Mitochondrial DNA: Distribution, Mutations, and Elimination



{kind=link}
{kind=link}
Abstract
1. Introduction
2. mtDNA Structure
3. mtDNA Mutation and Human Diseases
4. mtDNA Distribution
4.1. mtDNA Distribution and Mitochondrial Dynamics
4.2. mtDNA Distribution and Inner Membrane Structure
4.3. mtDNA Distribution and Cholesterol
5. mtDNA Release and Inflammasome
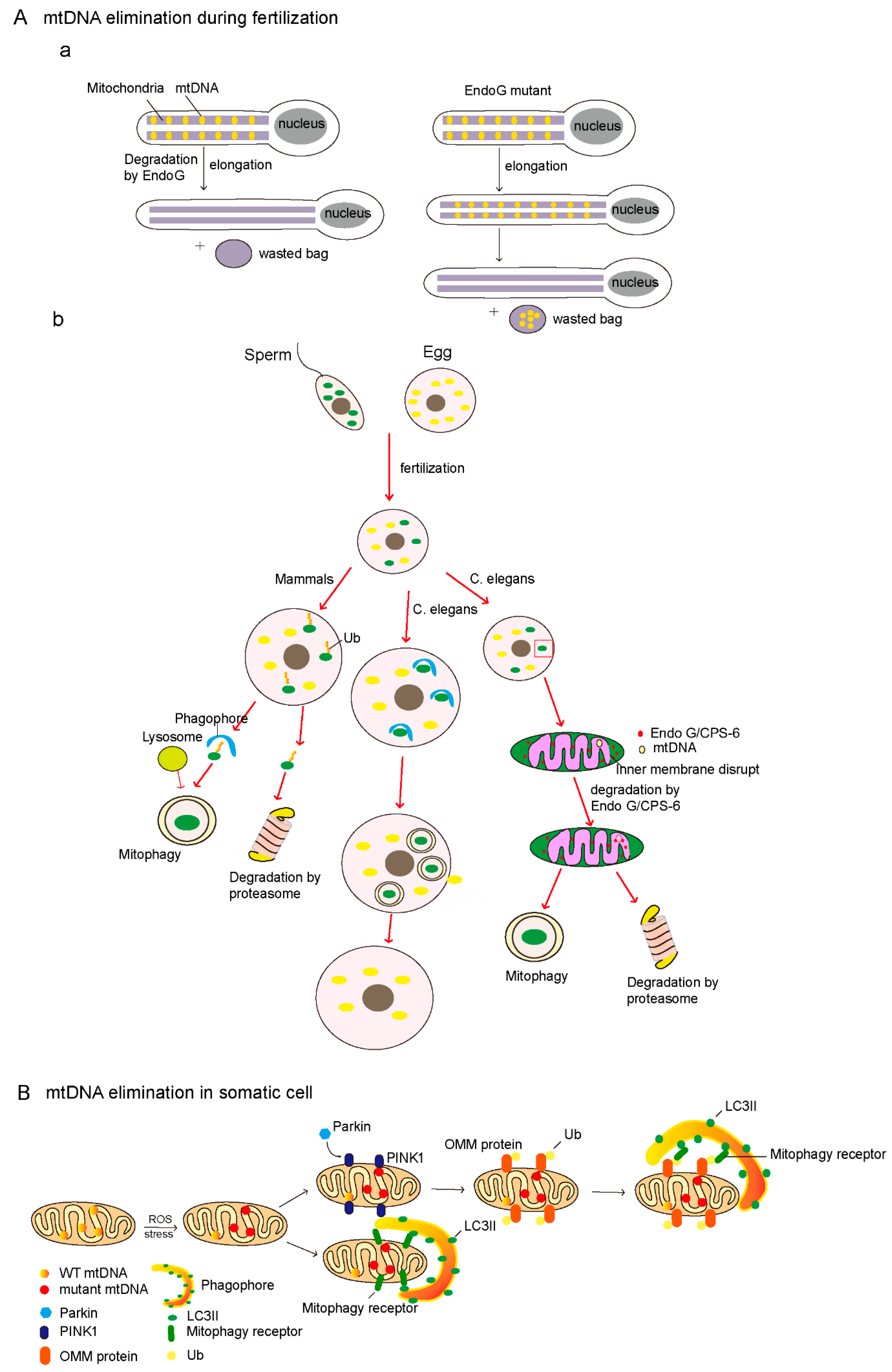
6. mtDNA Elimination
6.1. Endonuclease G-Mediated Degradation of Paternal mtDNA
6.2. Mitophagy-Mediated Degradation of Paternal mtDNA
6.3. The Clearance of Mutant mtDNA
7. Perspectives
Funding
Conflicts of Interest
References
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Martinou, J.C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000, 10, 369–377. [Google Scholar] [CrossRef]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, N.; Nishihama, T.; Owaki, H.; Kuramoto, Y.; Suenaga, M.; Miyoshi, E.; Emoto, Y.; Shibata, H.; Shono, M.; Higuti, T. Dynamics of mitochondria during the cell cycle. Biol. Pharm. Bull. 2006, 29, 1962–1965. [Google Scholar] [CrossRef]
- Finkel, T.; Hwang, P.M. The Krebs cycle meets the cell cycle: Mitochondria and the G(1)-S transition. Proc. Natl. Acad. Sci. USA 2009, 106, 11825–11826. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Maniura-Weber, K.; Goffart, S.; Garstka, H.L.; Montoya, J.; Weisner, R.J. Transient overexpression of mitochondrial transcription factor A (TFAM) is sufficient to stimulate mitochondrial DNA transcription, but not sufficient to increase mtDNA copy number in cultured cells. Nucleic Acids Res. 2004, 32, 6015–6027. [Google Scholar] [CrossRef] [PubMed]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Weidemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.-J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123, 1339–1348. [Google Scholar] [CrossRef]
- Stefano, G.B.; Kream, R.M. Mitochondrial DNA heteroplasmy in human health and disease. Biomed. Rep. 2016, 4, 259–262. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmüller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1727. [Google Scholar] [CrossRef]
- DeLuca, S.Z.; O’Farrell, P.H. Barriers to male transmission of mitochondrial DNA in sperm development. Dev. Cell 2012, 22, 660–668. [Google Scholar] [CrossRef]
- Chan, D.C.; Schon, E.A. Eliminating mitochondrial DNA from sperm. Dev. Cell 2012, 22, 469–470. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Li, H.M.; Li, H.Z.; Nakagawa, A.; Lin, L.J.L.; Lee, E.S.; Harry, B.L.; Skeen-Gaar, R.R.; Suehiro, Y.; William, D.; et al. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 2016, 353, 394–399. [Google Scholar] [CrossRef]
- Song, W.H.; Yi, Y.J.; Sutovsky, M.; Meyers, S.; Sutovsky, P. Autophagy and ubiquitin-proteasome system contribute to sperm mitophagy after mammalian fertilization. Proc. Natl. Acad. Sci. USA 2016, 113, E5261–E5270. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, T.; Chuang, C.-C. The Consequences of Damaged Mitochondrial DNA. In Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease; Buhlman, L.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 49–61. [Google Scholar]
