L.; Takemoto, J.K.; Martinez, S.E.; Davies, N.M. Chiral analytical method development and application to pre-clinical pharmacokinetics of pinocembrin. Biomed. Chromatogr. 2013, 27, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Sun, X.; Qi, Y.; Mei, C.; Sun, X.B.; Du, G.H. Uptake characteristics of pinocembrin and its effect on p-glycoprotein at the blood-brain barrier in in vitro cell experiments. J. Asian. Nat. Prod. Res. 2012, 14, 14–21. [Google Scholar] [CrossRef]
- Liu, R.; Wu, C.X.; Zhou, D.; Yang, F.; Tian, S.; Zhang, L.; Zhang, T.T.; Du, G.H. Pinocembrin protects against beta-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med. 2012, 10, 105. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.Z.; Song, J.K.; Sun, J.L.; Li, Y.J.; Zhou, S.B.; Zhang, T.T.; Du, G.H. Pinocembrin protects human brain microvascular endothelial cells against fibrillar amyloid-β1–40 injury by suppressing the MAPK/NF-κB inflammatory pathways. Biomed. Res. Int. 2014, 470393. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.Z.; Song, J.K.; Zhou, D.; Huang, C.; Bai, X.Y.; Xie, T.; Zhang, X.; Li, Y.J.; Wu, C.X.; et al. Pinocembrin improves cognition and protects the neurovascular unit in Alzheimer related deficits. Neurobiol. Aging 2014, 35, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, J.; Miao, Y.; Cui, Q.; Zhao, W.; Zhang, J.; Wang, H. Pinocembrin Protects SH-SY5Y Cells Against MPP(+)-Induced Neurotoxicity Through the Mitochondrial Apoptotic Pathway. J. Mol. Neurosci. 2014, 53, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Miao, Y.; Mir, A.Z.; Cheng, L.; Wang, L.; Zhao, L.; Cui, Q.; Zhao, W.; Wang, H. Inhibition of beta-amyloid-induced neurotoxicity by pinocembrin through Nrf2/HO-1 pathway in SH-SY5Y cells. J. Neurol. Sci. 2016, 368, 223–230. [Google Scholar] [CrossRef]
- Jin, X.; Liu, Q.; Jia, L.; Li, M.; Wang, X. Pinocembrin attenuates 6-OHDA-induced neuronal cell death through Nrf2/ARE pathway in SH-SY5Y cells. Cell. Mol. Neurobiol. 2015, 35, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Lue, L.F.; Paul, G.; Patel, A.; Sabbagh, M.N. Receptor for advanced glycation endproduct modulators: A new therapeutic target in Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 393–399. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Sahagan, B.; Nelson, R.B.; Selmer, J.; Rothlein, R.; Bell, J.M. The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr. Opin. Investig. Drugs 2009, 10, 672–680. [Google Scholar] [PubMed]
- Yan, S.S.; Chen, D.; Yan, S.; Guo, L.; Du, H.; Chen, J.X. RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front. Biosci. (Schol Ed). 2012, 4, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Emendato, A.; Milordini, G.; Zacco, E.; Sicorello, A.; Dal Piaz, F.; Guerrini, R.; Thorogate, R.; Picone, D.; Pastore, A. Glycation affects fibril formation of Aβ peptides. J. Biol. Chem. 2018, 293, 3100–13111. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Onyango, I.G.; Tuttle, J.B.; Bennett, J.P. Jr. Altered intracellular signaling and reduced viability of Alzheimer’s disease neuronal cybrids is reproduced by beta-amyloid peptide acting through receptor for advanced glycation end products (RAGE). Mol. Cell. Neurosci. 2005, 29, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Miranda, V.; Outeiro, T.F. The sour side of neurodegenerative disorders: The effects of protein glycation. J. Pathol. 2010, 221, 13–25. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef]
