Lipid Droplets: A Significant but Understudied Contributor of Host–Bacterial Interactions

Abstract
1. Introduction
2. Intracellular Bacteria
2.1. Obligate Intracellular Bacteria
2.1.1. Chlamydia spp.
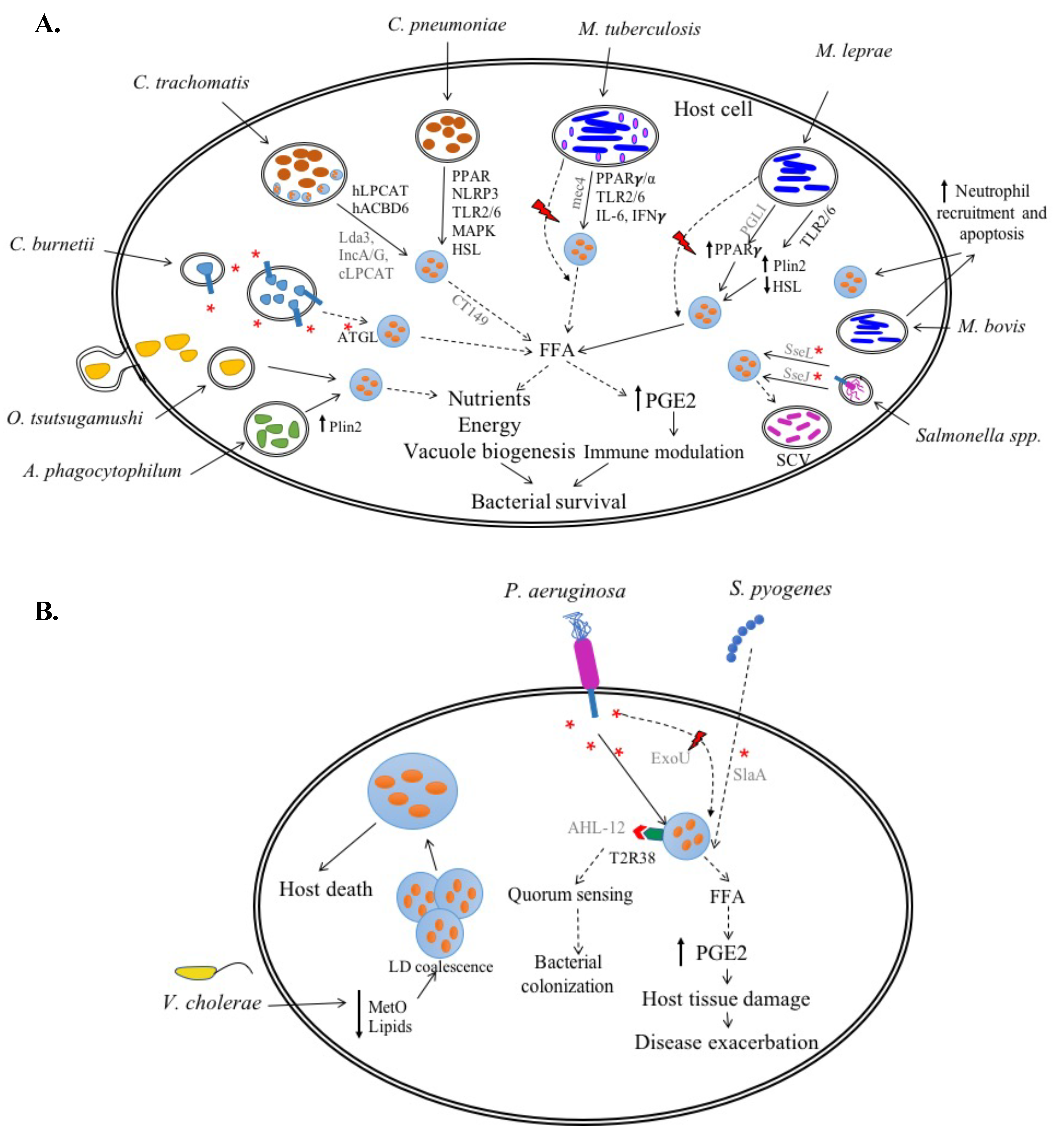
C. trachomatis
C. pneumoniae
2.1.2. Coxiella burnetii
2.1.3. Anaplasma phagocytophilum
2.1.4. Orientia tsutsugamushi
2.2. Facultative Intracellular Bacteria
2.2.1. Mycobacterium spp.
M. tuberculosis (Mtb)
M. bovis
M. leprae
2.2.2. Salmonella spp.
3. Extracellular Bacteria
3.1. Pseudomonas aeruginosa
3.2. Streptococcus pyogenes
3.3. Vibrio cholerae
4. Gut Microbiota
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Tauchi-Sato, K.; Ozeki, S.; Houjou, T.; Taguchi, R.; Fujimoto, T. The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J. Biol. Chem. 2002, 277, 44507–44512. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef]
- Pol, A.; Gross, S.P.; Parton, R.G. Review: Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Guo, Y.; Gross, S.P.; Welte, M.A. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol. 2006, 16, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef]
- Vaughan, M.; Berger, J.E.; Steinberg, D. Hormone-Sensitive Lipase and Monoglyceride Lipase Activities in Adipose Tissue. J. Biol. Chem. 1964, 239, 401–409. [Google Scholar]
- Guijas, C.; Rodriguez, J.P.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipase A2 regulation of lipid droplet formation. Biochim. Biophys. Acta 2014, 1841, 1661–1671. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Ann. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Krahmer, N.; Farese, R.V., Jr.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Pennetta, G.; Welte, M.A. Emerging Links between Lipid Droplets and Motor Neuron Diseases. Dev. Cell 2018, 45, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Cabirol-Pol, M.J.; Khalil, B.; Rival, T.; Faivre-Sarrailh, C.; Besson, M.T. Glial lipid droplets and neurodegeneration in a Drosophila model of complex I deficiency. Glia 2018, 66, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.C.; Weller, P.F. Lipid droplets in leukocytes: Organelles linked to inflammatory responses. Exp. Cell Res. 2016, 340, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Den Brok, M.H.; Raaijmakers, T.K.; Collado-Camps, E.; Adema, G.J. Lipid Droplets as Immune Modulators in Myeloid Cells. Trends Immunol. 2018, 39, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Ott, M. Emerging role of lipid droplets in host/pathogen interactions. J. Biol. Chem. 2012, 287, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Roingeard, P.; Melo, R.C. Lipid droplet hijacking by intracellular pathogens. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef]
- Saka, H.A.; Valdivia, R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Ann. Rev. Cell Dev. Biol. 2012, 28, 411–437. [Google Scholar] [CrossRef]
- Sorgi, C.A.; Secatto, A.; Fontanari, C.; Turato, W.M.; Belanger, C.; de Medeiros, A.I.; Kashima, S.; Marleau, S.; Covas, D.T.; Bozza, P.T.; et al. Histoplasma capsulatum cell wall {beta}-glucan induces lipid body formation through CD18, TLR2, and dectin-1 receptors: Correlation with leukotriene B4 generation and role in HIV-1 infection. J. Immunol. 2009, 182, 4025–4035. [Google Scholar] [CrossRef]
- Melo, R.C.; Dvorak, A.M. Lipid body-phagosome interaction in macrophages during infectious diseases: Host defense or pathogen survival strategy? PLoS Pathogens 2012, 8, e1002729. [Google Scholar] [CrossRef]
- Samanta, D.; Mulye, M.; Clemente, T.M.; Justis, A.V.; Gilk, S.D. Manipulation of Host Cholesterol by Obligate Intracellular Bacteria. Front. Cell. Infect. Microbiol. 2017, 7, 165. [Google Scholar] [CrossRef]
- Stehr, M.; Elamin, A.A.; Singh, M. Cytosolic lipid inclusions formed during infection by viral and bacterial pathogens. Microbes Infect. Institut. Pasteur. 2012, 14, 1227–1237. [Google Scholar] [CrossRef]
- Toledo, A.; Benach, J.L. Hijacking and use of host lipids by intracellular pathogens. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef]
- Vallochi, A.L.; Teixeira, L.; Oliveira, K.D.S.; Maya-Monteiro, C.M.; Bozza, P.T. Lipid Droplet, a Key Player in Host-Parasite Interactions. Front. Immunol. 2018, 9, 1022. [Google Scholar] [CrossRef]
- Lavie, M.; Dubuisson, J. Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimistry 2017, 141, 62–69. [Google Scholar] [CrossRef]
- Herker, E.; Ott, M. Unique ties between hepatitis C virus replication and intracellular lipids. Trends Endocrinol. Metab. 2011, 22, 241–248. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, Y.; Sanyal, S. Modulation of Lipid Droplet Metabolism-A Potential Target for Therapeutic Intervention in Flaviviridae Infections. Front. Microbiol. 2017, 8, 2286. [Google Scholar] [CrossRef]
- De Mattos, K.A.; Sarno, E.N.; Pessolani, M.C.; Bozza, P.T. Deciphering the contribution of lipid droplets in leprosy: Multifunctional organelles with roles in Mycobacterium leprae pathogenesis. Memórias do Instituto Oswaldo Cruz 2012, 107 (Suppl. 1), 156–166. [Google Scholar] [CrossRef]
- Barisch, C.; Soldati, T. Breaking fat! How mycobacteria and other intracellular pathogens manipulate host lipid droplets. Biochimie 2017, 141, 54–61. [Google Scholar] [CrossRef]
- Elamin, A.A.; Stehr, M.; Singh, M. Lipid Droplets and Mycobacterium leprae Infection. J. Pathog. 2012, 2012, 361374. [Google Scholar] [CrossRef]
- Suter, E. Interaction between phagocytes and pathogenic microorganisms. Bacteriol. Rev. 1956, 20, 94–132. [Google Scholar]