- Nicholls, T.J.; Gustafsson, C.M. Separating and Segregating the Human Mitochondrial Genome. Trends Biochem. Sci. 2018, 43, 869–881. [Google Scholar] [CrossRef]
- Sbisa, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian mitochondrial D-loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktov, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- van Oven, M.; Kayser, M. Updated Comprehensive Phylogenetic Tree of Global Human Mitochondrial DNA Variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morganhughes, J.A. Deletions of Muscle Mitochondrial-DNA in Patients with Mitochondrial Myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Zeviani, M.; Moraes, C.T.; DiMauro, S.; Nakase, H.; Bonilla, E.; Schon, E.A.; Rowland, L.P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988, 38, 1339. [Google Scholar] [CrossRef]
- Wallace, D.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Howell, N.; Bindoff, L.A.; McCullough, D.A.; Kubacka, I.; Poulton, J.; Mackey, D.; Taylor, L.; Turnbull, D.M. Leber hereditary optic neuropathy: Identification of the same mitochondrial ND1 mutation in six pedigrees. Am. J. Hum. Genet. 1991, 49, 939–950. [Google Scholar]
- Johns, D.R.; Neufeld, M.J.; Park, R.D. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1992, 187, 1551–1557. [Google Scholar] [CrossRef]
- de Vries, D.D.; van Engelen, B.G.; Gabreëls, F.J.; Ruitenbeek, W.; van Oost, B.A. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh’s syndrome. Ann. Neurol. 1993, 34, 410–412. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A New Mitochondrial Disease Associated with Mitochondrial-DNA Heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Silvestri, G.; Moraes, G.T.; Shanske, S.; Oh, S.J.; Di Mauro, S. A new mtDNA mutation in the tRNA(Lys) gene associated with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet. 1992, 51, 1213–1217. [Google Scholar]
- Ballinger, S.W.; Shoffner, J.M.; Hedaya, E.V.; Trounce, I.; Polak, M.A.; Koontz, D.A.; Wallace, D.C. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat. Genet. 1992, 1, 11–15. [Google Scholar] [CrossRef]
- Kadowaki, T.; Kadowaki, H.; Mori, Y.; Tobe, K.; Sakuta, R.; Suzuki, Y.; Tanabe, Y.; Sakura, H.; Awata, T.; Goto, Y.; et al. A Subtype of Diabetes Mellitus Associated with a Mutation of Mitochondrial DNA. N. Engl. J. Med. 1994, 330, 962–968. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA Mutations in Disease and Aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Howell, N.; Elson, J.L.; Chinnery, P.F.; Turnbull, D.M. mtDNA mutations and common neurodegenerative disorders. Trends Genet. 2005, 21, 583–586. [Google Scholar] [CrossRef]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Chen, H.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; Parman, Y.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Malka, F.; Lombes, A.; Rojo, M. Organization, dynamics and transmission of mitochondrial DNA: Focus on vertebrate nucleoids. Biochim. Biophys. Acta 2006, 1763, 463–472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Osman, C.; Noriega, T.R.; Okreglak, V.; Fung, J.C.; Walter, P. Integrity of the yeast mitochondrial genome, but not its distribution and inheritance, relies on mitochondrial fission and fusion. Proc. Natl. Acad. Sci. USA 2015, 112, E947–E956. [Google Scholar] [CrossRef]
