Myricetin Suppresses the Propagation of Hepatocellular Carcinoma via Down-Regulating Expression of YAP

 , and
, and 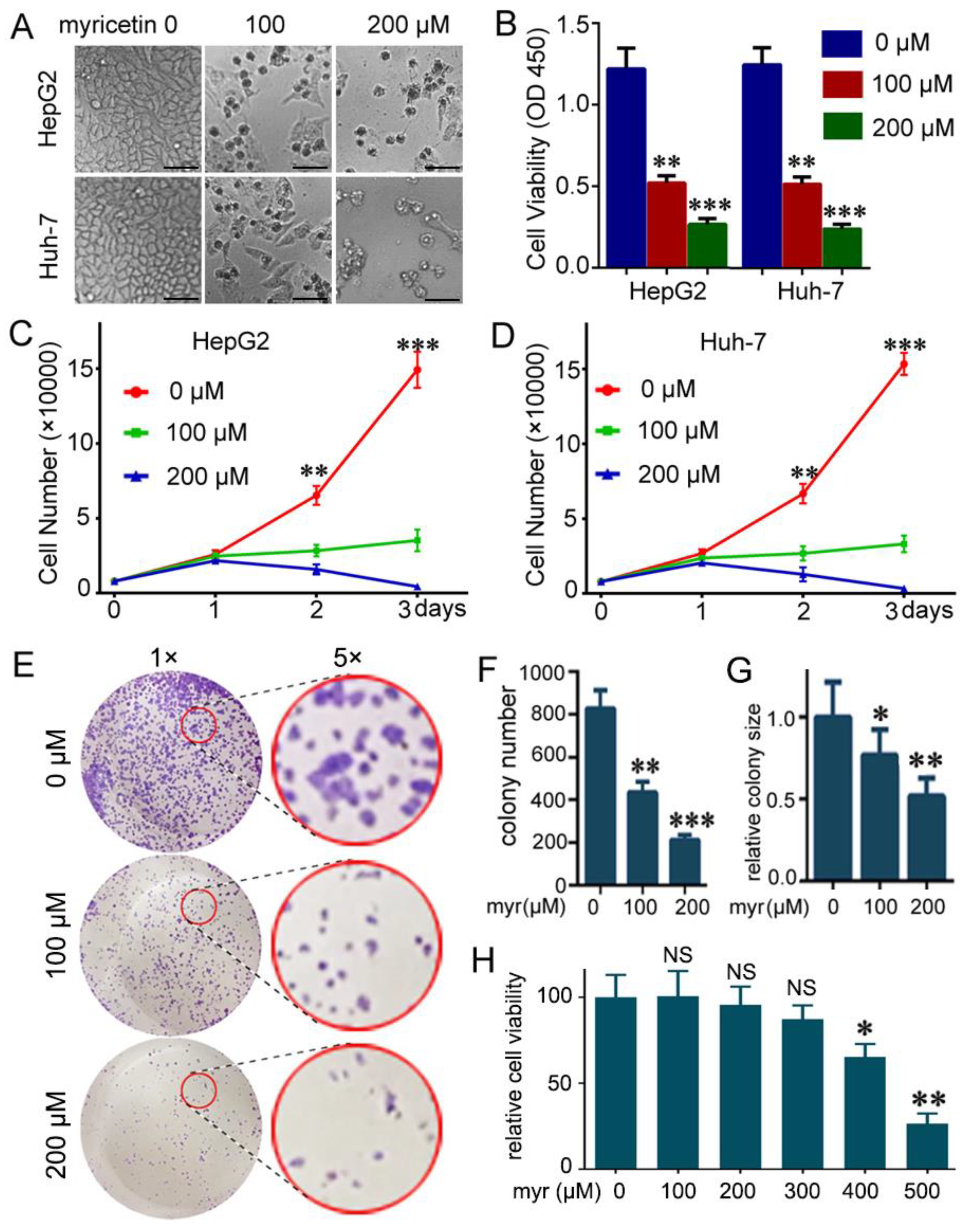{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Cell Growth Assay
2.4. Cell Proliferation/Apoptosis Assay
2.5. Colony Formation Assay
2.6. Quantitative Real-Time PCR
2.7. Lentivirus Construction and Infection
2.8. Western Blot
2.9. In Vitro Kinase Assays
2.10. Xenograft Tumor Growth Assay
2.11. DNA Fragmentation Detection
2.12. Statistical Analysis
3. Results
3.1. Myricetin Suppressed Viability and Colony Formation of HCC Cells
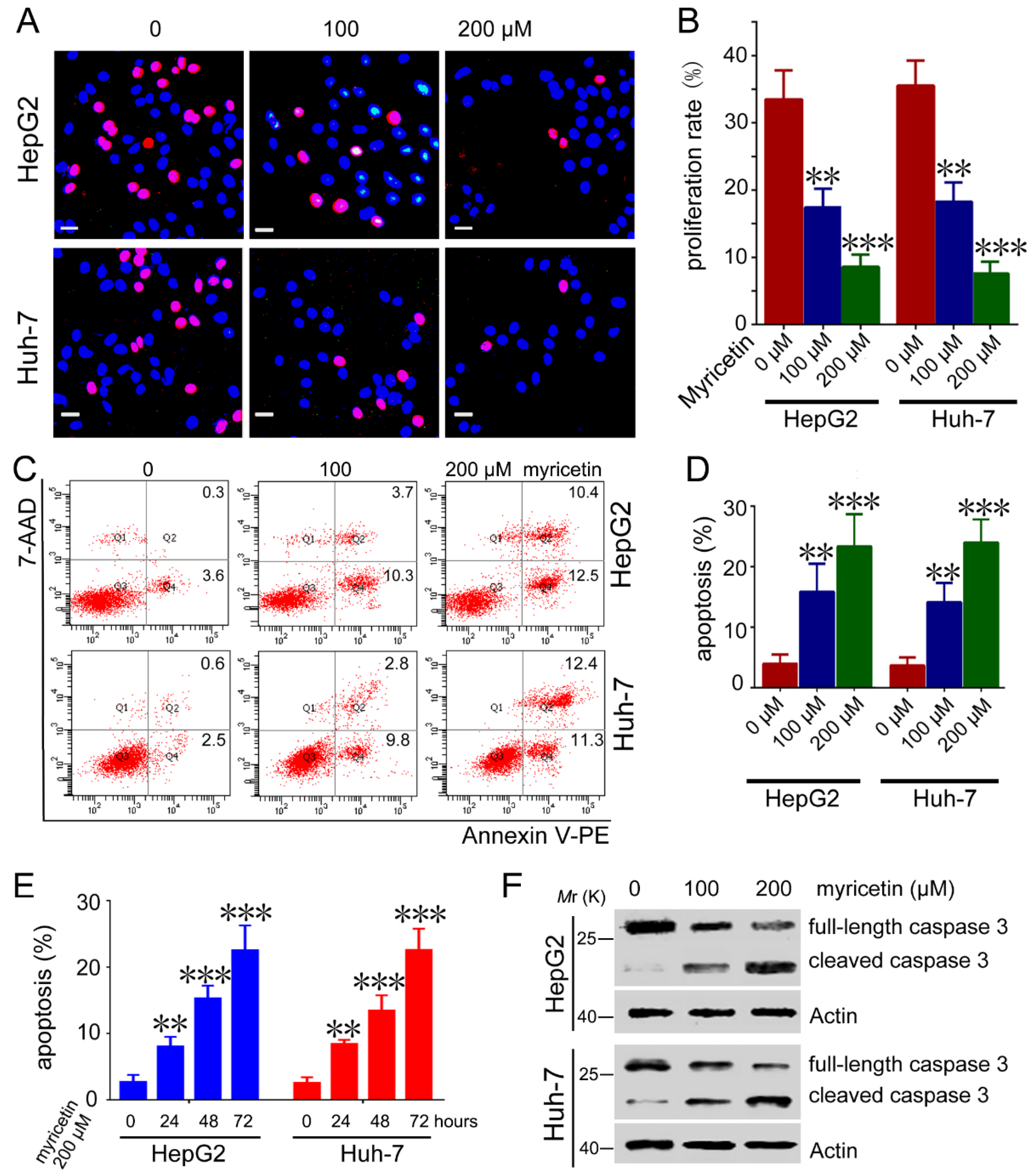
3.2. Myricetin Induced Apoptosis and Reduced Cell Proliferation of HCC Cells
3.3. Myricetin Decreased Expression of YAP and Its Target Genes
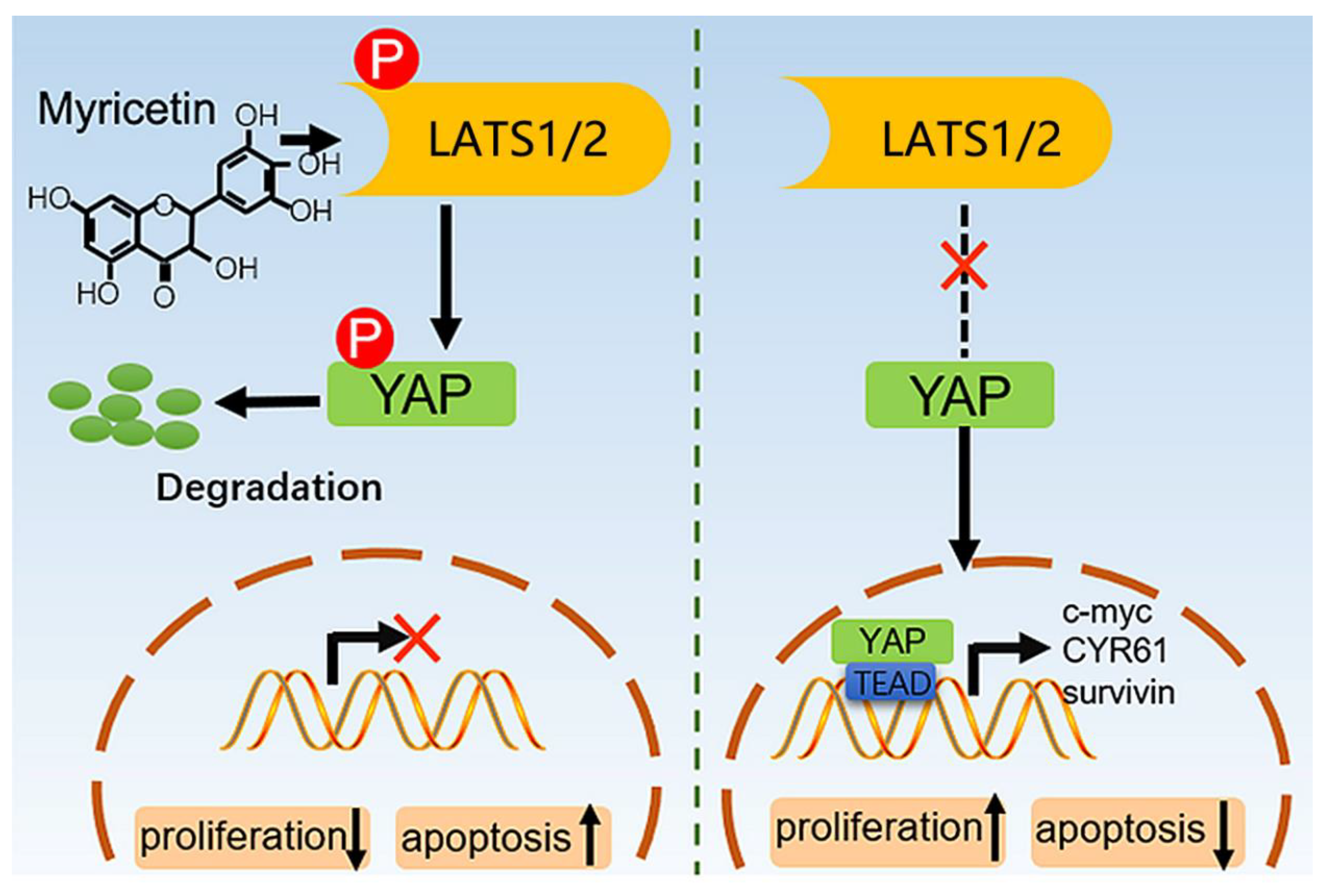
3.4. Myricetin Promotes YAP Degradation by Stimulating LATS1/2 Activation
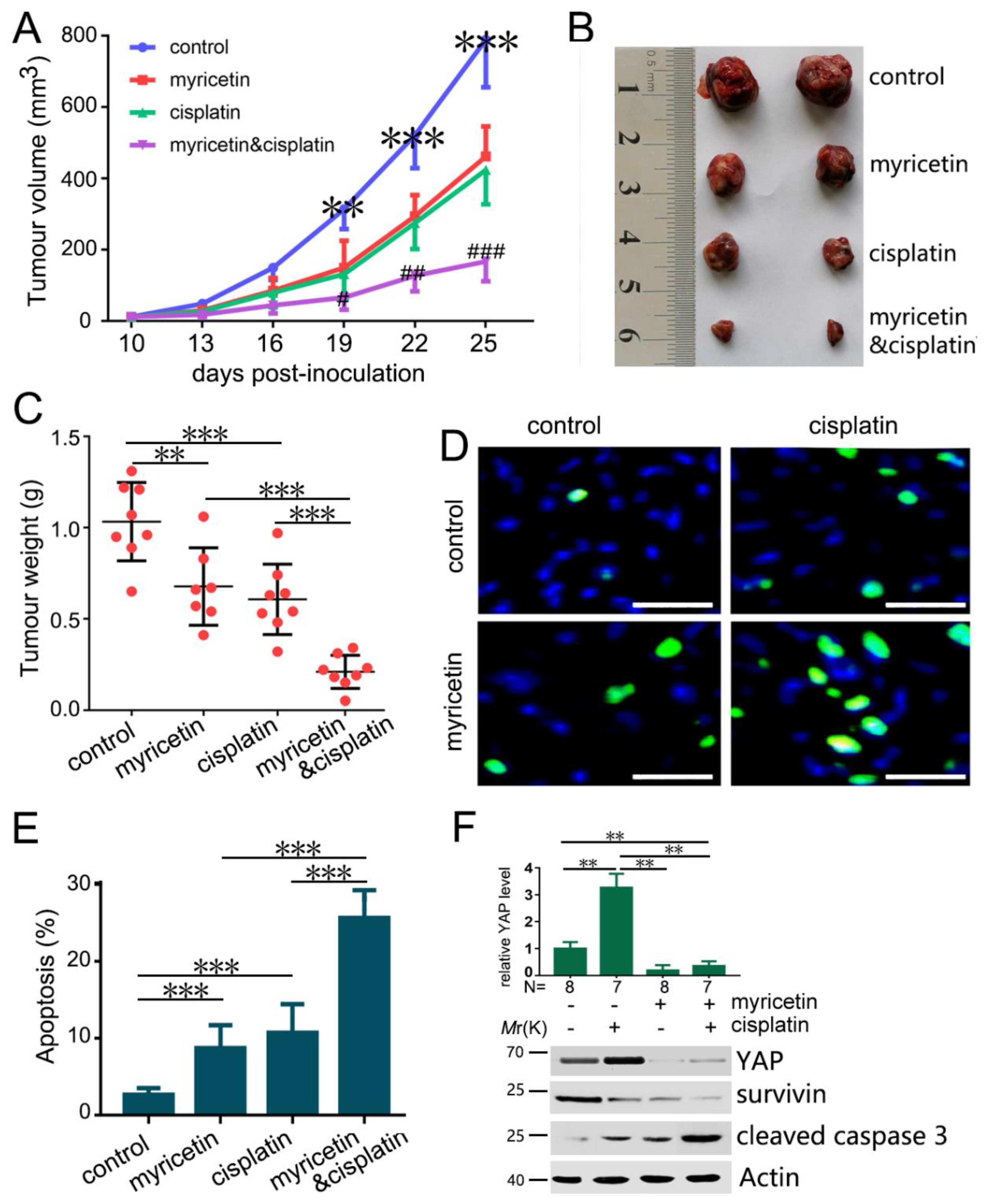
3.5. Down-Regulation of YAP by Myricetin Sensitized HCC Cells to Cisplatin
3.6. The Combination of Myricetin and Cisplatin Synergistically Inhibits Tumor Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Moukhadder, H.M.; Halawi, R.; Cappellini, M.D.; Taher, A.T. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: A comprehensive review. Cancer 2017, 123, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Vitale, A.; Peck-Radosavljevic, M.; Giannini, E.G.; Vibert, E.; Sieghart, W.; Van Poucke, S.; Pawlik, T.M. Personalized treatment of patients with very early hepatocellular carcinoma. J. Hepatol. 2017, 66, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Mahato, R.I. Recent advances in hepatocellular carcinoma therapy. Pharmacol. Ther. 2017, 173, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Afaq, F.; Mukhtar, H. Cancer chemoprevention through dietary antioxidants: Progress and promise. Antioxid. Redox Signal. 