- Moulder, J.W. Comparative biology of intracellular parasitism. Microbiol. Rev. 1985, 49, 298–337. [Google Scholar]
- Raupach, B.; Kaufmann, S.H. Immune responses to intracellular bacteria. Curr. Opin. Immunol. 2001, 13, 417–428. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Coordinating innate immune cells to optimize microbial killing. Immunity 2008, 29, 672–674. [Google Scholar] [CrossRef]
- Knodler, L.A.; Celli, J. Eating the strangers within: Host control of intracellular bacteria via xenophagy. Cell. Microbiol. 2011, 13, 1319–1327. [Google Scholar] [CrossRef]
- Walpole, G.F.W.; Grinstein, S.; Westman, J. The role of lipids in host-pathogen interactions. IUBMB Life 2018, 70, 384–392. [Google Scholar] [CrossRef]
- Becker, Y. Chlamydia. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Bastidas, R.J.; Elwell, C.A.; Engel, J.N.; Valdivia, R.H. Chlamydial intracellular survival strategies. Cold Spring Harb. Perspect. Med. 2013, 3, a010256. [Google Scholar] [CrossRef]
- Kumar, Y.; Cocchiaro, J.; Valdivia, R.H. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr. Biol. 2006, 16, 1646–1651. [Google Scholar] [CrossRef]
- Saka, H.A.; Thompson, J.W.; Chen, Y.S.; Dubois, L.G.; Haas, J.T.; Moseley, A.; Valdivia, R.H. Chlamydia trachomatis Infection Leads to Defined Alterations to the Lipid Droplet Proteome in Epithelial Cells. PLoS ONE 2015, 10, e0124630. [Google Scholar] [CrossRef]
- Sharma, M.; Recuero-Checa, M.A.; Fan, F.Y.; Dean, D. Chlamydia trachomatis regulates growth and development in response to host cell fatty acid availability in the absence of lipid droplets. Cell. Microbiol. 2018, 20. [Google Scholar] [CrossRef]
- Cocchiaro, J.L.; Kumar, Y.; Fischer, E.R.; Hackstadt, T.; Valdivia, R.H. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. USA 2008, 105, 9379–9384. [Google Scholar] [CrossRef]
- Rank, R.G.; Whittimore, J.; Bowlin, A.K.; Wyrick, P.B. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infect. Immun. 2011, 79, 3291–3301. [Google Scholar] [CrossRef]
- Hackstadt, T.; Scidmore-Carlson, M.A.; Shaw, E.I.; Fischer, E.R. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell. Microbiol. 1999, 1, 119–130. [Google Scholar] [CrossRef]
- Recuero-Checa, M.A.; Sharma, M.; Lau, C.; Watkins, P.A.; Gaydos, C.A.; Dean, D. Chlamydia trachomatis growth and development requires the activity of host Long-chain Acyl-CoA Synthetases (ACSLs). Sci. Rep. 2016, 6, 23148. [Google Scholar] [CrossRef]
- Peters, J.; Byrne, G.I. Chlamydia trachomatis growth depends on eukaryotic cholesterol esterification and is affected by Acyl-CoA:cholesterol acyltransferase inhibition. Pathog. Dis. 2015, 73, ftv028. [Google Scholar] [CrossRef][Green Version]
- Soupene, E.; Kuypers, F.A. Phosphatidylserine decarboxylase CT699, lysophospholipid acyltransferase CT775, and acyl-ACP synthase CT776 provide membrane lipid diversity to Chlamydia trachomatis. Sci. Rep. 2017, 7, 15767. [Google Scholar] [CrossRef]
- Soupene, E.; Rothschild, J.; Kuypers, F.A.; Dean, D. Eukaryotic protein recruitment into the Chlamydia inclusion: Implications for survival and growth. PLoS ONE 2012, 7, e36843. [Google Scholar] [CrossRef]
- Peters, J.; Onguri, V.; Nishimoto, S.K.; Marion, T.N.; Byrne, G.I. The Chlamydia trachomatis CT149 protein exhibits esterase activity in vitro and catalyzes cholesteryl ester hydrolysis when expressed in HeLa cells. Microbes Infect. 2012, 14, 1196–1204. [Google Scholar] [CrossRef][Green Version]
- Soupene, E.; Wang, D.; Kuypers, F.A. Remodeling of host phosphatidylcholine by Chlamydia acyltransferase is regulated by acyl-CoA binding protein ACBD6 associated with lipid droplets. Microbiologyopen 2015, 4, 235–251. [Google Scholar] [CrossRef]
- Fukuda, E.Y.; Lad, S.P.; Mikolon, D.P.; Iacobelli-Martinez, M.; Li, E. Activation of lipid metabolism contributes to interleukin-8 production during Chlamydia trachomatis infection of cervical epithelial cells. Infect. Immun. 2005, 73, 4017–4024. [Google Scholar] [CrossRef]
- Belland, R.J.; Ouellette, S.P.; Gieffers, J.; Byrne, G.I. Chlamydia pneumoniae and atherosclerosis. Cell. Microbiol. 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Campbell, L.A.; Kuo, C.C. Chlamydia pneumoniae—An infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2004, 2, 23–32. [Google Scholar] [CrossRef]
- Mori, M.; Itabe, H.; Higashi, Y.; Fujimoto, Y.; Shiomi, M.; Yoshizumi, M.; Ouch, Y.; Takano, T. Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. J. Lipid Res. 2001, 42, 1771–1781. [Google Scholar]
- Bobryshev, Y.V.; Killingsworth, M.C.; Tran, D.; Lord, R. Amalgamation of Chlamydia pneumoniae inclusions with lipid droplets in foam cells in human atherosclerotic plaque. Virchows Arch. 2008, 453, 69–77. [Google Scholar] [CrossRef]
- Cheng, B.; Wu, X.; Sun, S.; Wu, Q.; Mei, C.; Xu, Q.; Wu, J.; He, P. MAPK-PPARalpha/gamma signal transduction pathways are involved in Chlamydia pneumoniae-induced macrophage-derived foam cell formation. Microb. Pathog. 2014, 1–8. [Google Scholar] [CrossRef]
- He, P.; Mei, C.; Cheng, B.; Liu, W.; Wang, Y.; Wan, J. Chlamydia pneumoniae induces macrophage-derived foam cell formation by up-regulating acyl-coenzyme A: Cholesterol acyltransferase 1. Microbes Infect. 2009, 11, 157–163. [Google Scholar] [CrossRef]
- Mei, C.L.; He, P.; Cheng, B.; Liu, W.; Wang, Y.F.; Wan, J.J. Chlamydia pneumoniae induces macrophage-derived foam cell formation via PPAR alpha and PPAR gamma-dependent pathways. Cell Biol. Int. 2009, 33, 301–308. [Google Scholar] [CrossRef]