- Jayashankar, V.; Rafelski, S.M. Integrating mitochondrial organization and dynamics with cellular architecture. Curr. Opin. Cell Biol. 2014, 26, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; de Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy plus phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011, 21, 12–20. [Google Scholar] [CrossRef]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef]
- Nakada, K.; Inoue, K.; Ono, T.; Isobe, K.; Ogura, A.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001, 7, 934–940. [Google Scholar] [CrossRef]
- Murley, A.; Lackner, L.L.; Osman, C.; West, M.; Voeltz, G.K.; Walter, P.; Nunnari, J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife 2013, 2, e00422. [Google Scholar] [CrossRef]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef]
- Vogel, F.; Bornhövd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247. [Google Scholar] [CrossRef]
- Yamamoto, H.; Esaki, M.; Kanamori, T.; Tamura, Y.; Nishikawa, S.; Endo, T. Tim50, a new subunit of the TIM23 complex that links mitochondrial protein translocation across the outer membrane and that across the inner membrane. Mol. Biol. Cell 2002, 13, 128a. [Google Scholar]
- Donzeau, M.; Káldi, K.; Adam, A.; Paschen, S.; Wanner, G.; Guiard, B.; Bauer, M.F.; Neupert, W.; Brunner, M. Tim23 links the inner and outer mitochondrial membranes. Cell 2000, 101, 401–412. [Google Scholar] [CrossRef]
- Schorr, S.; van der Laan, M. Integrative functions of the mitochondrial contact site and cristae organizing system. Semin. Cell Dev. Biol. 2018, 76, 191–200. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Renken, C.; Siragusa, G.; Perkins, G.; Washington, L.; Nulton, J.; Salamon, P.; Frey, T.G. A thermodynamic model describing the nature of the crista junction: A structural motif in the mitochondrion. J. Struct. Biol. 2002, 138, 137–144. [Google Scholar] [CrossRef]
- van der Laan, M.; Horvath, S.E.; Pfanner, N. Mitochondrial contact site and cristae organizing system. Curr. Opin. Cell Biol. 2016, 41, 33–42. [Google Scholar] [CrossRef]
- Pfanner, N.; van der Laan, M.; Amati, P.; Capaldi, R.A.; Caudy, A.A.; Chacinska, A.; Darshi, M.; Deckers, M.; Hoppins, S.; Icho, T.; et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014, 204, 1083–1086. [Google Scholar] [CrossRef]
- Itoh, K.; Tamura, Y.; Iijima, M.; Sesaki, H. Effects of Fcj1-Mos1 and mitochondrial division on aggregation of mitochondrial DNA nucleoids and organelle morphology. Mol. Biol. Cell 2013, 24, 1842–1851. [Google Scholar] [CrossRef]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef]
- Kozjak-Pavlovic, V. The MICOS complex of human mitochondria. Cell Tissue Res. 2017, 367, 83–93. [Google Scholar] [CrossRef]
