Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. ROS Activity Measurement
2.3. Mitochondria Mass Measurement
2.4. Animals
2.5. Histology, Immunohistochemistry and Immunofluorescence
2.6. Oil Red O Stain
2.7. Auto-Fluorescence Detection of Lipofuscin
2.8. Plasma Aminotransferase (ALT) Analysis
2.9. Plasma and Hepatic Triglyceride (TG) Measurement
2.10. Western Blot Analysis
2.11. RNA Isolation and Real-Time PCR Analysis
2.12. Statistical Analysis
3. Results
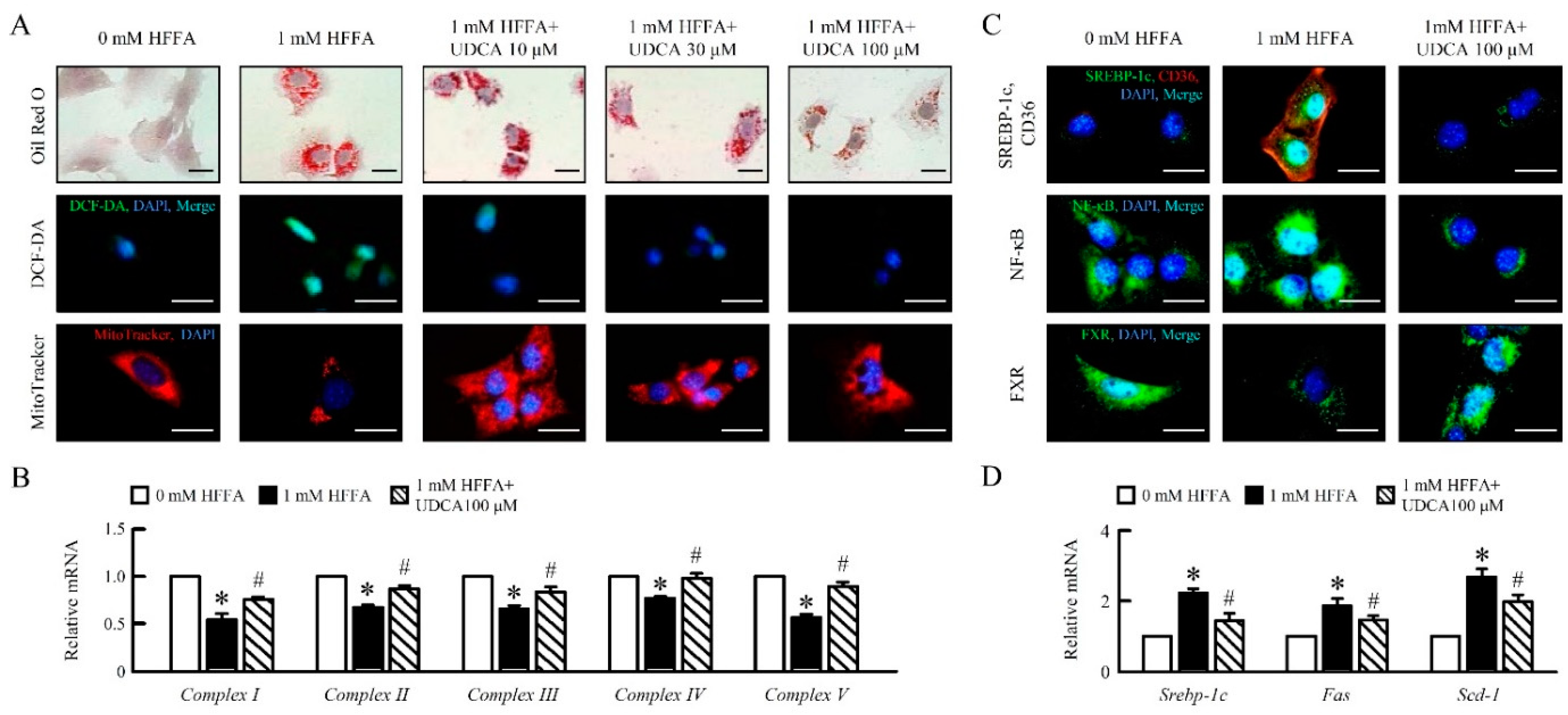
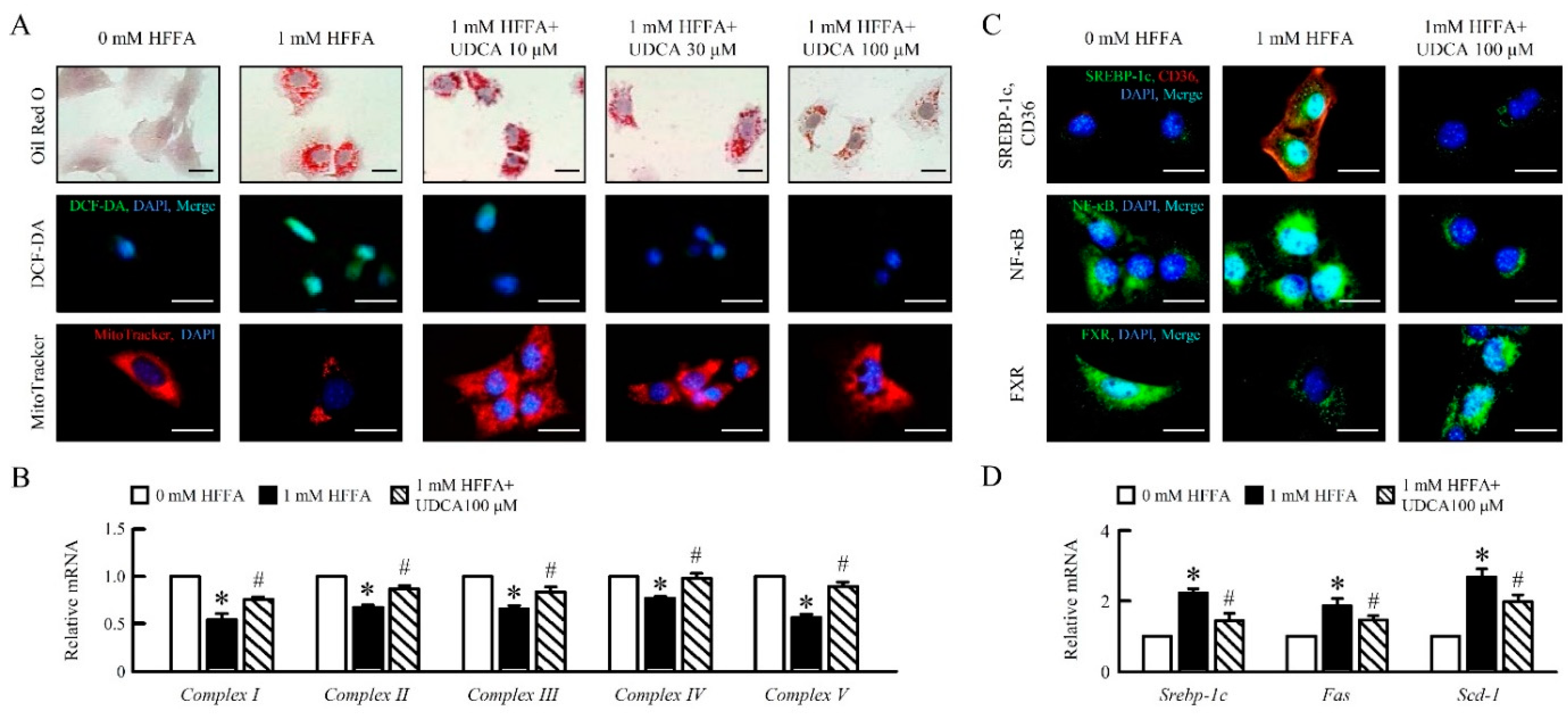
3.1. UDCA Attenuates High Free Fatty Acid (HFFA) Induced-Lipid Accumulation and Mitochondrial Dysfunction of in AML12 Cells
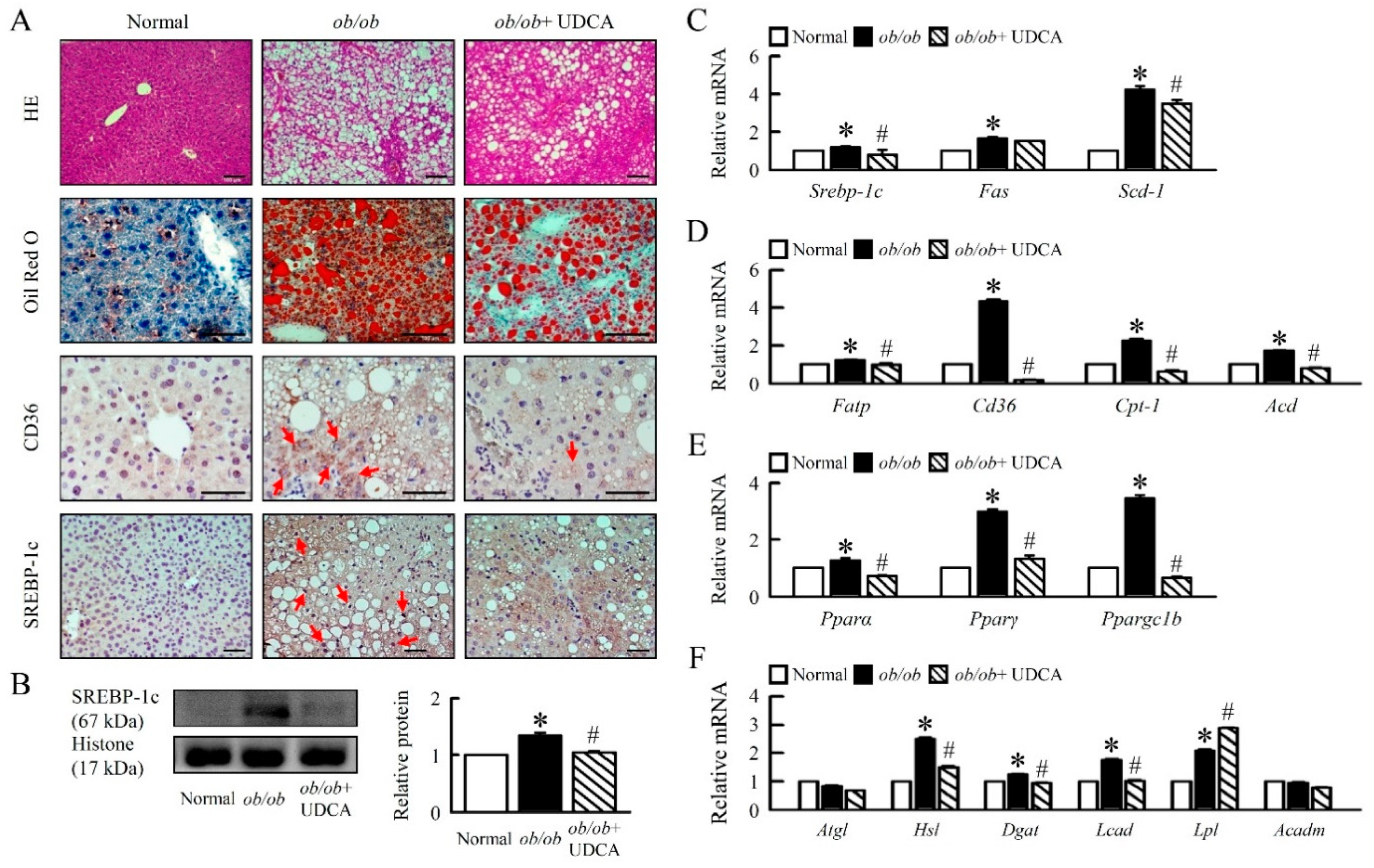
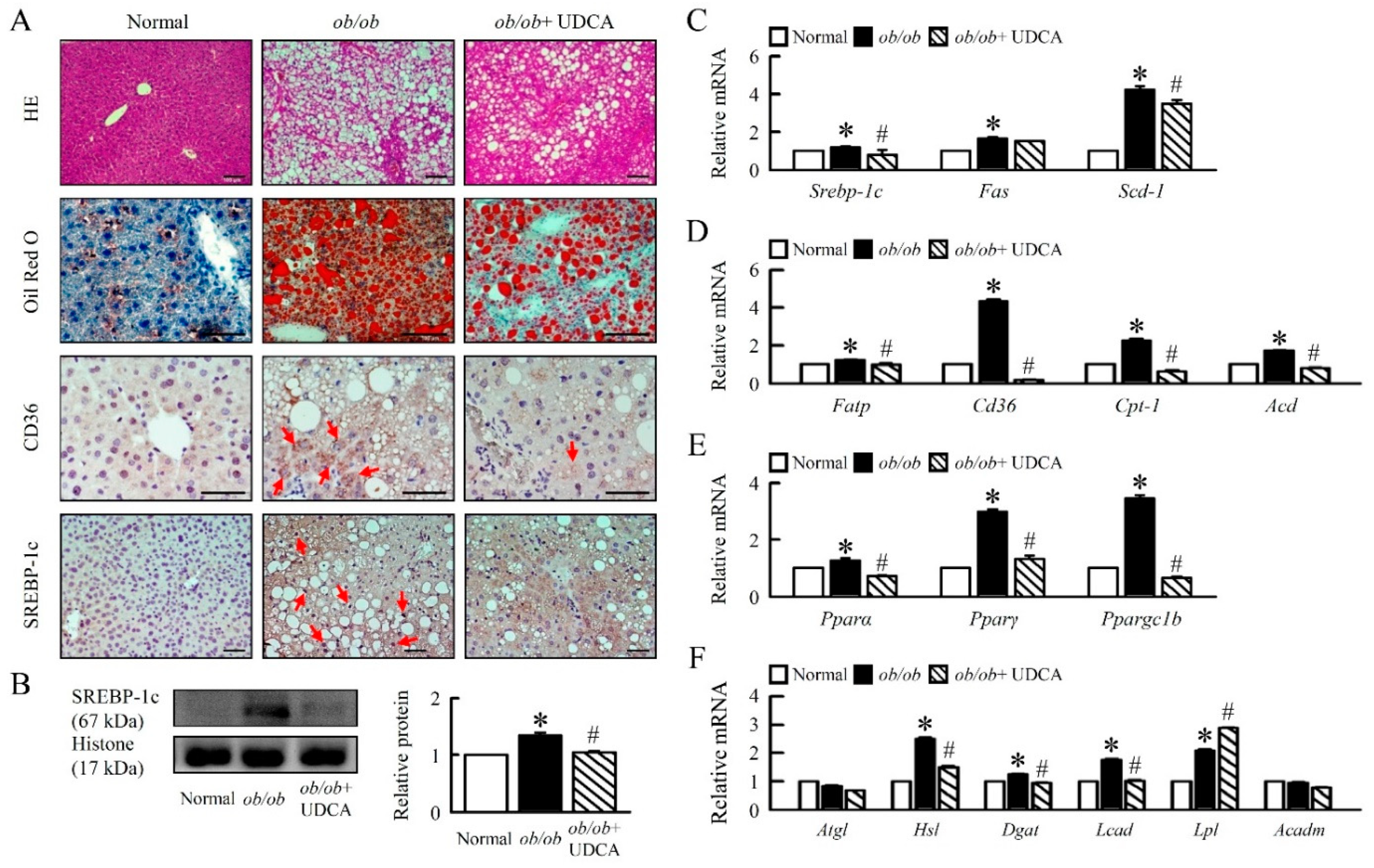
3.2. UDCA Attenuates Hepatosteatosis by Reducing Lipogenesis and β-Oxidation in ob/ob Mice
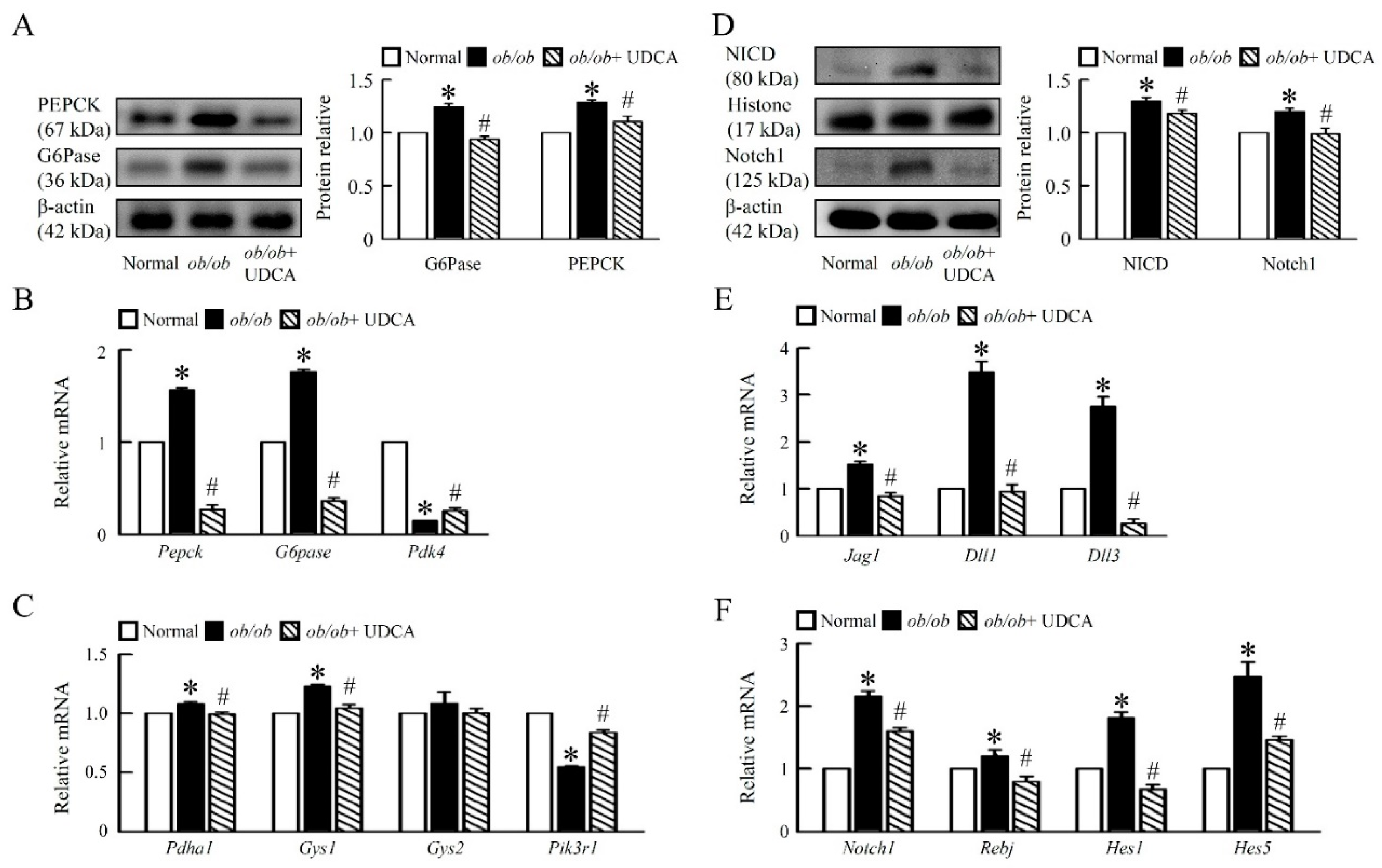
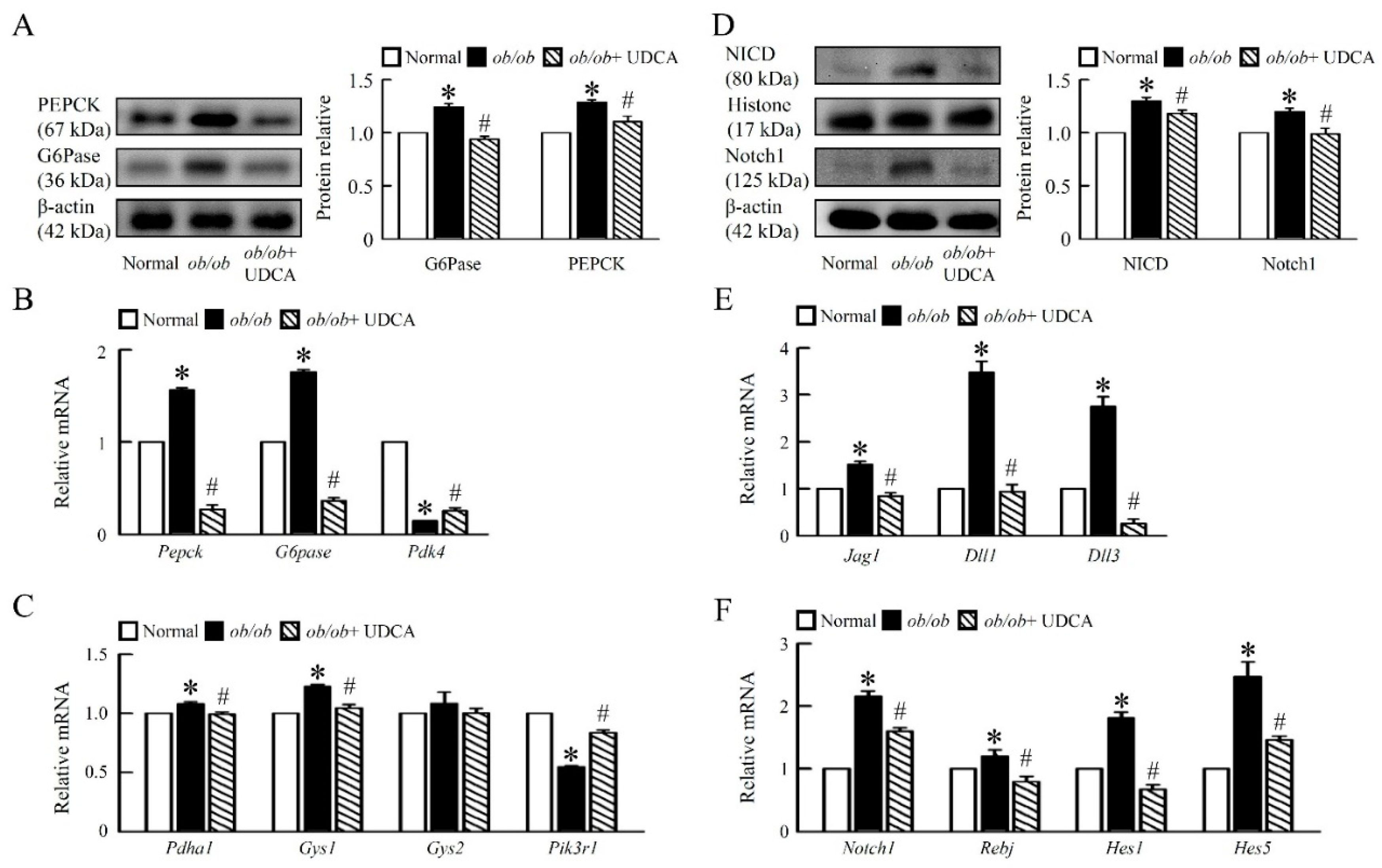
3.3. UDCA Ameliorates Hepatic Glucose Metabolism Disorder and Notch1 Signaling in ob/ob Mice
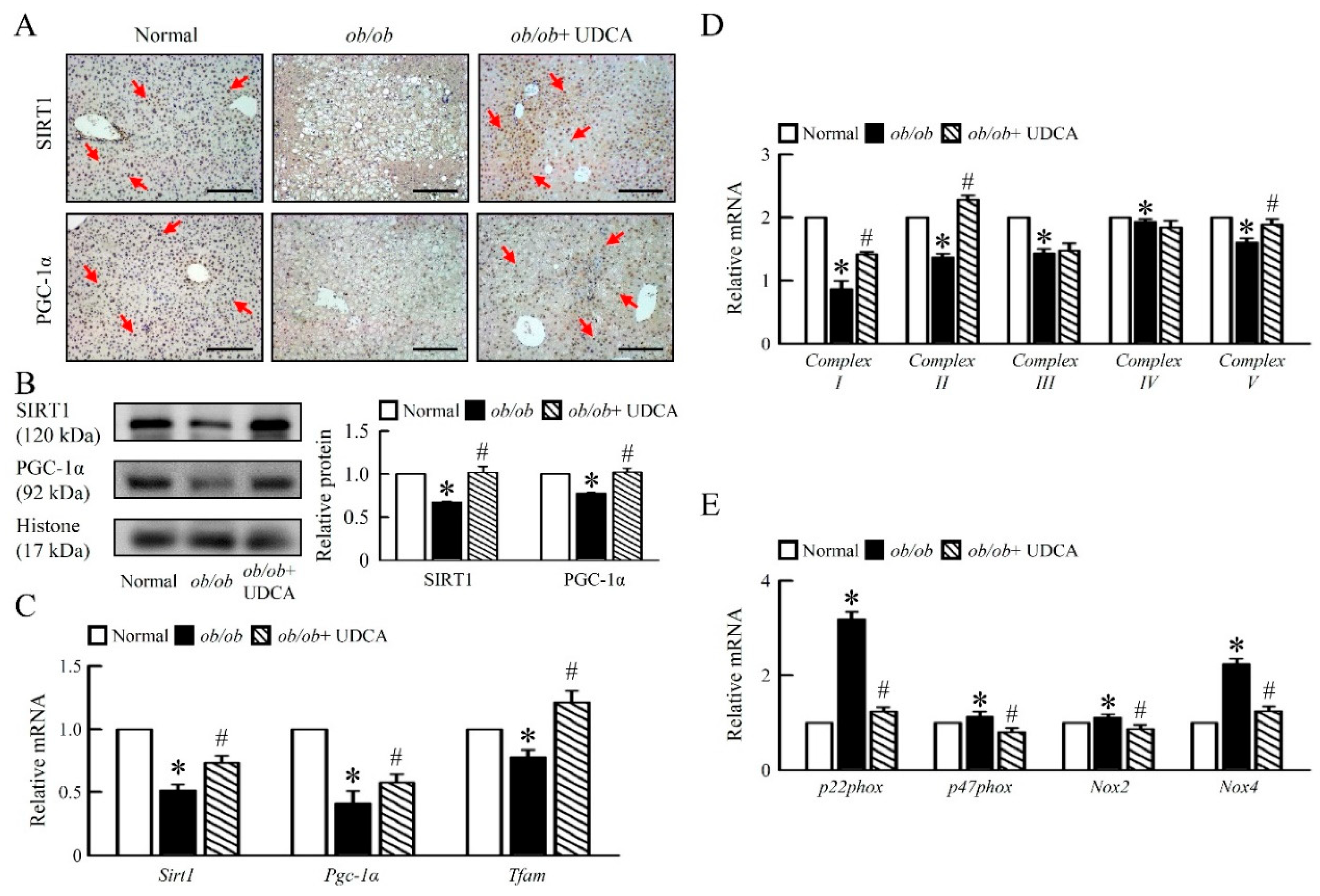
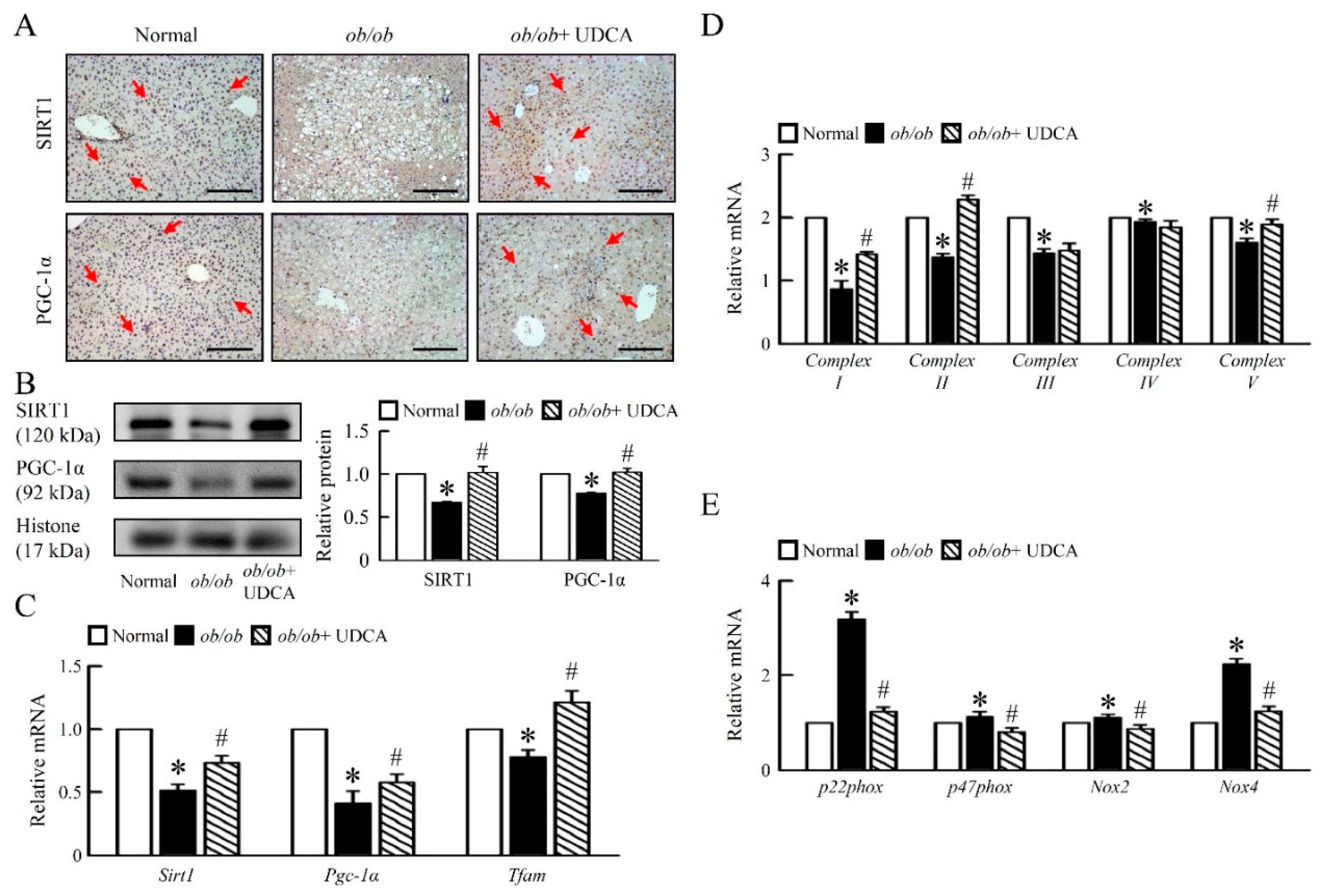
3.4. UDCA Improves Hepatic Mitochondrial Dysfunction and Biogenesis in ob/ob Mice
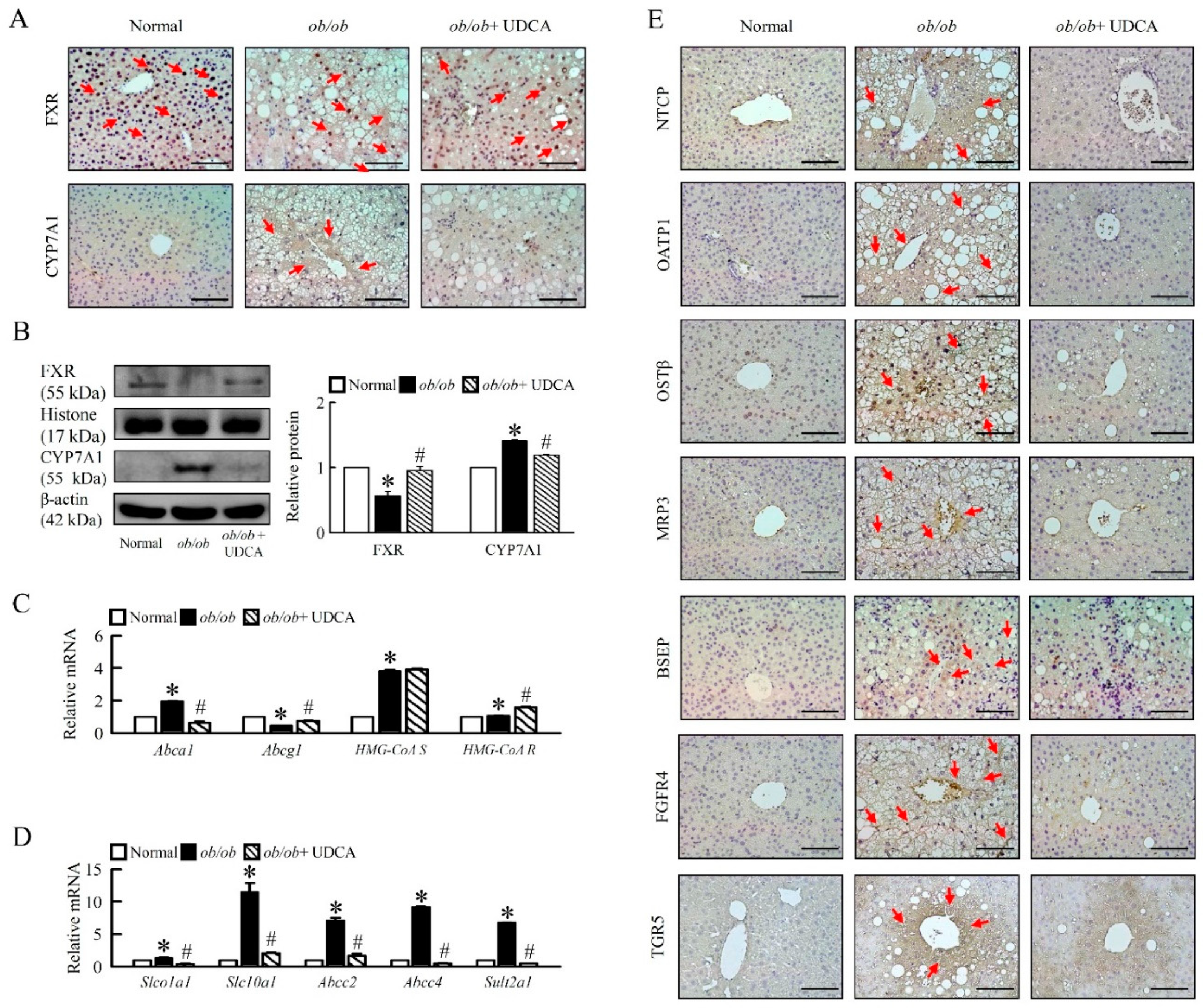
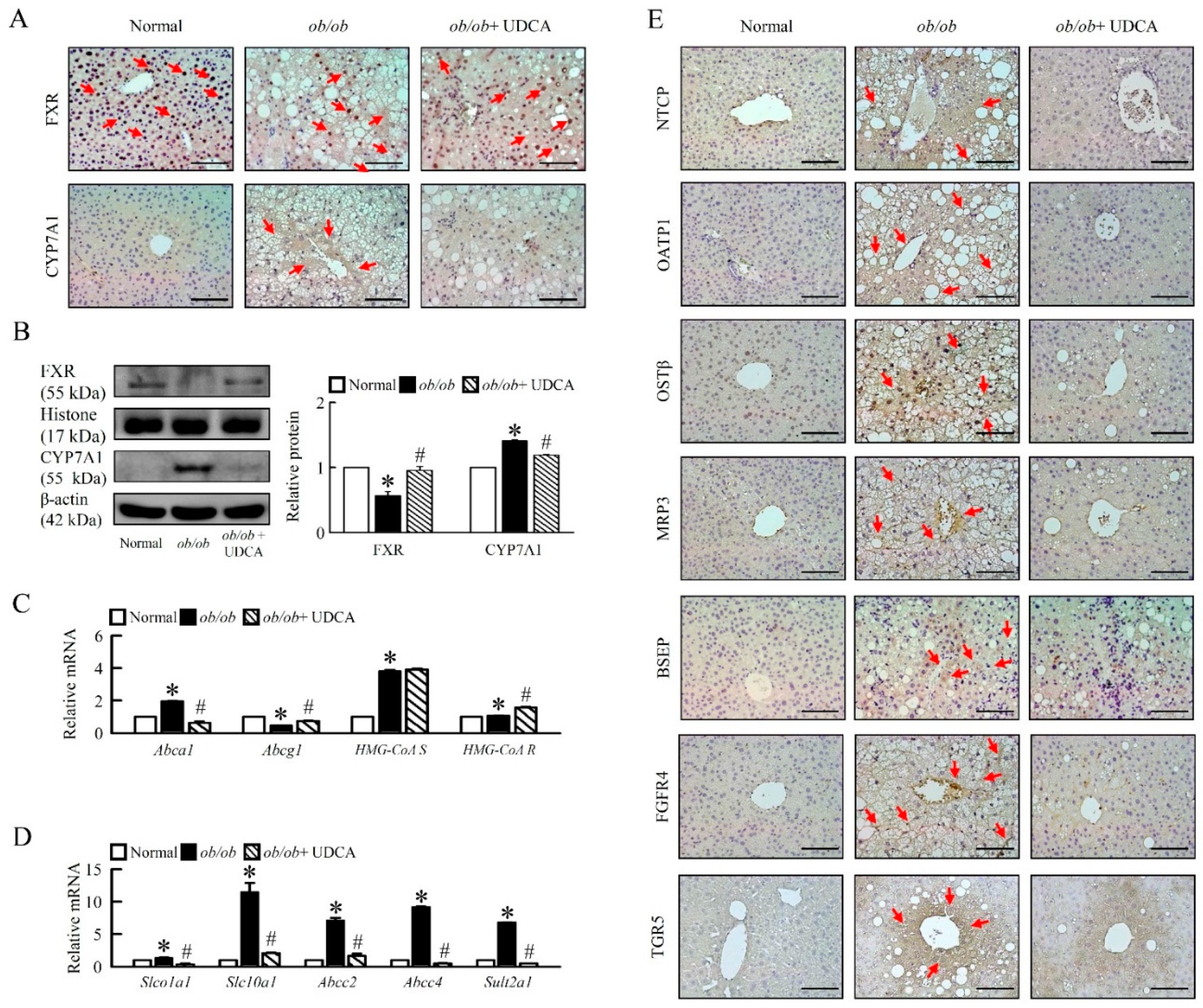
3.5. Effects of UDCA on Regulates Bile Acid Metabolism and Alters Transporters Expression in ob/ob Mice
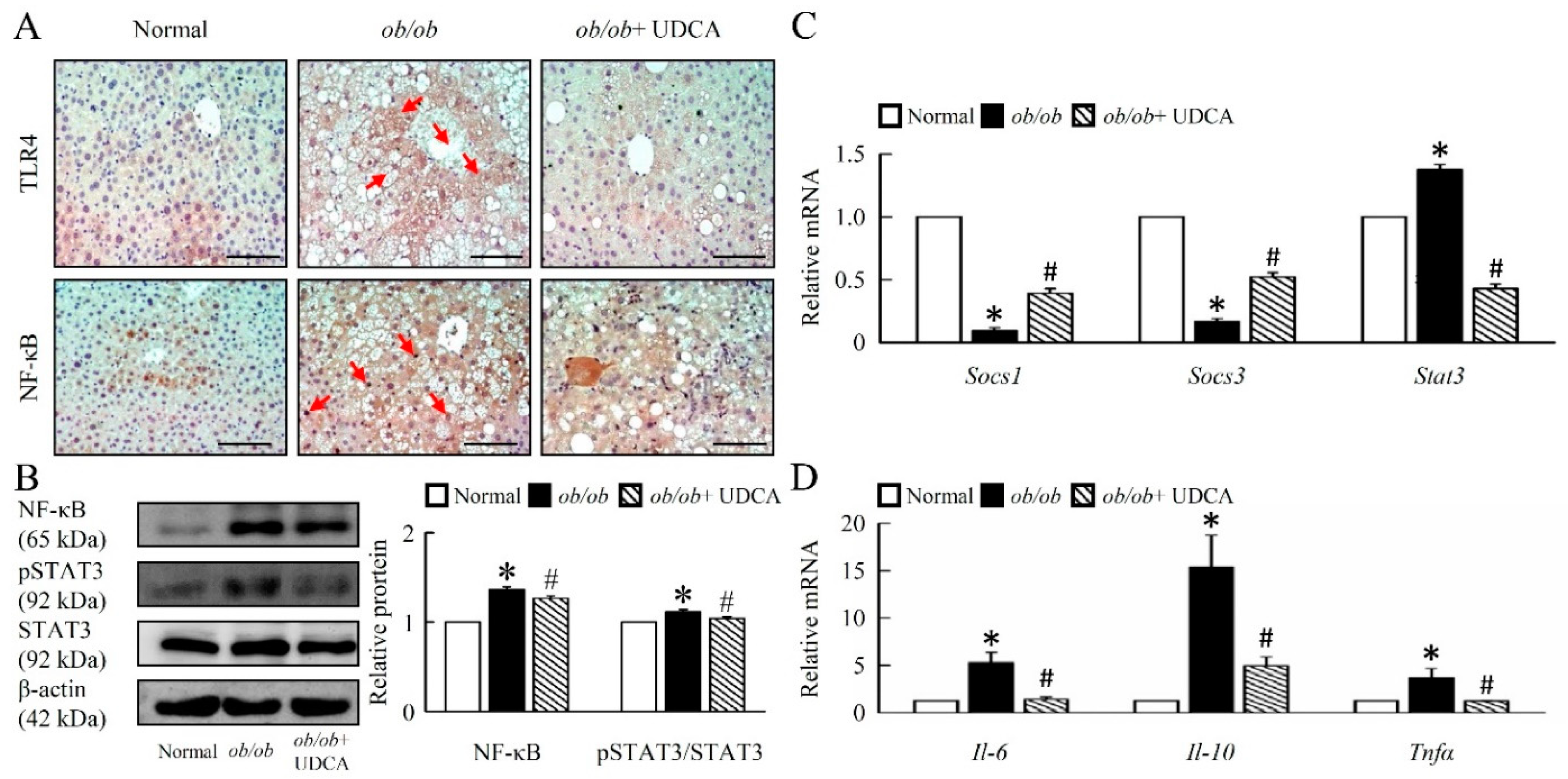
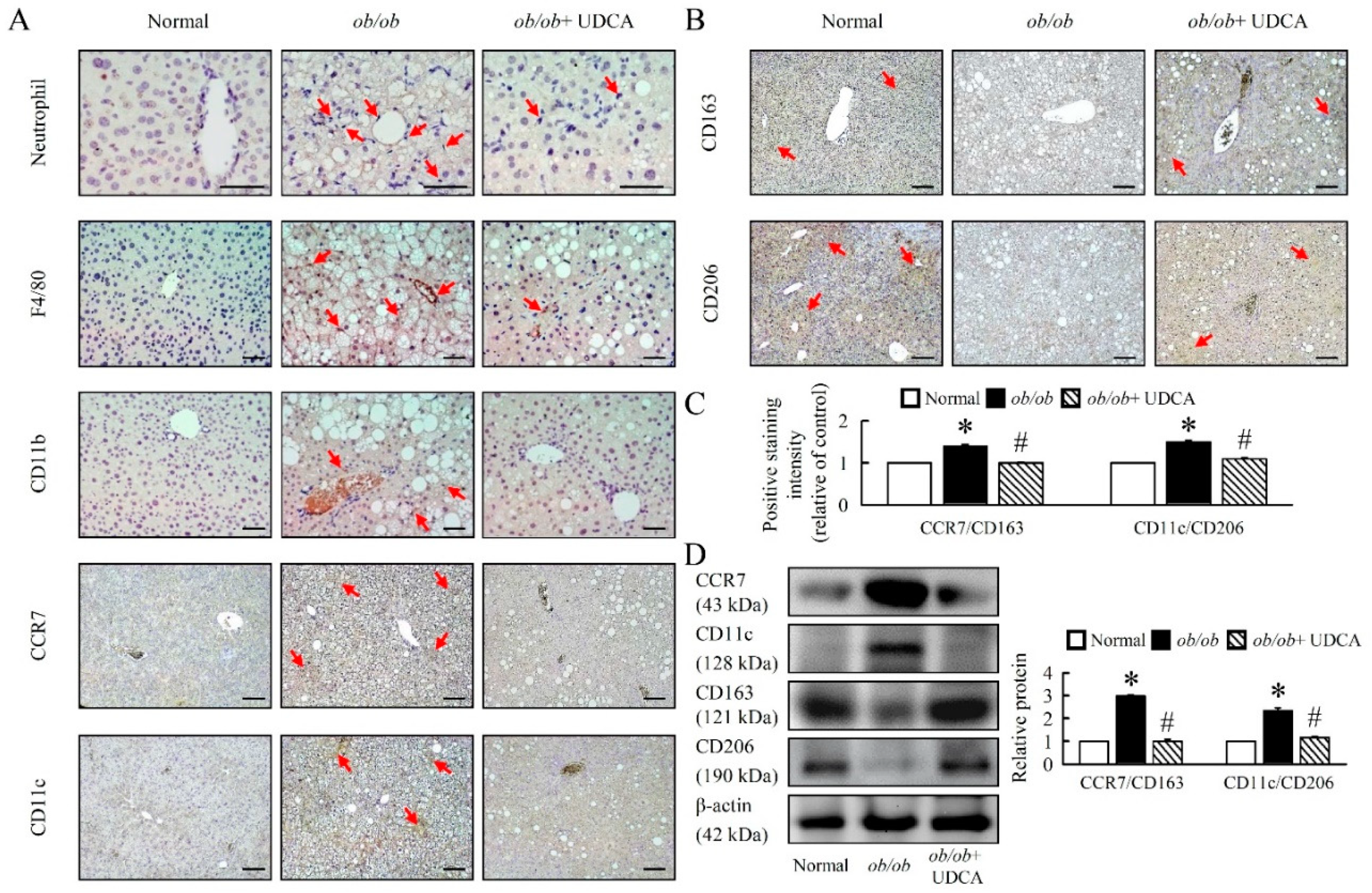
3.6. UDCA Attenuates Hepatic Inflammation and Alternative Macrophage Activation in ob/ob Mice
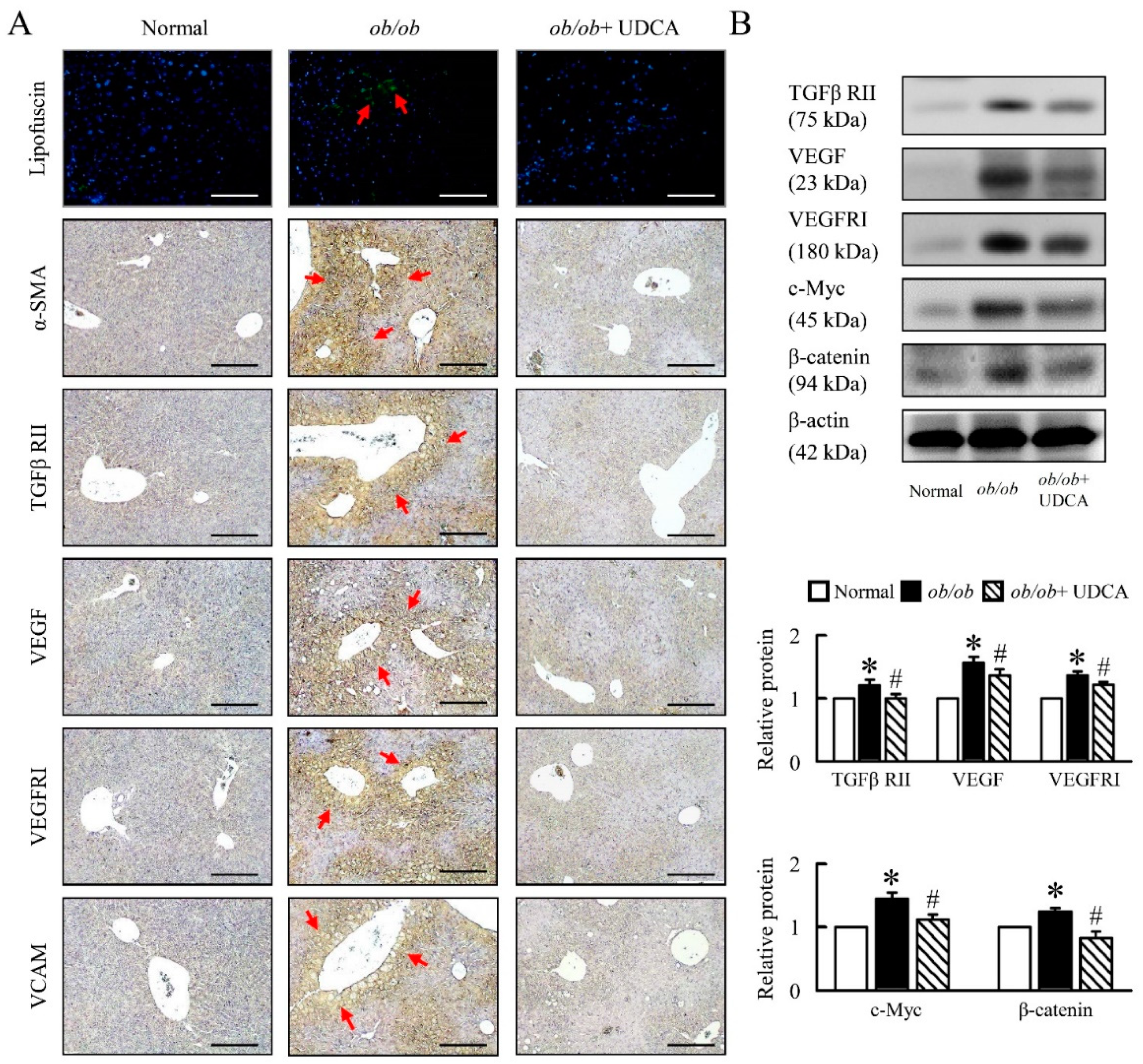
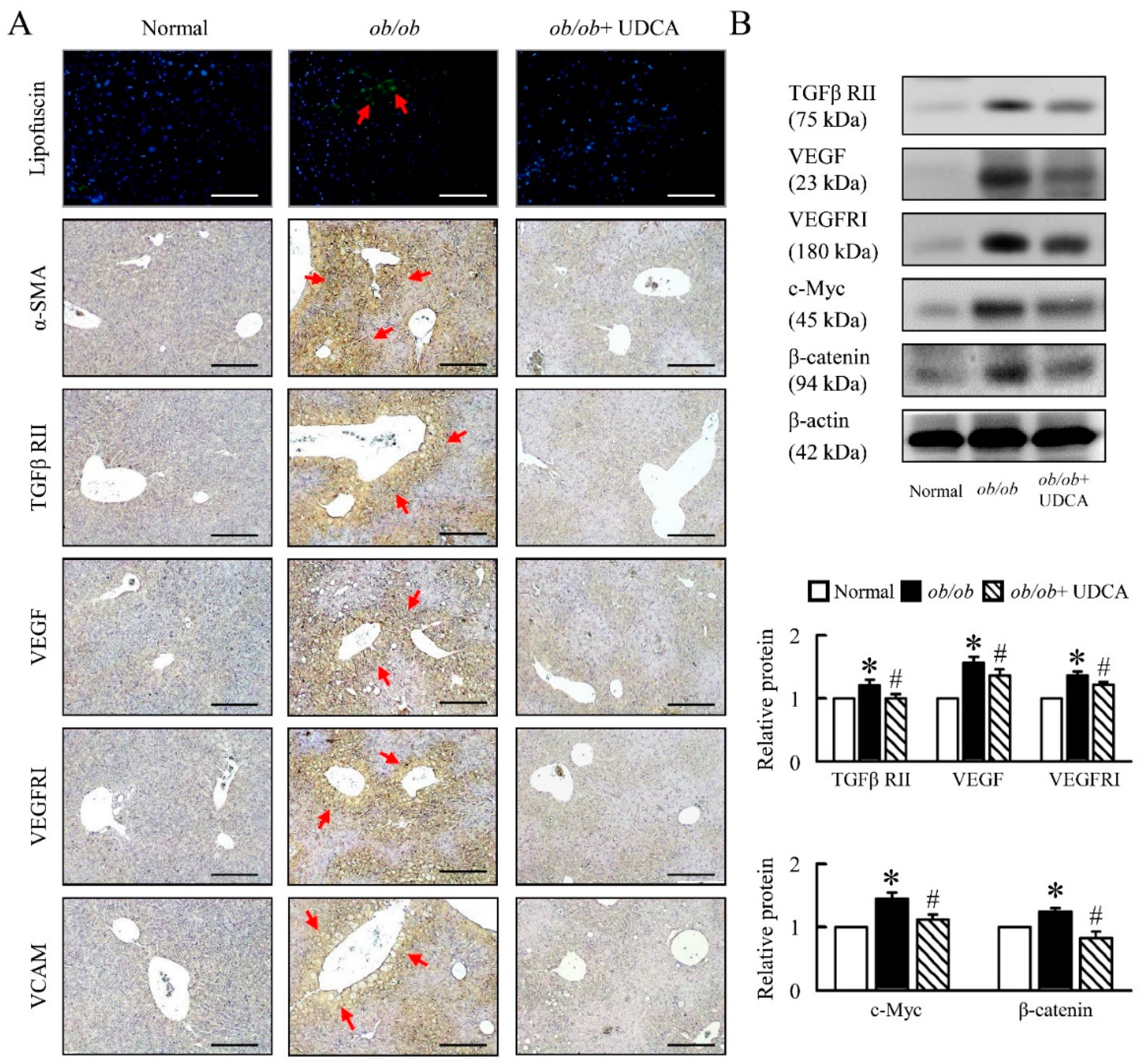
3.7. UDCA Partially Ameliorates Inflammation-Induced Hepatic Angiogenesis in ob/ob Mice
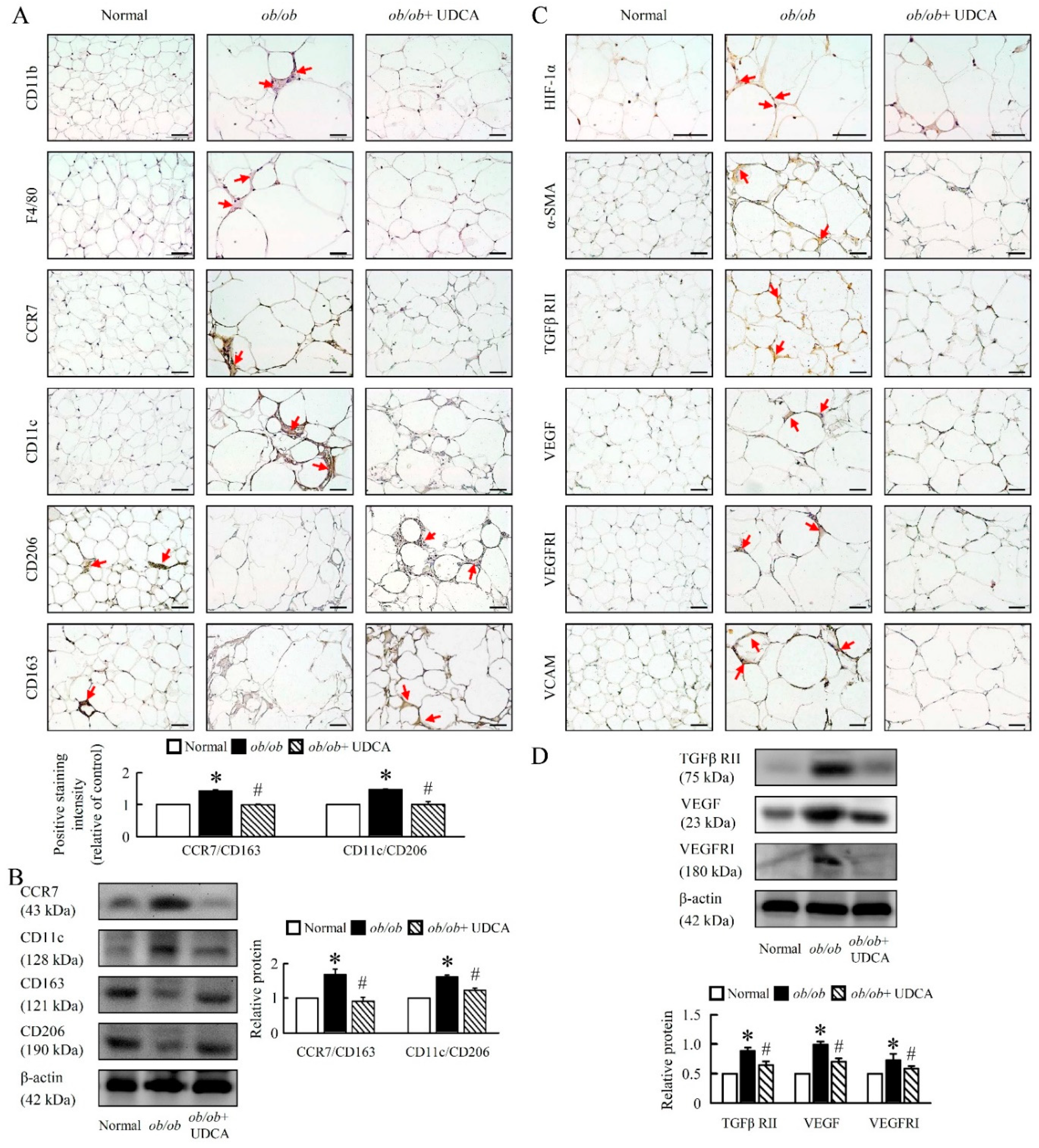
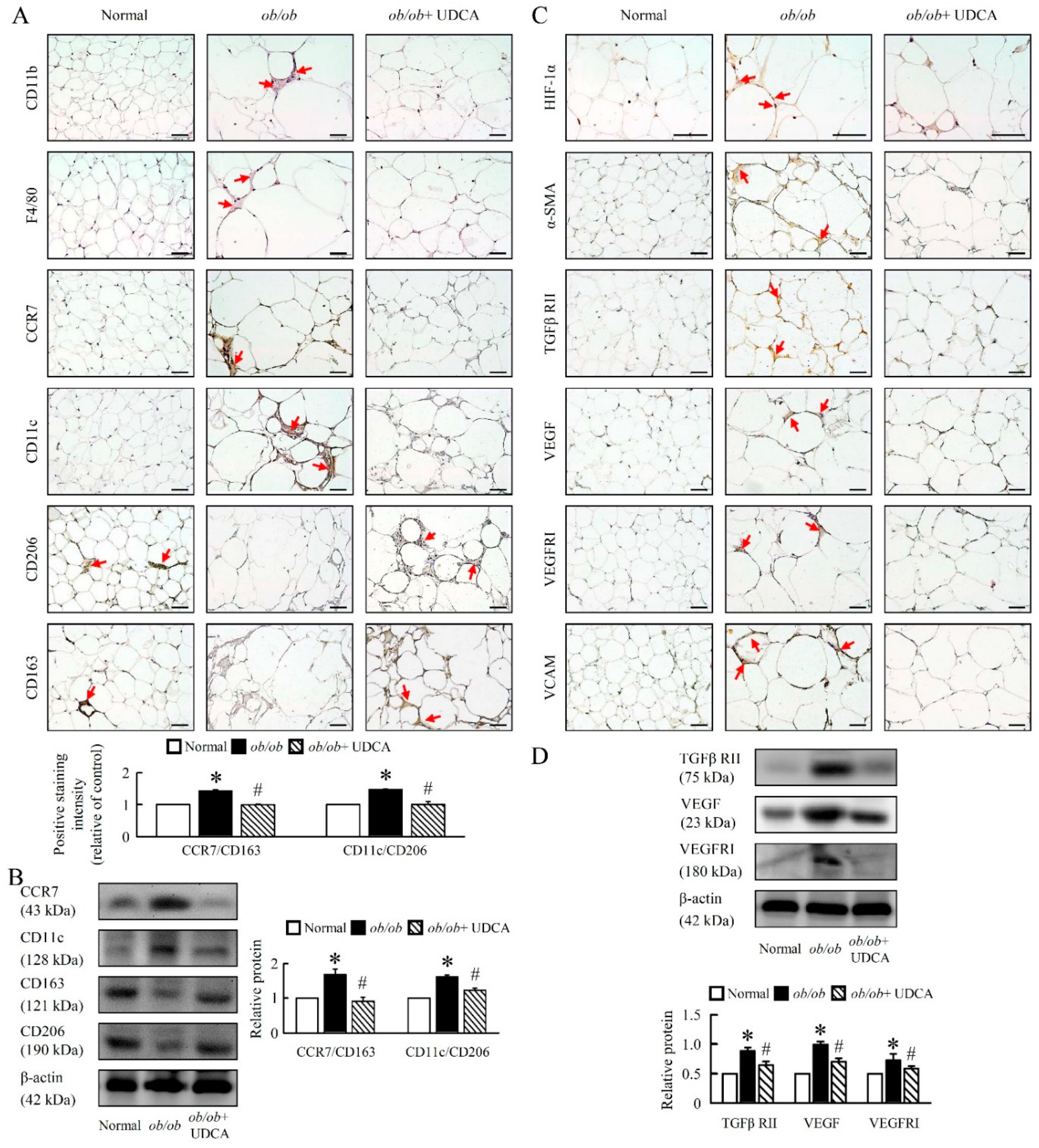