- Walenna, N.F.; Kurihara, Y.; Chou, B.; Ishii, K.; Soejima, T.; Itoh, R.; Shimizu, A.; Ichinohe, T.; Hiromatsu, K. Chlamydia pneumoniae exploits adipocyte lipid chaperone FABP4 to facilitate fat mobilization and intracellular growth in murine adipocytes. Biochem. Biophys. Res. Commun. 2018, 495, 353–359. [Google Scholar] [CrossRef]
- Barbier, O.; Torra, I.P.; Duguay, Y.; Blanquart, C.; Fruchart, J.C.; Glineur, C.; Staels, B. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Evans, R.M. Peroxisome proliferator-activated receptor-gamma in macrophage lipid homeostasis. Trends Endocrinol. Metab. 2002, 13, 331–335. [Google Scholar] [CrossRef]
- Castrillo, A.; Tontonoz, P. Nuclear receptors in macrophage biology: At the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 2004, 20, 455–480. [Google Scholar] [CrossRef]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Mei, S.; Gu, H.; Ward, A.; Yang, X.; Guo, H.; He, K.; Liu, Z.; Cao, W. p38 mitogen-activated protein kinase (MAPK) promotes cholesterol ester accumulation in macrophages through inhibition of macroautophagy. J. Biol. Chem. 2012, 287, 11761–11768. [Google Scholar] [CrossRef] [PubMed]
- Itoh, R.; Murakami, I.; Chou, B.; Ishii, K.; Soejima, T.; Suzuki, T.; Hiromatsu, K. Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem. Biophys. Res. Commun. 2014, 452, 689–694. [Google Scholar] [CrossRef]
- Cao, F.; Castrillo, A.; Tontonoz, P.; Re, F.; Byrne, G.I. Chlamydia pneumoniae—Induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect. Immun. 2007, 75, 753–759. [Google Scholar] [CrossRef]
- Rupp, J.; Berger, M.; Reiling, N.; Gieffers, J.; Lindschau, C.; Haller, H.; Dalhoff, K.; Maass, M. Cox-2 inhibition abrogates Chlamydia pneumoniae-induced PGE2 and MMP-1 expression. Biochem. Biophys. Res. Commun. 2004, 320, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Van Schaik, E.J.; Chen, C.; Mertens, K.; Weber, M.M.; Samuel, J.E. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat. Rev. Microbiol. 2013, 11, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Heinzen, R.A. Lounging in a lysosome: The intracellular lifestyle of Coxiella burnetii. Cell. Microbiol. 2007, 9, 829–840. [Google Scholar] [CrossRef]
- Beare, P.A.; Gilk, S.D.; Larson, C.L.; Hill, J.; Stead, C.M.; Omsland, A.; Cockrell, D.C.; Howe, D.; Voth, D.E.; Heinzen, R.A. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Godi, A. PI-loting membrane traffic. Nat. Cell Biol. 2004, 6, 487–492. [Google Scholar] [CrossRef]
- Howe, D.; Heinzen, R.A. Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell. Microbiol. 2006, 8, 496–507. [Google Scholar] [CrossRef]
- Gilk, S.D.; Cockrell, D.C.; Luterbach, C.; Hansen, B.; Knodler, L.A.; Ibarra, J.A.; Steele-Mortimer, O.; Heinzen, R.A. Bacterial colonization of host cells in the absence of cholesterol. PLoS Pathog. 2013, 9, e1003107. [Google Scholar] [CrossRef]
- Beare, P.A.; Unsworth, N.; Andoh, M.; Voth, D.E.; Omsland, A.; Gilk, S.D.; Williams, K.P.; Sobral, B.W.; Kupko, J.J., 3rd; Porcella, S.F.; et al. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect. Immun. 2009, 77, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Mulye, M.; Samanta, D.; Winfree, S.; Heinzen, R.A.; Gilk, S.D. Elevated Cholesterol in the Coxiella burnetii Intracellular Niche Is Bacteriolytic. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Ayoubi, P.; Shaw, E.I. Coxiella burnetii Nine Mile II proteins modulate gene expression of monocytic host cells during infection. BMC Microbiol. 2010, 10, 244. [Google Scholar] [CrossRef]
- Ren, Q.; Robertson, S.J.; Howe, D.; Barrows, L.F.; Heinzen, R.A. Comparative DNA microarray analysis of host cell transcriptional responses to infection by Coxiella burnetii or Chlamydia trachomatis. Ann. N. Y. Acad. Sci. 2003, 990, 701–713. [Google Scholar] [CrossRef]
- Mulye, M.; Zapata, B.; Gilk, S.D. Altering lipid droplet homeostasis affects Coxiella burnetii intracellular growth. PLoS ONE 2018, 13, e0192215. [Google Scholar] [CrossRef]
- Brouqui, P.; Dumler, J.S.; Raoult, D. Immunohistologic demonstration of Coxiella burnetii in the valves of patients with Q fever endocarditis. Am. J. Med. 1994, 97, 451–458. [Google Scholar] [CrossRef]
- Graham, J.G.; MacDonald, L.J.; Hussain, S.K.; Sharma, U.M.; Kurten, R.C.; Voth, D.E. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell. Microbiol. 2013, 15, 1012–1025. [Google Scholar] [CrossRef]
- Sandoz, K.M.; Valiant, W.G.; Eriksen, S.G.; Hruby, D.E.; Allen, R.D., 3rd; Rockey, D.D. The broad-spectrum antiviral compound ST-669 restricts chlamydial inclusion development and bacterial growth and localizes to host cell lipid droplets within treated cells. Antimicrob. Agents Chemother. 2014, 58, 3860–3866. [Google Scholar] [CrossRef]
- Stead, C.M.; Cockrell, D.C.; Beare, P.A.; Miller, H.E.; Heinzen, R.A. A Coxiella burnetii phospholipase A homolog pldA is required for optimal growth in macrophages and developmental form lipid remodeling. BMC Microbiol. 2018, 18, 33. [Google Scholar] [CrossRef]
- Koster, F.T.; Williams, J.C.; Goodwin, J.S. Cellular immunity in Q fever: Modulation of responsiveness by a suppressor T cell-monocyte circuit. J. Immunol. 1985, 135, 1067–1072. [Google Scholar]
- Shannon, J.G.; Heinzen, R.A. Adaptive immunity to the obligate intracellular pathogen Coxiella burnetii. Immunol Res. 2009, 43, 138–148. [Google Scholar] [CrossRef]