2008, 10, 475–510. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Rajavel, T.; Habtemariam, S.; Nabavi, S.F.; Nabavi, S.M. Molecular mechanisms underlying anticancer effects of myricetin. Life Sci. 2015, 142, 19–25. [Google Scholar] [CrossRef]
- Zheng, A.W.; Chen, Y.Q.; Zhao, L.Q.; Feng, J.G. Myricetin induces apoptosis and enhances chemosensitivity in ovarian cancer cells. Oncol. Lett. 2017, 13, 4974–4978. [Google Scholar] [CrossRef] [Green Version]
- Jiao, D.; Zhang, X.D. Myricetin suppresses p21-activated kinase 1 in human breast cancer mcf-7 cells through downstream signaling of the beta-catenin pathway. Oncol. Rep. 2016, 36, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, X.; Wang, Y.; Du, Y.; Sun, Q.; Zang, W.; Zhao, G. Myricetin inhibits proliferation and induces apoptosis and cell cycle arrest in gastric cancer cells. Mol. Cell Biochem. 2015, 408, 163–170. [Google Scholar] [CrossRef]
- Jung, S.K.; Lee, K.W.; Byun, S.; Kang, N.J.; Lim, S.H.; Heo, Y.S.; Bode, A.M.; Bowden, G.T.; Lee, H.J.; Dong, Z. Myricetin suppresses uvb-induced skin cancer by targeting fyn. Cancer Res. 2008, 68, 6021–6029. [Google Scholar] [CrossRef]
- Yang, C.; Lim, W.; Bazer, F.W.; Song, G. Myricetin suppresses invasion and promotes cell death in human placental choriocarcinoma cells through induction of oxidative stress. Cancer Lett. 2017, 399, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Zou, Z.Q.; Xu, C.W.; Shen, Y.Z.; Li, D. Myricetin induces g2/m phase arrest in hepg2 cells by inhibiting the activity of the cyclin b/cdc2 complex. Mol. Med. Rep. 2011, 4, 273–277. [Google Scholar]