3.8. UDCA Alters EWAT M1 and M2 Macrophage Polarization and Ameliorates Initial Angiogenesis in ob/ob Mice
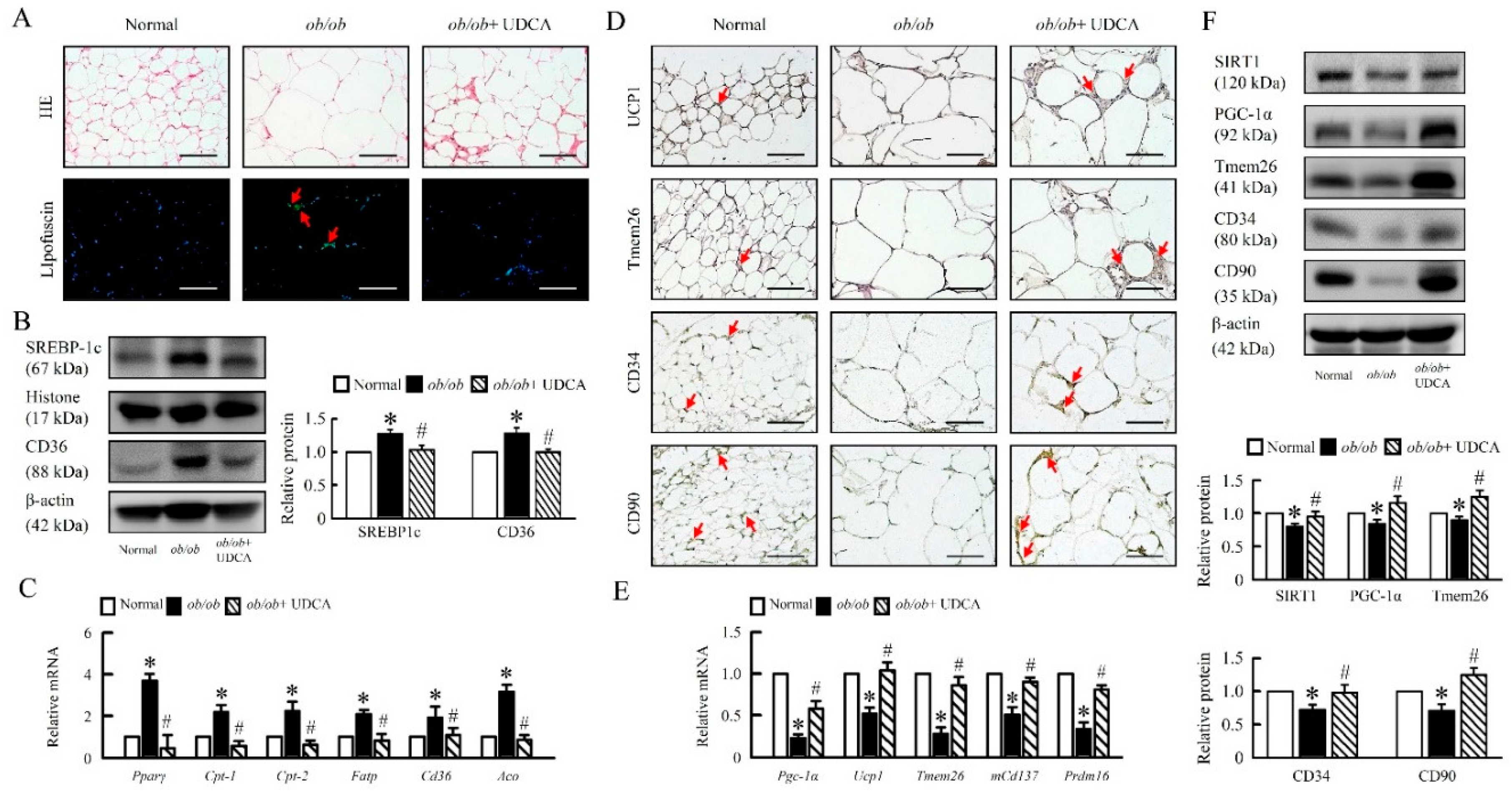
3.9. UDCA Attenuates Lipogenesis and Induces Browning in EWAT of ob/ob Mice
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Non-alcoholic Fatty Liver Disease and Non-alcoholic Steatohepatitis. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Harlow, K.E.; Africa, J.A.; Wells, A.; Belt, P.H.; Behling, C.A.; Jain, A.K.; Molleston, J.P.; Newton, K.P.; Rosenthal, P.; Vos, M.B.; et al. Clinically Actionable Hypercholesterolemia and Hypertriglyceridemia in Children with Nonalcoholic Fatty Liver Disease. J. Pediatr. 2018, 198, 76–83. [Google Scholar] [CrossRef]
- Jinjuvadia, R.; Antaki, F.; Lohia, P.; Liangpunsakul, S. The Association between Nonalcoholic Fatty Liver Disease and Metabolic Abnormalities in The United States Population. J. Clin. Gastroenterol. 2017, 51, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; de Winther, M.P. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. J. Vis. Exp. 2015, 105, 1–7. [Google Scholar] [CrossRef]
- Fischer, K.; Ruiz, H.H.; Jhun, K.; Finan, B.; Oberlin, D.J.; van der Heide, V.; Kalinovich, A.V.; Petrovic, N.; Wolf, Y.; Clemmensen, C.; et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 2017, 23, 623–630. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Qiu, Y.; Cui, X.; Goh, Y.P.; Mwangi, J.; David, T.; Mukundan, L.; Brombacher, F.; Locksley, R.M.; Chawla, A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011, 480, 104–108. [Google Scholar] [CrossRef]