- Izzo, A.A.; Marmion, B.P. Variation in interferon-gamma responses to Coxiella burnetii antigens with lymphocytes from vaccinated or naturally infected subjects. Clin. Exp. Immunol. 1993, 94, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.R.; Rosenthal, K.S.; Pfaller, M.A. Medical Microbiology, 8th ed.; Philadelphia, P.A., Ed.; Elsevier: Philadelphia, PA, USA, 2016. [Google Scholar]
- Dumler, J.S.; Barbet, A.F.; Bekker, C.P.; Dasch, G.A.; Palmer, G.H.; Ray, S.C.; Rikihisa, Y.; Rurangirwa, F.R. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: Unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ’HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 2001, 51, 2145–2165. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Rikihisa, Y. Obligatory intracellular parasitism by Ehrlichia chaffeensis and Anaplasma phagocytophilum involves caveolae and glycosylphosphatidylinositol-anchored proteins. Cell. Microbiol. 2003, 5, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Lin, M.; Rikihisa, Y. Cholesterol-dependent Anaplasma phagocytophilum exploits the low-density lipoprotein uptake pathway. PLoS Pathog. 2009, 5, e1000329. [Google Scholar] [CrossRef]
- Lin, M.; Rikihisa, Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect. Immun. 2003, 71, 5324–5331. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, J.; Ayoubi, P.; Blouin, E.F.; Almazan, C.; Naranjo, V.; Kocan, K.M. Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell. Microbiol. 2005, 7, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Roman, R.; Almazan, C.; Naranjo, V.; Blouin, E.F.; Kocan, K.M.; de la Fuente, J. Expression of perilipin in human promyelocytic cells in response to Anaplasma phagocytophilum infection results in modified lipid metabolism. J. Med. Microbiol. 2008, 57, 159–163. [Google Scholar] [CrossRef]
- Niu, H.; Kozjak-Pavlovic, V.; Rudel, T.; Rikihisa, Y. Anaplasma phagocytophilum Ats-1 is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog. 2010, 6, e1000774. [Google Scholar] [CrossRef]
- Beyer, A.R.; Truchan, H.K.; May, L.J.; Walker, N.J.; Borjesson, D.L.; Carlyon, J.A. The Anaplasma phagocytophilum effector AmpA hijacks host cell SUMOylation. Cell. Microbiol. 2015, 17, 504–519. [Google Scholar] [CrossRef]
- Moore, H.P.; Silver, R.B.; Mottillo, E.P.; Bernlohr, D.A.; Granneman, J.G. Perilipin targets a novel pool of lipid droplets for lipolytic attack by hormone-sensitive lipase. J. Biol. Chem. 2005, 43109–43120. [Google Scholar] [CrossRef]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef]
- Rikihisa, Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: Subversive manipulators of host cells. Nat. Rev. Microbiol. 2010, 8, 328–339. [Google Scholar] [CrossRef]
- McPherson, R.A.; Pincus, M.R. Henry’s Clinical Diagnosis and Management by Laboratory Methods, 23rd ed.; Elsevier: St. Louis, MI, USA, 2017. [Google Scholar]
- Seong, S.Y.; Choi, M.S.; Kim, I.S. Orientia tsutsugamushi infection: Overview and immune responses. Microbes Infect. 2001, 3, 11–21. [Google Scholar] [CrossRef]
- Chu, H.; Lee, J.H.; Han, S.H.; Kim, S.Y.; Cho, N.H.; Kim, I.S.; Choi, M.S. Exploitation of the endocytic pathway by Orientia tsutsugamushi in nonprofessional phagocytes. Infect. Immun. 2006, 74, 4246–4253. [Google Scholar] [CrossRef]
- Ewing, E.P., Jr.; Takeuchi, A.; Shirai, A.; Osterman, J.V. Experimental infection of mouse peritoneal mesothelium with scrub typhus rickettsiae: An ultrastructural study. Infect. Immun. 1978, 19, 1068–1075. [Google Scholar]
- Kim, M.J.; Kim, M.K.; Kang, J.S. Involvement of lipid rafts in the budding-like exit of Orientia tsutsugamushi. Microb. Pathog. 2013, 63, 37–43. [Google Scholar] [CrossRef]
- Ogawa, M.; Fukasawa, M.; Satoh, M.; Hanada, K.; Saijo, M.; Uchiyama, T.; Ando, S. The intracellular pathogen Orientia tsutsugamushi responsible for scrub typhus induces lipid droplet formation in mouse fibroblasts. Microbes Infect. 2014, 16, 962–966. [Google Scholar] [CrossRef]
- Dennis, E.A. Introduction to Thematic Review Series: Phospholipases: Central Role in Lipid Signaling and Disease. J. Lipid Res. 2015, 56, 1245–1247. [Google Scholar] [CrossRef]
- Housley, N.A.; Winkler, H.H.; Audia, J.P. The Rickettsia prowazekii ExoU homologue possesses phospholipase A1 (PLA1), PLA2, and lyso-PLA2 activities and can function in the absence of any eukaryotic cofactors in vitro. J. Bacteriol. 2011, 193, 4634–4642. [Google Scholar] [CrossRef]
- Rahman, M.S.; Gillespie, J.J.; Kaur, S.J.; Sears, K.T.; Ceraul, S.M.; Beier-Sexton, M.; Azad, A.F. Rickettsia typhi possesses phospholipase A2 enzymes that are involved in infection of host cells. PLoS Pathog. 2013, 9, e1003399. [Google Scholar] [CrossRef]
- Guillemot, L.; Medina, M.; Pernet, E.; Leduc, D.; Chignard, M.; Touqui, L.; Wu, Y. Cytosolic phospholipase A2alpha enhances mouse mortality induced by Pseudomonas aeruginosa pulmonary infection via interleukin 6. Biochimie 2014, 107, 95–104. [Google Scholar] [CrossRef]
- Hurley, B.P.; Pirzai, W.; Mumy, K.L.; Gronert, K.; McCormick, B.A. Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, 286–294. [Google Scholar] [CrossRef]
- Saliba, A.M.; Nascimento, D.O.; Silva, M.C.; Assis, M.C.; Gayer, C.R.; Raymond, B.; Coelho, M.G.; Marques, E.A.; Touqui, L.; Albano, R.M.; et al. Eicosanoid-mediated proinflammatory activity of Pseudomonas aeruginosa ExoU. Cell. Microbiol. 2005, 7, 1811–1822. [Google Scholar] [CrossRef]
- Rydkina, E.; Sahni, A.; Baggs, R.B.; Silverman, D.J.; Sahni, S.K. Infection of human endothelial cells with spotted Fever group rickettsiae stimulates cyclooxygenase 2 expression and release of vasoactive prostaglandins. Infect. Immun. 2006, 74, 5067–5074. [Google Scholar] [CrossRef]