- Seydi, E.; Rasekh, H.R.; Salimi, A.; Mohsenifar, Z.; Pourahmad, J. Myricetin selectively induces apoptosis on cancerous hepatocytes by directly targeting their mitochondria. Basic Clin. Pharmacol. Toxicol. 2016, 119, 249–258. [Google Scholar] [CrossRef]
- Zhang, X.H.; Chen, S.Y.; Tang, L.; Shen, Y.Z.; Luo, L.; Xu, C.W.; Liu, Q.; Li, D. Myricetin induces apoptosis in hepg2 cells through akt/p70s6k/bad signaling and mitochondrial apoptotic pathway. Anticancer Agents Med. Chem. 2013, 13, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Haza, A.I. Selective apoptotic effects of piceatannol and myricetin in human cancer cells. J. Appl. Toxicol. 2012, 32, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. Yap/taz at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, Q.; Zhang, J.; Xu, X.; Liu, X.; Wang, Q.; Yi, R.; Xie, X.; Yao, L.; Liu, W.; et al. Functional and clinical evidence that taz is a candidate oncogene in hepatocellular carcinoma. J. Cell Biochem. 2015, 116, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jiang, N.; Zhou, B.; Liu, Q.; Du, C. Taz regulates cell proliferation and epithelial-mesenchymal transition of human hepatocellular carcinoma. Cancer Sci. 2015, 106, 151–159. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. Mtorc2 promotes type i insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mtor. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yin, Y.; Li, C.; Chen, J.; Xie, J.; Lu, Z.; Li, M.; Wang, Y.; Zhang, C.C. Acer3 supports development of acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2016, 478, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ji, X.; Zhang, H.; Zhou, Q.; Cao, X.; Tang, M.; Si, Y.; Yan, H.; Li, L.; Liang, T.; et