- Tsiloulis, T.; Carey, A.L.; Bayliss, J.; Canny, B.; Meex, R.C.R.; Watt, M.J. No evidence of white adipocyte browning after endurance exercise training in obese men. Int. J. Obes. 2018, 42, 721–727. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Dyson, J.K.; Alexander, G.J.M.; Chapman, M.H.; Collier, J.; Hubscher, S.; Patanwala, I.; Pereira, S.P.; Thain, C.; Thorburn, D.; et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut 2018, 67, 1568–1594. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.H.; Tabibian, J.H.; Carey, E.J.; Lindor, K.D. Emerging drugs for the treatment of Primary Biliary Cholangitis. Expert Opin. Emerg. Drugs 2016, 21, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; de Ledinghen, V.; Oberti, F.; Mathurin, P.; Wartelle-Bladou, C.; Renou, C.; Sogni, P.; Maynard, M.; Larrey, D.; Serfaty, L.; et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J. Hepatol. 2011, 54, 1011–1019. [Google Scholar] [CrossRef]
- Tsuchida, T.; Shiraishi, M.; Ohta, T.; Sakai, K.; Ishii, S. Ursodeoxycholic acid improves insulin sensitivity and hepatic steatosis by inducing the excretion of hepatic lipids in high-fat diet-fed KK-Ay mice. Metabolism 2012, 61, 944–953. [Google Scholar] [CrossRef]
- Shaheen, A.A.; Kaplan, G.G.; Almishri, W.; Vallerand, I.; Frolkis, A.D.; Patten, S.; Swain, M.G. The impact of depression and antidepressant usage on primary biliary cholangitis clinical outcomes. PLoS ONE 2018, 13, e0194839. [Google Scholar] [CrossRef]
- Dosa, P.I.; Ward, T.; Castro, R.E.; Rodrigues, C.M.; Steer, C.J. Synthesis and evaluation of water-soluble prodrugs of ursodeoxycholic acid (UDCA), an anti-apoptotic bile acid. ChemMedChem 2013, 8, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S. Immunomodulating and anti-apoptotic action of ursodeoxycholic acid: Where are we and where should we go? Eur. J. Gastroenterol. Hepatol. 2005, 17, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Dilger, K.; Hohenester, S.; Winkler-Budenhofer, U.; Bastiaansen, B.A.; Schaap, F.G.; Rust, C.; Beuers, U. Effect of ursodeoxycholic acid on bile acid profiles and intestinal detoxification machinery in primary biliary cirrhosis and health. J. Hepatol. 2012, 57, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med. 1998, 4, 165–178. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef]