- Walker, T.S.; Brown, J.S.; Hoover, C.S.; Morgan, D.A. Endothelial prostaglandin secretion: Effects of typhus rickettsiae. J. Infect. Dis. 1990, 162, 1136–1144. [Google Scholar] [CrossRef]
- Knodler, L.A.; Vallance, B.A.; Celli, J.; Winfree, S.; Hansen, B.; Montero, M.; Steele-Mortimera, O. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. USA 2010, 107, 17733–17738. [Google Scholar] [CrossRef]
- Silva, M.T. Classical labeling of bacterial pathogens according to their lifestyle in the host: Inconsistencies and alternatives. Front. Microbiol. 2012, 3, 71. [Google Scholar] [CrossRef]
- Casadevall, A. Evolution of intracellular pathogens. Annu. Rev. Microbiol. 2008, 62, 19–33. [Google Scholar] [CrossRef]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef]
- Almeida, P.E.; Carneiro, A.B.; Silva, A.R.; Bozza, P.T. PPARgamma Expression and Function in Mycobacterial Infection: Roles in Lipid Metabolism, Immunity, and Bacterial Killing. PPAR Res. 2012, 2012, 383829. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Zumla, A.; Chakaya, J.; Centis, R.; D’Ambrosio, L.; Mwaba, P.; Bates, M.; Kapata, N.; Nyirenda, T.; Chanda, D.; Mfinanga, S.; et al. Tuberculosis treatment and management—An update on treatment regimens, trials, new drugs, and adjunct therapies. Lancet Respir. Med. 2015, 3, 220–234. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Weber, S.S.; Ragaz, C.; Hilbi, H. Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 2009, 71, 1341–1352. [Google Scholar] [CrossRef]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar] [CrossRef]
- Woo, M.; Wood, C.; Kwon, D.; Park, K.P.; Fejer, G.; Delorme, V. Mycobacterium tuberculosis Infection and Innate Responses in a New Model of Lung Alveolar Macrophages. Front. Immunol. 2018, 9, 438. [Google Scholar] [CrossRef]
- Salamon, H.; Bruiners, N.; Lakehal, K.; Shi, L.; Ravi, J.; Yamaguchi, K.D.; Gennaro, M.L. Cutting edge: Vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J. Immunol. 2014, 193, 30–34. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef]
- Daniel, J.; Sirakova, T.; Kolattukudy, P. An acyl-CoA synthetase in Mycobacterium tuberculosis involved in triacylglycerol accumulation during dormancy. PLoS ONE 2014, 9, e114877. [Google Scholar] [CrossRef]
- Daniel, J.; Kapoor, N.; Sirakova, T.; Sinha, R.; Kolattukudy, P. The perilipin-like PPE15 protein in Mycobacterium tuberculosis is required for triacylglycerol accumulation under dormancy-inducing conditions. Mol. Microbiol. 2016, 101, 784–794. [Google Scholar] [CrossRef]
- Elamin, A.A.; Stehr, M.; Spallek, R.; Rohde, M.; Singh, M. The Mycobacterium tuberculosis Ag85A is a novel diacylglycerol acyltransferase involved in lipid body formation. Mol. Microbiol. 2011, 81, 1577–1592. [Google Scholar] [CrossRef]
- Armstrong, R.M.; Adams, K.L.; Zilisch, J.E.; Bretl, D.J.; Sato, H.; Anderson, D.M.; Zahrt, T.C. Rv2744c Is a PspA Ortholog That Regulates Lipid Droplet Homeostasis and Nonreplicating Persistence in Mycobacterium tuberculosis. J. Bacteriol. 2016, 198, 1645–1661. [Google Scholar] [CrossRef]
- Daniel, J.; Maamar, H.; Deb, C.; Sirakova, T.D.; Kolattukudy, P.E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011, 7, e1002093. [Google Scholar] [CrossRef]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Knight, M.; Braverman, J.; Asfaha, K.; Gronert, K.; Stanley, S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-gamma/HIF-1alpha signaling and supports host defense. PLoS Pathog. 2018, 14, e1006874. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef]
- Dunn, P.L.; North, R.J. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect. Immun. 1995, 63, 3428–3437. [Google Scholar]
- Jaisinghani, N.; Dawa, S.; Singh, K.; Nandy, A.; Menon, D.; Bhandari, P.D.; Khare, G.; Tyagi, A.; Gandotra, S. Necrosis Driven Triglyceride Synthesis Primes Macrophages for Inflammation During Mycobacterium tuberculosis Infection. Front. Immunol. 2018, 9, 1490. [Google Scholar] [CrossRef]
- Almeida, P.E.; Silva, A.R.; Maya-Monteiro, C.M.; Torocsik, D.; D’Avila, H.; Dezso, B.; Magalhães, K.G.; Castro-Faria-Neto, H.C.; Nagy, L.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guerin infection induces TLR2-dependent peroxisome proliferator-activated receptor gamma expression and activation: Functions in inflammation, lipid metabolism, and pathogenesis. J. Immunol. 2009, 183, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Spiropoulos, J.; Cooley, W.; Khara, J.S.; Gladstone, C.A.; Asai, M.; Bossé, J.T.; Robertson, B.D.; Newton, S.M.; Langford, P.R. Galleria mellonella—A novel infection model for the Mycobacterium tuberculosis complex. Virulence 2018, 9, 1126–1137. [Google Scholar] [CrossRef]
- D’Avila, H.; Melo, R.C.; Parreira, G.G.; Werneck-Barroso, E.; Castro-Faria-Neto, H.C.; Bozza, P.T. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: Intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 2006, 176, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Pean, C.B.; Schiebler, M.; Tan, S.W.; Sharrock, J.A.; Kierdorf, K.; Brown, K.P.; Maserumule, M.C.; Menezes, S.; Pilátová, M.; Bronda, K.; et al. Regulation of phagocyte triglyceride by a STAT-ATG2 pathway controls mycobacterial infection. Nat. Commun. 2017, 8, 14642. [Google Scholar] [CrossRef]