al. Deubiquitylase usp9x suppresses tumorigenesis by stabilizing large tumor suppressor kinase 2 (lats2) in the hippo pathway. J. Biol. Chem. 2017, 293, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by lats and ck1 regulates yap stability through scf(beta-trcp). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, J. Role of yap/taz transcriptional regulators in resistance to anti-cancer therapies. Cell Mol. Life Sci. 2017, 74, 1457–1474. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Kang, N.J.; Rogozin, E.A.; Kim, H.G.; Cho, Y.Y.; Bode, A.M.; Lee, H.J.; Surh, Y.J.; Bowden, G.T.; Dong, Z. Myricetin is a novel natural inhibitor of neoplastic cell transformation and mek1. Carcinogenesis 2007, 28, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Fujii, M.; Hou, D.X. Myricetin directly targets jak1 to inhibit cell transformation. Cancer Lett. 2009, 275, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Yang, H.J.; Youn, H.; Yun, Y.J.; Seong, K.M.; Youn, B. Myricetin inhibits akt survival signaling and induces bad-mediated apoptosis in a low dose ultraviolet (uv)-b-irradiated hacat human immortalized keratinocytes. J. Radiat. Res. 2010, 51, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the tead-yap complex suppresses the oncogenic activity of yap. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking vgll4 function acts as a yap antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cao, Y.; Xu, J.; Wang, Y.; Li, W.; Wang, Q.; Hu, Z.; Hao, Y.; Hu, L.; Sun, Y.; et al. Yap transcriptionally regulates cox-2 expression and gccsysm-4 (g-4), a dual yap/cox-2 inhibitor, overcomes drug resistance in colorectal cancer. J. Exp. Clin. Cancer Res. 2017, 36, 144. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.L.; Ciavattone, N.; Grun, D.; Adhikary, G.; Eckert, R.L. Sulforaphane reduces yap/np63alpha signaling to reduce cancer stem cell survival and tumor formation. Oncotarget 2017, 8, 73407–73418. [Google Scholar] [CrossRef] [PubMed]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The hippo effector yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of yap oncoprotein by the hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Iyer, S.C.; Gopal, A.; Halagowder, D. Myricetin induces apoptosis by inhibiting p21 activated kinase 1 (pak1) signaling cascade in hepatocellular carcinoma. Mol. Cell Biochem. 2015, 407, 223–237. [Google Scholar] [CrossRef]
- Chakraborty, S.; Njah, K.; Pobbati, A.V.; Lim, Y.B.; Raju, A.; Lakshmanan, M.; Tergaonkar, V.; Lim, C.T.; Hong, W. Agrin as a mechanotransduction signal regulating yap through the hippo pathway. Cell Rep. 2017, 18, 2464–2479. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. Yap and taz: A nexus for hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Khanal, P.; Savage, P.; She, Y.M.; Cyr, T.D.; Yang, X. Yap-induced resistance of cancer cells to antitubulin drugs is modulated by a hippo-independent pathway. Cancer Res. 2014, 74, 4493–4503. [Google Scholar] [CrossRef] [PubMed]
- Ciamporcero, E.; Shen, H.; Ramakrishnan, S.; Yu Ku, S.; Chintala, S.; Shen, L.; Adelaiye, R.; Miles, K.M.; Ullio, C.; Pizzimenti, S.; et al. Yap activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene 2016, 35, 1541–1553. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, Z.; Rodriguez-Barrueco, R.; Borczuk, A.; Liu, H.; Yu, J.; Silva, J.M.; Cheng, S.K.; Perez-Soler, R.; Halmos, B. Functional genomics screen identifies yap1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget 2016, 7, 28976–28988. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.W.; Shao, G.L.; Luo, J.; Hao, W.Y.; Yao, Z.; Zheng, J.P. Yap regulates the proliferation and modifies the sensitivity to sorafenib in hepatocellular carcinoma cells. Zhonghua zhong liu za zhi 2018, 40, 818–823. [Google Scholar]
- Maronpot, R.R.; Koyanagi, M.; Davis, J.; Recio, L.; Marbury, D.; Boyle, M.; Hayashi, S.M. Safety assessment and single-dose toxicokinetics of the flavouring agent myricitrin in sprague-dawley rats. Food Addit. Contam. Part. A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 1799–1809. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Chen, J.; Yu, X.; Xu, S.; Li, D.; Zheng, Q.; Yin, Y. Myricetin Suppresses the Propagation of Hepatocellular Carcinoma via Down-Regulating Expression of YAP. Cells 2019, 8, 358. https://doi.org/10.3390/cells8040358
Li M, Chen J, Yu X, Xu S, Li D, Zheng Q, Yin Y. Myricetin Suppresses the Propagation of Hepatocellular Carcinoma via Down-Regulating Expression of YAP. Cells. 2019; 8(4):358. https://doi.org/10.3390/cells8040358
Chicago/Turabian StyleLi, Minjing, Jinliang Chen, Xiaofei Yu, Sen Xu, Defang Li, Qiusheng Zheng, and Yancun Yin. 2019. "Myricetin Suppresses the Propagation of Hepatocellular Carcinoma via Down-Regulating Expression of YAP" Cells 8, no. 4: 358. https://doi.org/10.3390/cells8040358
APA StyleLi, M., Chen, J., Yu, X., Xu, S., Li, D., Zheng, Q., & Yin, Y. (2019). Myricetin Suppresses the Propagation of Hepatocellular Carcinoma via Down-Regulating Expression of YAP. Cells, 8(4), 358. https://doi.org/10.3390/cells8040358