- Manley, S.; Ding, W. Role of farnesoid X receptor and bile acids in alcoholic liver disease. Acta Pharm. Sin. B 2015, 5, 158–167. [Google Scholar] [CrossRef]
- Sombetzki, M.; Fuchs, C.D.; Fickert, P.; Osterreicher, C.H.; Mueller, M.; Claudel, T.; Loebermann, M.; Engelmann, R.; Langner, C.; Sahin, E.; et al. 24-nor-ursodeoxycholic acid ameliorates inflammatory response and liver fibrosis in a murine model of hepatic schistosomiasis. J. Hepatol. 2015, 62, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.J.; Chang, H.H.; Tsai, T.H.; Lee, T.Y. Curcumin ameliorates mitochondrial dysfunction associated with inhibition of gluconeogenesis in free fatty acid-mediated hepatic lipoapoptosis. Int. J. Mol. Med. 2012, 30, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Poot, M.; Zhang, Y.Z.; Kramer, J.A.; Wells, K.S.; Jones, L.J.; Hanzel, D.K.; Lugade, A.G.; Singer, V.L.; Haugland, R.P. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J. Histochem. Cytochem. 1996, 44, 1363–1372. [Google Scholar] [CrossRef]
- Romijn, H.J.; van Uum, J.F.; Breedijk, I.; Emmering, J.; Radu, I.; Pool, C.W. Double immunolabeling of neuropeptides in the human hypothalamus as analyzed by confocal laser scanning fluorescence microscopy. J. Histochem. Cytochem. 1999, 47, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Abidi, P.; Kim, A.; Chen, W.; Huang, T.T.; Kraemer, F.B.; Liu, J. Transcriptional activation of hepatic ACSL3 and ACSL5 by oncostatin m reduces hypertriglyceridemia through enhanced beta-oxidation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2198–2205. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlstrom, A.; Marschall, H.U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef]