- Wang, D.; Shi, L.; Xin, W.; Xu, J.; Xu, J.; Li, Q.; Xu, Z.; Wang, J.; Wang, G.; Yao, W.; et al. Activation of PPARgamma inhibits pro-inflammatory cytokines production by upregulation of miR-124 in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 486, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Mattos, K.A.; D’Avila, H.; Rodrigues, L.S.; Oliveira, V.G.; Sarno, E.N.; Atella, G.C.; Pereira, G.M.; Bozza, P.T.; Pessolani, M.C. Lipid droplet formation in leprosy: Toll-like receptor-regulated organelles involved in eicosanoid formation and Mycobacterium leprae pathogenesis. J. Leukoc. Biol. 2010, 87, 371–384. [Google Scholar] [CrossRef]
- Mattos, K.A.; Lara, F.A.; Oliveira, V.G.; Rodrigues, L.S.; D’Avila, H.; Melo, R.C.; Manso, P.P.; Sarno, E.N.; Bozza, P.T.; Pessolani, M.C. Modulation of lipid droplets by Mycobacterium leprae in Schwann cells: A putative mechanism for host lipid acquisition and bacterial survival in phagosomes. Cell. Microbiol. 2011, 13, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, K.; Suzuki, K.; Nakamura, K.; Akama, T.; Kawashima, A.; Wu, H.; Hayashi, M.; Takahashi, S.; Ikuyama, S.; Ito, T.; et al. Expression of adipose differentiation-related protein (ADRP) and perilipin in macrophages infected with Mycobacterium leprae. FEMS Microbiol. Lett. 2008, 289, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.H.; An, S.K.; Lee, S.B. The formation of lipid droplets favors intracellular Mycobacterium leprae survival in SW-10, non-myelinating Schwann cells. PLoS Negl. Trop. Dis. 2017, 11, e0005687. [Google Scholar] [CrossRef]
- Diaz Acosta, C.C.; Dias, A.A.; Rosa, T.; Batista-Silva, L.R.; Rosa, P.S.; Toledo-Pinto, T.G.; Costa, F.D.M.R.; Lara, F.A.; Rodrigues, L.S.; Mattos, K.A.; et al. PGL I expression in live bacteria allows activation of a CD206/PPARgamma cross-talk that may contribute to successful Mycobacterium leprae colonization of peripheral nerves. PLoS Pathog. 2018, 14, e1007151. [Google Scholar] [CrossRef]
- Degang, Y.; Akama, T.; Hara, T.; Tanigawa, K.; Ishido, Y.; Gidoh, M.; Makino, M.; Ishii, N.; Suzuki, K. Clofazimine modulates the expression of lipid metabolism proteins in Mycobacterium leprae-infected macrophages. PLoS Negl. Trop. Dis. 2012, 6, e1936. [Google Scholar] [CrossRef]
- Mattos, K.A.; Oliveira, V.G.; D’Avila, H.; Rodrigues, L.S.; Pinheiro, R.O.; Sarno, E.N.; Pessolani, M.C.; Bozza, P.T. TLR6-driven lipid droplets in Mycobacterium leprae-infected Schwann cells: Immunoinflammatory platforms associated with bacterial persistence. J. Immunol. 2011, 187, 2548–2558. [Google Scholar] [CrossRef]
- Cruz, D.; Watson, A.D.; Miller, C.S.; Montoya, D.; Ochoa, M.T.; Sieling, P.A.; Gutierrez, M.A.; Navab, M.; Reddy, S.T.; Witztum, J.L.; et al. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J. Clin. Invest. 2008, 118, 2917–2928. [Google Scholar] [CrossRef]
- Cole, S.T.; Eiglmeier, K.; Parkhill, J.; James, K.D.; Thomson, N.R.; Wheeler, P.R.; Honoré, N.; Garnier, T.; Churcher, C.; Harris, D.; et al. Massive gene decay in the leprosy bacillus. Nature 2001, 409, 1007–1011. [Google Scholar] [CrossRef]
- Pang, T.; Bhutta, Z.A.; Finlay, B.B.; Altwegg, M. Typhoid fever and other salmonellosis: A continuing challenge. Trends Microbiol. 1995, 3, 253–255. [Google Scholar] [CrossRef]
- Boyle, E.C.; Bishop, J.L.; Grassl, G.A.; Finlay, B.B. Salmonella: From pathogenesis to therapeutics. J. Bacteriol. 2007, 189, 1489–1495. [Google Scholar] [CrossRef]
- Ohl, M.E.; Miller, S.I. Salmonella: A model for bacterial pathogenesis. Annu. Rev. Med. 2001, 52, 259–274. [Google Scholar] [CrossRef]
- Cossart, P.; Sansonetti, P.J. Bacterial invasion: The paradigms of enteroinvasive pathogens. Science 2004, 304, 242–248. [Google Scholar] [CrossRef]
- Buckley, J.T. Substrate specificity of bacterial glycerophospholipid:cholesterol acyltransferase. Biochemistry 1982, 21, 6699–6703. [Google Scholar] [CrossRef]
- MacIntyre, S.; Buckley, J.T. Presence of glycerophospholipid: Cholesterol acyltransferase and phospholipase in culture supernatant of Aeromonas hydrophila. J. Bacteriol. 1978, 135, 402–407. [Google Scholar]
- Nawabi, P.; Catron, D.M.; Haldar, K. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol. Microbiol. 2008, 68, 173–185. [Google Scholar] [CrossRef]
- Ruiz-Albert, J.; Yu, X.J.; Beuzon, C.R.; Blakey, A.N.; Galyov, E.E.; Holden, D.W. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 2002, 44, 645–661. [Google Scholar] [CrossRef]
- Lawley, T.D.; Chan, K.; Thompson, L.J.; Kim, C.C.; Govoni, G.R.; Monack, D.M. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2006, 2, e11. [Google Scholar] [CrossRef]
- Freeman, J.A.; Ohl, M.E.; Miller, S.I. The Salmonella enterica serovar typhimurium translocated effectors SseJ and SifB are targeted to the Salmonella-containing vacuole. Infect. Immun. 2003, 71, 418–427. [Google Scholar] [CrossRef]
- Arena, E.T.; Auweter, S.D.; Antunes, L.C.; Vogl, A.W.; Han, J.; Guttman, J.A.; Croxen, M.A.; Menendez, A.; Covey, S.D.; Borchers, C.H.; et al. The deubiquitinase activity of the Salmonella pathogenicity island 2 effector, SseL, prevents accumulation of cellular lipid droplets. Infect. Immun. 2011, 79, 4392–4400. [Google Scholar] [CrossRef]
- Antunes, L.C.; Andersen, S.K.; Menendez, A.; Arena, E.T.; Han, J.; Ferreira, R.B.; Borchers, C.H.; Finlay, B.B. Metabolomics reveals phospholipids as important nutrient sources during Salmonella growth in bile in vitro and in vivo. J. Bacteriol. 2011, 193, 4719–4725. [Google Scholar] [CrossRef]
- Conner, J.G.; Teschler, J.K.; Jones, C.J.; Yildiz, F.H. Staying Alive: Vibrio cholerae’s Cycle of Environmental Survival, Transmission, and Dissemination. Microbiol Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Carbonetti, N.H. Bordetella pertussis: New concepts in pathogenesis and treatment. Curr. Opin. Infect. Dis. 2016, 29, 287–294. [Google Scholar] [CrossRef]