- Van Puyvelde, K.; Mets, T.; Njemini, R.; Beyer, I.; Bautmans, I. Effect of advanced glycation end product intake on inflammation and aging: A systematic review. Nutr. Rev. 2014, 72, 638–650. [Google Scholar] [CrossRef]
- Salahuddin, P.; Rabbani, G.; Khan, R.H. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef]
- de Vos, L.C.; Lefrandt, J.D.; Dullaart, R.P.; Zeebregts, C.J.; Smit, A.J. Advanced glycation end products: An emerging biomarker for adverse outcome in patients with peripheral artery disease. Atherosclerosis 2016, 254, 291–299. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell. Death. Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent. Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta 2018, 1864, 3631–3643. [Google Scholar] [CrossRef]
- Bastos, D.H.M.; Gugliucci, A. Contemporary and controversial aspects of the Maillard reaction products. Curr. Opin. Food Sci. 2015, 1, 13–20. [Google Scholar] [CrossRef]
- Leone, S.; Fonderico, J.; Melchiorre, C.; Carpentieri, A.; Picone, D. Structural effects of methylglyoxal glycation, a study on the model protein MNEI. Mol Cell Biochem. 2019, 451, 165–171. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Prevention of protein glycation by natural compounds. Molecules 2015, 20, 3309–3334. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Irace, G.; Cammarota, M.; Di Maro, A.; Sirangelo, I. Vanillin affects amyloid aggregation and non-enzymatic glycation in human insulin. Sci. Rep. 2017, 7, 15086. [Google Scholar] [CrossRef]
- Yeh, W.J.; Hsia, S.M.; Lee, W.H.; Wu, C.H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food. Drug. Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Carafa, V.; Altucci, L.; Vitiello, M.; Balestrieri, M.L.; Ricci, G.; Irace, G.; Sirangelo, I. D-ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts. Biochim. Biophys. Acta 2016, 1862, 93–104. [Google Scholar] [CrossRef]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [PubMed]
- Smirnovas, V.; Winter, R. Revealing different aggregation pathways of amyloidogenic proteins by ultrasound velocimetry. Biophys. J. 2008, 94, 3241–3246. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Maritato, R.; Irace, G.; Sirangelo, I. Glycation Accelerates Fibrillization of the Amyloidogenic W7FW14F Apomyoglobin. PloS ONE 2013, 8, e80768. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Borriello, M.; D’Agostino, A.; Cimini, D.; Schiraldi, C.; Sirangelo, I. Protective effect of extractive and biotechnological chondroitin in insulin amyloid and advanced glycation end product-induced toxicity. J. Cell. Physiol. 2019, 234, 3814–3828. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Carafa, V.; Altucci, L.; Irace, G.; Borriello, M.; Vinciguerra, R.; Sirangelo, I. Glycation of Wild-Type Apomyoglobin Induces Formation of Highly Cytotoxic Oligomeric Species. J. Cell. Physiol. 2015, 230, 2807–2820. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Vella, F.M.; Irace, G.; Manco, G.; Iannuzzi, C. Glycation in Demetalated Superoxide Dismutase 1 Prevents Amyloid Aggregation and Produces Cytotoxic Ages Adducts. Front. Mol. Biosci. 