- Tomkin, G.H.; Owens, D. Obesity diabetes and the role of bile acids in metabolism. J. Transl. Int. Med. 2016, 4, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, D.; Rudling, M.; Angelin, B.; Bjorkhem, I.; Forsell, P.; Nilsell, K. Hepatic cholesterol metabolism in human obesity. Hepatology 1997, 25, 1447–1450. [Google Scholar] [CrossRef]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 2013, 58, 155–168. [Google Scholar] [CrossRef]
- Chen, H.; Liu, X.; Chen, H.; Cao, J.; Zhang, L.; Hu, X.; Wang, J. Role of SIRT1 and AMPK in mesenchymal stem cells differentiation. Ageing Res. Rev. 2014, 13, 55–64. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, C.; Liu, X.; Li, M.; Gao, M.; Fang, F.; Chang, Y. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Mol. Metab. 2017, 6, 138–147. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 2011, 17, 961–967. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Qiang, L.; Kangsamaksin, T.; Kitajewski, J.; Ginsberg, H.N.; Accili, D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med. 2013, 19, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chi, F.; Guo, T.; Punj, V.; Lee, W.N.; French, S.W.; Tsukamoto, H. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J. Clin. Investig. 2015, 125, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Reddy, J.K. PPARalpha in the pathogenesis of fatty liver disease. Hepatology 2004, 40, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Lukivskaya, O.; Patsenker, E.; Buko, V.U. Protective effect of ursodeoxycholic acid on liver mitochondrial function in rats with alloxan-induced diabetes: Link with oxidative stress. Life