- Peterson, J.W. Bacterial Pathogenesis. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Ostberg, Y.; Berg, S.; Comstedt, P.; Wieslander, A.; Bergstrom, S. Functional analysis of a lipid galactosyltransferase synthesizing the major envelope lipid in the Lyme disease spirochete Borrelia burgdorferi. FEMS Microbiol. Lett. 2007, 272, 22–29. [Google Scholar] [CrossRef][Green Version]
- Wunder, C.; Churin, Y.; Winau, F.; Warnecke, D.; Vieth, M.; Lindner, B.; Zähringer, U.; Mollenkopf, H.J.; Heinz, E.; Meyer, T.F. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat. Med. 2006, 12, 1030–1038. [Google Scholar] [CrossRef]
- Sarabhai, S.K.A.; Capalash, N.; Sharma, P. Quorum Sensing in Pseudomonas aeruginosa: Mechanism and Regulation of Virulence. In Pseudomonas: Molecular and Applied Biology; Kahlon, R., Ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Cohen, T.P.D.; Prince, A. Pseudomonas aeruginosa Host Immune Evasion; Ramos, J.L., Goldberg, J., Eds.; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar]
- D’Agata, E. Pseudomonas aeruginosa and Other Pseudomonas Species. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Barbieri, J.T.; Sun, J. Pseudomonas aeruginosa ExoS and ExoT. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 79–92. [Google Scholar] [CrossRef]
- Ochoa, C.D.; Alexeyev, M.; Pastukh, V.; Balczon, R.; Stevens, T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J. Bacteriol. 2012, 287, 25407–25418. [Google Scholar] [CrossRef]
- Deng, Q.; Barbieri, J.T. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 2008, 62, 271–288. [Google Scholar] [CrossRef]
- Anderson, D.M.; Frank, D.W. Five mechanisms of manipulation by bacterial effectors: A ubiquitous theme. PLoS Pathog. 2012, 8, e1002823. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Zeng, H.; Azim, A.C.; Joo, M.; Dey, S.K.; Breyer, R.M.; Peebles, R.S.; Blackwell, T.S.; Christman, J.W. Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur. J. Immunol. 2007, 37, 1001–1009. [Google Scholar] [CrossRef]
- Aronoff, D.M.; Bergin, I.L.; Lewis, C.; Goel, D.; O’Brien, E.; Peters-Golden, M.; Mancuso, P. E-prostanoid 2 receptor signaling suppresses lung innate immunity against Streptococcus pneumoniae. Prostaglandins Other Lipid Mediat. 2012, 98, 23–30. [Google Scholar] [CrossRef]
- Plotkowski, M.C.; Brandao, B.A.; de Assis, M.C.; Feliciano, L.F.; Raymond, B.; Freitas, C.; Saliba, A.M.; Zahm, J.M.; Touqui, L.; Bozza, P.T. Lipid body mobilization in the ExoU-induced release of inflammatory mediators by airway epithelial cells. Microb. Pathog. 2008, 45, 30–37. [Google Scholar] [CrossRef]
- Phillips, R.M.; Six, D.A.; Dennis, E.A.; Ghosh, P. In vivo phospholipase activity of the Pseudomonas aeruginosa cytotoxin ExoU and protection of mammalian cells with phospholipase A2 inhibitors. J. Biol. Chem. 2003, 278, 41326–41332. [Google Scholar] [CrossRef]
- Maurer, S.; Wabnitz, G.H.; Kahle, N.A.; Stegmaier, S.; Prior, B.; Giese, T.; Gaida, M.M.; Samstag, Y.; Hänsch, G.M. Tasting Pseudomonas aeruginosa Biofilms: Human Neutrophils Express the Bitter Receptor T2R38 as Sensor for the Quorum Sensing Molecule N-(3-Oxododecanoyl)-l-Homoserine Lactone. Front. Immunol. 2015, 6, 369. [Google Scholar] [CrossRef]
- Vikstrom, E.; Magnusson, K.E.; Pivoriunas, A. The Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone stimulates phagocytic activity in human macrophages through the p38 MAPK pathway. Microbes Infect. 2005, 7, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Wagner, C.; Muller, W.; Brenner-Weiss, G.; Hug, F.; Prior, B.; Obst, U.; Hänsch, G.M. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone. Infect. Immun. 2006, 74, 5687–5692. [Google Scholar] [CrossRef]
- Wagner, C.; Zimmermann, S.; Brenner-Weiss, G.; Hug, F.; Prior, B.; Obst, U.; Hänsch, G.M. The quorum-sensing molecule N-3-oxododecanoyl homoserine lactone (3OC12-HSL) enhances the host defence by activating human polymorphonuclear neutrophils (PMN). Anal. Bioanal. Chem. 2007, 387, 481–487. [Google Scholar] [CrossRef]
- Welte, M.A. Proteins under new management: Lipid droplets deliver. Trends Cell Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef]
- Cunningham, M.W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef]
- Sitkiewicz, I.; Nagiec, M.J.; Sumby, P.; Butler, S.D.; Cywes-Bentley, C.; Musser, J.M. Emergence of a bacterial clone with enhanced virulence by acquisition of a phage encoding a secreted phospholipase A2. Proc. Natl. Acad. Sci. USA 2006, 103, 16009–16014. [Google Scholar] [CrossRef]
- Blaschke, U.; Beineke, A.; Klemens, J.; Medina, E.; Goldmann, O. Induction of Cyclooxygenase 2 by Streptococcus pyogenes Is Mediated by Cytolysins. J. Innate Immun. 2017, 9, 587–597. [Google Scholar] [CrossRef]
- Goldmann, O.; Hertzen, E.; Hecht, A.; Schmidt, H.; Lehne, S.; Norrby-Teglund, A.; Medina, E. Inducible cyclooxygenase released prostaglandin E2 modulates the severity of infection caused by Streptococcus pyogenes. J. Immunol. 2010, 185, 2372–2381. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, A.S.; Hang, S.; Vijayakumar, V.; Wong, A.C.; Asara, J.M.; Watnick, P.I. Vibrio cholerae ensures function of host proteins required for virulence through consumption of luminal methionine sulfoxide. PLoS Pathog. 2017, 13, e1006428. [Google Scholar] [CrossRef]