2016, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Lages, A.; Gomes, R.A.; Neves, H.; Família, C.; Coelho, A.V.; Quintas, A. Insulin glycation by methylglyoxal results in native-like aggregation and inhibition of fibril formation. BMC Biochem. 2011, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Miyata, T. Early- and advanced non-enzymatic glycation in diabetic vascular complications: The search for therapeutics. Amino Acids 2012, 42, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G. Vascular AGE-ing by methylglyoxal: The past, the present and the future. Diabetologia 2015, 58, 1715–1719. [Google Scholar] [CrossRef]
- Tóbon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Target. 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Matiacevich, S.B.; Buera, M.P. A critical evaluation of fluorescence as a potential marker for the Maillard reaction. Food Chem. 2006, 95, 423–430. [Google Scholar] [CrossRef]
- Saad, M.A.; Abdel Salam, M.; Kenawy, S.A.; Attia, A.S. Pinocembrin attenuates hippocampal inflammation, oxidative perturbations and apoptosis in a rat model of global cerebral ischemia reperfusion. Pharmacol. Rep. 2015, 67, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.T.; Wang, K.J.; Li, L.; Li, H.; Geng, M. Pinocembrin inhibits lipopolysaccharide-induced inflammatory mediators production in BV2 microglial cells through suppression of PI3K/Akt/NF-κB pathway. Eur. J. Pharmacol. 2015, 761, 211–216. [Google Scholar] [CrossRef]
- Gu, X.; Zhang, Q.; Du, Q.; Shen, H.; Zhu, Z. Pinocembrin attenuates allergic airway inflammation via inhibition of NF-κB pathway in mice. Int. Immunopharmacol. 2017, 53, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A., Jr. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 6809703. [Google Scholar] [CrossRef]
- Guedes, S.; Vitorino, R.; Domingues, M.R.; Amado, F.; Domingues, P. Mass spectrometry characterization of theglycation sites of bovine insulin by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 1319–1326. [Google Scholar] [CrossRef]
- Pal, S.; Saha, C. A review on structure-affinity relationship of dietary flavonoids with serum albumins. J. Biomol. Struct. Dyn. 2014, 32, 1132–1147. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Galiniak, S.; Bartosz, G. Kinetics of glycoxidation of bovine serum albumin by glucose, fructose and ribose and its prevention by food components. Molecules 2014, 19, 18828–18849. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef]

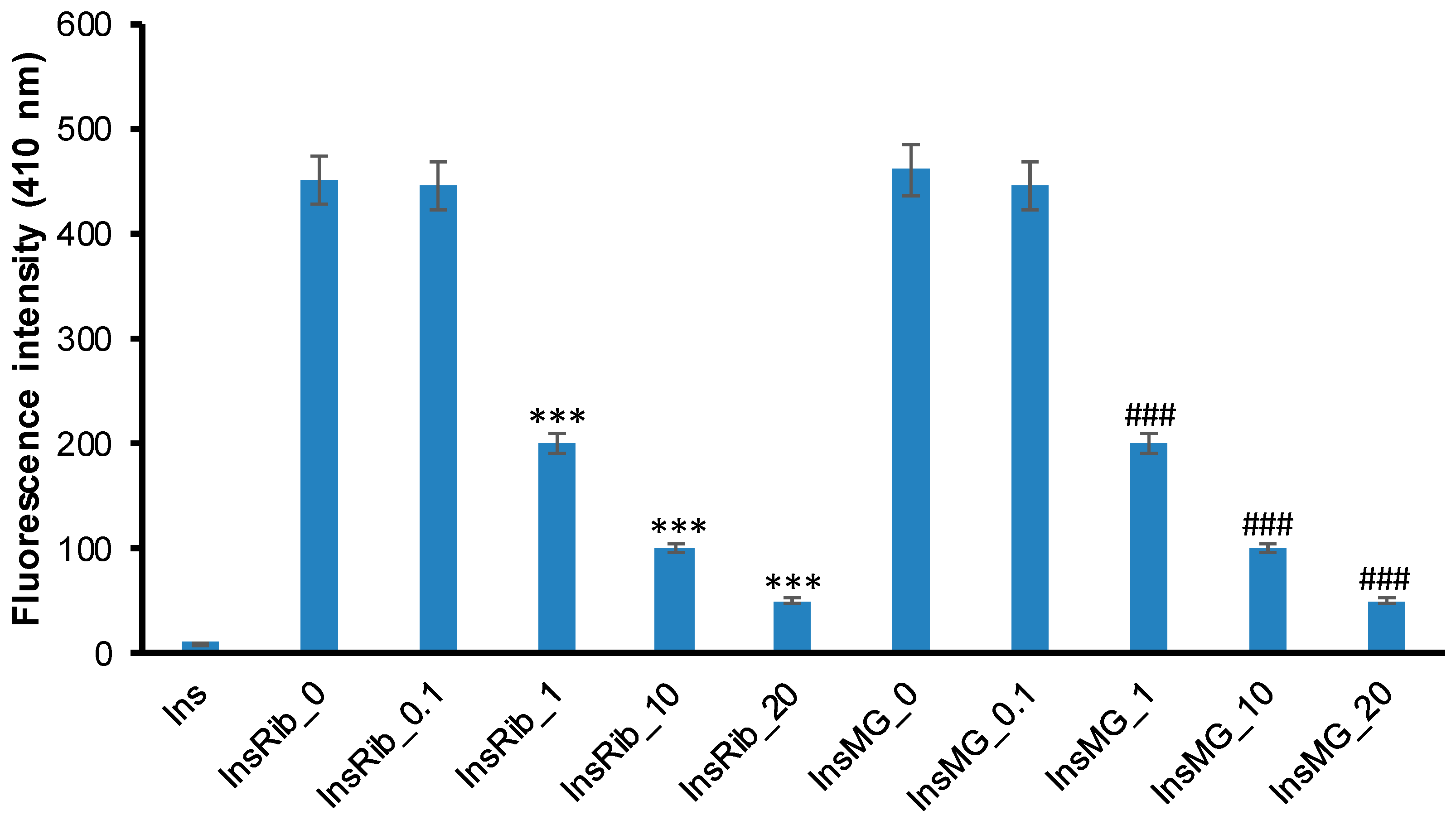
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borriello, M.; Iannuzzi, C.; Sirangelo, I. Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin. Cells 2019, 8, 385. https://doi.org/10.3390/cells8050385
Borriello M, Iannuzzi C, Sirangelo I. Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin. Cells. 2019; 8(5):385. https://doi.org/10.3390/cells8050385
Chicago/Turabian StyleBorriello, Margherita, Clara Iannuzzi, and Ivana Sirangelo. 2019. "Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin" Cells 8, no. 5: 385. https://doi.org/10.3390/cells8050385
APA StyleBorriello, M., Iannuzzi, C., & Sirangelo, I. (2019). Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin. Cells, 8(5), 385. https://doi.org/10.3390/cells8050385