Sci. 2007, 80, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; San Martin, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Schaeffler, A.; Gross, P.; Buettner, R.; Bollheimer, C.; Buechler, C.; Neumeier, M.; Kopp, A.; Schoelmerich, J.; Falk, W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology 2009, 126, 233–245. [Google Scholar] [CrossRef]
- Vieira-Potter, V.J. Inflammation and macrophage modulation in adipose tissues. Cell Microbiol. 2014, 16, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Iwashita, M.; Nagayasu, S.; Yamashita, A.; Shinjo, T.; Hashikata, A.; Asano, T.; Kushiyama, A.; Ishimaru, N.; Takahama, Y.; et al. Protection from diet-induced obesity and insulin resistance in mice lacking CCL19-CCR7 signaling. Obesity 2015, 23, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, V.; Cardellini, M.; Cinti, F.; Corgosinho, F.; Cardolini, I.; D’Adamo, M.; Zingaretti, M.C.; Bellia, A.; Lauro, D.; Gentileschi, P.; et al. Omental adipose tissue fibrosis and insulin resistance in severe obesity. Nutr. Diabetes 2015, 5, e175. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Haring, H.U. The role of hepatokines in metabolism. Nat. Rev. Endocrinol. 2013, 9, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef]
- Oh, A.R.; Bae, J.S.; Lee, J.; Shin, E.; Oh, B.C.; Park, S.C.; Cha, J.Y. Ursodeoxycholic acid decreases age-related adiposity and inflammation in mice. BMB Rep. 2016, 49, 105–110. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Number | Company | Dilution |
|---|---|---|---|
| α-SMA | A5228 | Sigma, USA | IHC 1:80 |
| β-actin | MAB1501 | Millipore, Germany | WB 1:1000 |
| β-catenin | ab6302 | Abcam, UK | WB 1:500 |
| BSEP | ab217532 | Abcam, UK | IHC 1:80 |
| c-Myc | ab32 | Abcam, UK | WB 1:500 |
| CCR7 | ab1657 | Abcam, UK | WB 1:500; IHC 1:80 |