- Jian, F.; Chen, D.; Chen, L.; Yan, C.; Lu, B.; Zhu, Y.; Chen, S.; Shi, A.; Chan, D.C.; Song, Z. Sam50 Regulates PINK1-Parkin-Mediated Mitophagy by Controlling PINK1 Stability and Mitochondrial Morphology. Cell Rep. 2018, 23, 2989–3005. [Google Scholar] [CrossRef]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 2002, 110, 597–603. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Gerhold, J.M.; Cansiz-Arda, Ş.; Lõhmus, M.; Engberg, O.; Reyes, A.; van Rennes, H.; Sanz, A.; Holt, I.J.; Cooper, H.M.; Spelbrink, J.N. Human Mitochondrial DNA-Protein Complexes Attach to a Cholesterol-Rich Membrane Structure. Sci. Rep. 2015, 5, 15292. [Google Scholar] [CrossRef]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef]
- Fujimoto, M.; Hayashi, T.; Su, T.P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef]
- He, J.; Mao, C.C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Merle, N.; Charton, R.; Brandolin, G.; Rousseau, D. Topological analysis of ATAD3A insertion in purified human mitochondria. J. Bioenerg. Biomembr. 2010, 42, 143–150. [Google Scholar] [CrossRef]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef]
- Peralta, S.; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismäki, J.; Moraes, C.T. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 2018, 131, jcs217075. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef]
- Wang, X.D. Hot papers—Apoptosis—Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked by J. Yang, X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai, T.I. Peng, D.P. Jones, and X. Wang—Comments. Scientist 1999, 13, 16. [Google Scholar]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.J.; Chen, Z.J.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immun. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.H.; Chen, Z.J.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta 2013, 1833, 1979–1984. [Google Scholar] [CrossRef]
- Gyllensten, U.; Wharton, D.; Josefsson, A.; Wilson, A.C. Paternal inheritance of mitochondrial DNA in mice. Nature 1991, 352, 255–257. [Google Scholar] [CrossRef]
- Yu, Z.; O’Farrell, P.H.; Yakubovich, N.; DeLuca, S.Z. The Mitochondrial DNA Polymerase Promotes Elimination of Paternal Mitochondrial Genomes. Curr. Biol. 2017, 27, 1033–1039. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef]
- Parrish, J.; Li, L.; Klotz, K.; Ledwich, D.; Wang, X.; Xue, D. Mitochondrial endonuclease G is important for apoptosis in C. elegans. Nature 2001, 412, 90–94. [Google Scholar] [CrossRef]
- Parrish, J.Z.; Yang, C.; Shen, B.; Xue, D. CRN-1, a Caenorhabditis elegans FEN-1 homologue, cooperates with CPS-6/EndoG to promote apoptotic DNA degradation. EMBO J. 2003, 22, 3451–3460. [Google Scholar] [CrossRef]
- Song, W.H.; Ballard, J.W.; Yi, Y.J.; Sutovsky, P. Regulation of mitochondrial genome inheritance by autophagy and ubiquitin-proteasome system: Implications for health, fitness, and fertility. Biomed. Res. Int. 2014, 2014, 981867. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef]
- Sutovsky, P.; McCauley, T.C.; Sutovsky, M.; Day, B.N. Early degradation of paternal mitochondria in domestic pig (Sus scrofa) is prevented by selective proteasomal inhibitors lactacystin and MG132. Biol. Reprod. 2003, 68, 1793–1800. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA Degradation of paternal mitochondria by allogeneic organelle autophagy, allophagy. Autophagy 2012, 8, 424–425. [Google Scholar] [CrossRef]