- Moravec, A.R.; Siv, A.W.; Hobby, C.R.; Lindsay, E.N.; Norbash, L.V.; Shults, D.J.; Symes, S.J.K.; Giles, D.K. Exogenous Polyunsaturated Fatty Acids Impact Membrane Remodeling and Affect Virulence Phenotypes among Pathogenic Vibrio Species. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Giles, D.K.; Hankins, J.V.; Guan, Z.; Trent, M.S. Remodelling of the Vibrio cholerae membrane by incorporation of exogenous fatty acids from host and aquatic environments. Mol. Microbiol. 2011, 79, 716–728. [Google Scholar] [CrossRef]
- Hang, S.; Purdy, A.E.; Robins, W.P.; Wang, Z.; Mandal, M.; Chang, S.; Mekalanos, J.J.; Watnick, P.I. The acetate switch of an intestinal pathogen disrupts host insulin signaling and lipid metabolism. Cell Host Microbe 2014, 16, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.E. Colonic bacterial metabolites and human health. Curr. Opin. Microbiol. 2013, 16, 246–254. [Google Scholar] [CrossRef]
- Sheng, Y.; Ren, H.; Limbu, S.M.; Sun, Y.; Qiao, F.; Zhai, W.; Du, Z.-Y.; Zhang, M. The Presence or Absence of Intestinal Microbiota Affects Lipid Deposition and Related Genes Expression in Zebrafish (Danio rerio). Front. Microbiol. 2018, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Semova, I.; Carten, J.D.; Stombaugh, J.; Mackey, L.C.; Knight, R.; Farber, S.A.; Rawls, J.F. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 2012, 12, 277–288. [Google Scholar] [CrossRef]
- Zanni, E.; Laudenzi, C.; Schifano, E.; Palleschi, C.; Perozzi, G.; Uccelletti, D.; Devirgiliis, C. Impact of a Complex Food Microbiota on Energy Metabolism in the Model Organism Caenorhabditis elegans. Biomed. Res. Int. 2015, 2015, 621709. [Google Scholar] [CrossRef]
- Tazi, A.; Araujo, J.R.; Mulet, C.; Arena, E.T.; Nigro, G.; Pedron, T.; Sansonetti, P.J. Disentangling Host-Microbiota Regulation of Lipid Secretion by Enterocytes: Insights from Commensals Lactobacillus paracasei and Escherichia coli. MBio 2018, 9. [Google Scholar] [CrossRef]
- Bougneres, L.; Helft, J.; Tiwari, S.; Vargas, P.; Chang, B.H.; Chan, L.; Campisi, L.; Lauvau, G.; Hugues, S.; Kumar, P.; et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 2009, 31, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Reinicke, A.T.; Omilusik, K.D.; Basha, G.; Jefferies, W.A. Dendritic cell cross-priming is essential for immune responses to Listeria monocytogenes. PLoS ONE 2009, 4, e7210. [Google Scholar] [CrossRef]
- Winau, F.; Weber, S.; Sad, S.; de Diego, J.; Hoops, S.L.; Breiden, B.; Sandhoff, K.; Brinkmann, V.; Kaufmann, S.H.; Schaible, U.E. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 2006, 24, 105–117. [Google Scholar] [CrossRef]
- Yrlid, U.; Wick, M.J. Salmonella-induced apoptosis of infected macrophages results in presentation of a bacteria-encoded antigen after uptake by bystander dendritic cells. J. Exp. Med. 2000, 191, 613–624. [Google Scholar] [CrossRef]
- Anand, P.; Cermelli, S.; Li, Z.; Kassan, A.; Bosch, M.; Sigua, R.; Huang, L.; Ouellette, A.J.; Pol, A.; Welte, M.A.; et al. A novel role for lipid droplets in the organismal antibacterial response. Elife 2012, 1, e00003. [Google Scholar] [CrossRef]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: An update. Prostaglandins Leukot Essent Fatty Acids 2011, 85, 205–213. [Google Scholar] [CrossRef]
- Nicolaou, G.; Goodall, A.H.; Erridge, C. Diverse bacteria promote macrophage foam cell formation via Toll-like receptor-dependent lipid body biosynthesis. J. Atheroscler. Thromb. 2012, 19, 137–148. [Google Scholar] [CrossRef]
- Chen, X.; Alonzo, F., 3rd. Bacterial lipolysis of immune-activating ligands promotes evasion of innate defenses. Proc. Natl. Acad. Sci. USA 2019, 116, 3764–3773. [Google Scholar] [CrossRef]
 —bacterial phospholipases.
—bacterial phospholipases.
—bacterial phospholipases.
—bacterial phospholipases.

{kind=link}
| Obligate Intracellular Bacteria (Vacuolar) | |
| Chlamydia trachomatis | LD formation required for optimal growth |
| Chlamydia pneumoniae | LD formation required for growth |
| Coxiella burnetii | LD breakdown essential for growth |
| Anaplasma phagocytophilum | LD formation increases growth |
| Obligate Intracellular Bacteria (Cytoplasmic) | |
| Orientia tsutsugamushi | LD formation increases growth |
| Facultative Intracellular Bacteria | |
| Mycobacterium tuberculosis | Contrasting roles |
| Mycobacterium bovis | Contrasting roles |
| Mycobacterium leprae | LD formation essential for growth |
| Salmonella spp. | Unknown |
| Extracellular Bacteria | |
| Pseudomonas aeruginosa | Unknown |
| Vibrio cholerae | Unknown |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libbing, C.L.; McDevitt, A.R.; Azcueta, R.-M.P.; Ahila, A.; Mulye, M. Lipid Droplets: A Significant but Understudied Contributor of Host–Bacterial Interactions. Cells 2019, 8, 354. https://doi.org/10.3390/cells8040354
Libbing CL, McDevitt AR, Azcueta R-MP, Ahila A, Mulye M. Lipid Droplets: A Significant but Understudied Contributor of Host–Bacterial Interactions. Cells. 2019; 8(4):354. https://doi.org/10.3390/cells8040354
Chicago/Turabian StyleLibbing, Cassandra L., Adam R. McDevitt, Rea-Mae P. Azcueta, Ahila Ahila, and Minal Mulye. 2019. "Lipid Droplets: A Significant but Understudied Contributor of Host–Bacterial Interactions" Cells 8, no. 4: 354. https://doi.org/10.3390/cells8040354
APA StyleLibbing, C. L., McDevitt, A. R., Azcueta, R.-M. P., Ahila, A., & Mulye, M. (2019). Lipid Droplets: A Significant but Understudied Contributor of Host–Bacterial Interactions. Cells, 8(4), 354. https://doi.org/10.3390/cells8040354