| CD11b | ab133357 | Abcam, UK | IHC 1:80 |
| CD11c | ab11029 | Abcam, UK | IHC 1:80 |
| CD11c | ab52632 | Abcam, UK | WB 1:500 |
| CD163 | ab182422 | Abcam, UK | WB 1:500; IHC 1:80 |
| CD206 | ab64693 | Abcam, UK | WB 1:500; IHC 1:80 |
| CD34 | ab8158 | Abcam, UK | WB 1:500; IHC 1:80 |
| CD36 | SC-70644 | Santz cruz, USA | WB 1:500; IHC 1:80 |
| CD36, Alexa Fluor 647 | 102609 | BioLegend, USA | IF 1:100 |
| CD90 | ab3105 | Abcam, UK | WB 1:500; IHC 1:80 |
| CYP7A1 | SC-518007 | Santz cruz, USA | WB 1:500; IHC 1:80 |
| F4/80 | 123102 | BioLegend, CA | IHC 1:80 |
| FGFR4 | ab44971 | Abcam, UK | IHC 1:80 |
| FXR | SC-13063 | Santz cruz, USA | WB 1:500; IHC 1:80; IF 1:80 |
| G6Pase | SC-25840 | Santz cruz, USA | WB 1:500 |
| HIF-1α | ab1 | Abcam, UK | IHC 1:60 |
| Histone | SC-56616 | Santz cruz, USA | WB 1:1000 |
| MRP3 | ab3375 | Abcam, UK | IHC 1:80 |
| Neutrophil | ab2557 | Abcam, UK | IHC 1:80 |
| NF-κB | ab16502 | Abcam, UK | WB 1:500; IHC 1:80; IF 1:80 |
| Notch1 | ab52627 | Abcam, UK | WB 1:500 |
| NICD | ab8925 | Abcam, UK | WB 1:500 |
| NTCP | ab131084 | Abcam, UK | IHC 1:80 |
| OATP1 | ab203036 | Abcam, USA | IHC 1:80 |
| OSTβ | orb1964 | Biorbyt, USA | IHC 1:80 |
| PEPCK | SC-377027 | Santz cruz, USA | WB 1:500 |
| PGC-1α | ab54481 | Abcam, UK | WB 1:500; IHC 1:80 |
| SIRT1 | ab110304 | Abcam, UK | WB 1:500; IHC 1:80 |
| SREBP-1c | SC-366 | Santz cruz, USA | WB 1:500; IHC 1:100; IF 1:80 |
| p-STAT3 | 05-485 | Millipore, Germany | WB 1:500 |
| STAT3 | 06-596 | Millipore, Germany | WB 1:500 |
| TGFβ RII | SC-17791 | Santz cruz, USA | WB 1:500; IHC 1:100 |
| TGR5 | ab72608 | Abcam, UK | IHC 1:80 |
| TLR4 | ab13556 | Abcam, UK | IHC 1:80 |
| Tmem26 | ab186640 | Abcam, UK | WB 1:500; IHC 1:80 |
| UCP1 | ab10983 | Abcam, UK | WB 1:500; IHC 1:80 |
| VCAM | SC-8304 | Santz cruz, USA | IHC 1:80 |