- Thompson, W.E.; Ramalho-Santos, J.; Sutovsky, P. Ubiquitination of Prohibitin in Mammalian Sperm Mitochondria: Possible Roles in the Regulation of Mitochondrial Inheritance and Sperm Quality Control1. Biol. Reprod. 2003, 69, 254–260. [Google Scholar] [CrossRef]
- Sutovsky, P.; van Leyen, K.; McCauley, T.; Day, B.N.; Sutovsky, M. Degradation of paternal mitochondria after fertilization: Implications for heteroplasmy, assisted reproductive technologies and mtDNA inheritance. Reprod. Biomed. Online 2004, 8, 24–33. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Degradation of Paternal Mitochondria by Fertilization-Triggered Autophagy in C. elegans Embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Thorburn, D.R.; Samuels, D.C.; White, S.L.; Dahl, H.M.; Turnbull, D.M.; Lightowlers, R.N.; Howell, N. The inheritance of mitochondrial DNA heteroplasmy: Random drift, selection or both? Trends Genet. 2000, 16, 500–505. [Google Scholar] [CrossRef]
- Kandul, N.P.; Zhang, T.; Hay, B.A.; Guo, M. Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat. Commun. 2016, 7, 13100. [Google Scholar] [CrossRef]
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938. [Google Scholar] [CrossRef]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Tanaka, M.; Borgeld, H.J.; Zhang, J.; Muramatsu, S.; Gong, J.S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002, 9, 534–541. [Google Scholar]
- Bayona-Bafaluy, M.P.; Blits, B.; Battersby, B.J.; Shoubridge, E.A.; Moraes, C.T. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 14392–14397. [Google Scholar] [CrossRef]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J.; et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015, 161, 459–469. [Google Scholar] [CrossRef]
- Ding, C.; Wu, Z.; Huang, L.; Wang, Y.; Xue, J.; Chen, S.; Deng, Z.; Wang, L.; Song, Z.; Chen, S. Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci. Rep. 2015, 5, 16064. [Google Scholar] [CrossRef]
- Sastri, M.; Darshi, M.; Mackey, M.; Ramachandra, R.; Ju, S.; Phan, S.; Adams, S.; Stein, K.; Douglas, C.R.; Kim, J.J.; et al. Sub-mitochondrial localization of the genetic-tagged mitochondrial intermembrane space-bridging components Mic19, Mic60 and Sam50. J. Cell Sci. 2017, 130, 3248–3260. [Google Scholar] [CrossRef]
- Gilkerson, R.; Bravo, L.; Garcia, I.; Gaytan, N.; Herrera, A.; Maldonado, A.; Quintanilla, B. The Mitochondrial Nucleoid: Integrating Mitochondrial DNA into Cellular Homeostasis. Cold Spring Harb. Perspect. Biol. 2013, 5, a011080. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. https://doi.org/10.3390/cells8040379
Yan C, Duanmu X, Zeng L, Liu B, Song Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells. 2019; 8(4):379. https://doi.org/10.3390/cells8040379
Chicago/Turabian StyleYan, Chaojun, Xiaoying Duanmu, Ling Zeng, Bing Liu, and Zhiyin Song. 2019. "Mitochondrial DNA: Distribution, Mutations, and Elimination" Cells 8, no. 4: 379. https://doi.org/10.3390/cells8040379
APA StyleYan, C., Duanmu, X., Zeng, L., Liu, B., & Song, Z. (2019). Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells, 8(4), 379. https://doi.org/10.3390/cells8040379