| VEGF | SC-80434 | Santz cruz, USA | IHC 1:80 |
| VEGF | ab69479 | Abcam, UK | WB 1:500 |
| VEGFRI | 1303-1 | Epitomics, USA | WB 1:500; IHC 1:80 |
| DAPI | 62248 | Thermo Fisher Scientific, USA | IF 1:10000 |
| Mouse Secondary Antibody | G-21040 | Thermo Fisher Scientific, USA | WB 1:3000; IHC 1:100 |
| Rabbit Secondary Antibody | G-21234 | Thermo Fisher Scientific, USA | WB 1:3000; IHC 1:100 |
| Goat Secondary Antibody | 31402 | Thermo Fisher Scientific, USA | WB 1:3000; IHC 1:100 |
| Rat Secondary Antibody | 31470 | Thermo Fisher Scientific, USA | WB 1:3000; IHC 1:100 |
| Mouse Secondary Antibody, Alexa Fluor 488 | A-11029 | Thermo Fisher Scientific, USA | IF 1:100 |
| Rabbit Secondary Antibody, Alexa Fluor 488 | A11034 | Thermo Fisher Scientific, USA | IF 1:100 |
| Rabbit Secondary Antibody, Alexa Fluor Plus 647 | A32795 | Thermo Fisher Scientific, USA | IF 1:100 |
| Gene | Forward | Reverse |
|---|---|---|
| SREBP-1c | 5′ actgtcttggttgttgatgagctggagcat 3′ | 5′ atcggcgcggaagctgtcggggtagcgtc 3′ |
| FAS | 5′ tgtcattggcctcctcaaaaagggcgtcca 3′ | 5′ tcaccactgtgggctctgcagagaagcgag 3′ |
| SCD-1 | 5′ ccggagaccccttagatcga 3′ | 5′ tagcctgtaaaagatttctgcaaacc 3′ |
| FATP | 5′ gcttcaacagccgtatcctc 3′ | 5′ tcttcttgttggtggcactg 3′ |
| CD36 | 5′ gcaaaacgactgcaggtcaac 3′ | 5′ tggtcccagtctcatttagcca 3′ |
| CPT-1 | 5′ ggacagagactgtgcgttcct 3′ | 5′ gcgatatccaacagtgcttga 3′ |
| CPT-2 | 5′ caaggccctggctgatgatgtg 3′ | 5′ agtctctgtccgcccctctcg 3′ |
| ACO | 5′ atgaatcccgatctgcgcaaggagc 3′ | 5′ aaaggcatgtaacccgtagcactcc 3′ |
| PPARα | 5′ cgtacggcaatggctttatc 3′ | 5′ aacggcttcctcaggttctt 3′ |
| PPARγ | 5′ cctcaaacttggcaatactc 3′ | 5′- agcaacaacataagcgtcat 3′ |
| Ppargc1b | 5′ cagccagtacagccccgatg 3′ | 5′ ggtgtgtcgccttcatccag 3′ |
| ATGL | 5′ aacaccagcatccagttcaa 3′ | 5′ ggttcagtaggccattcctc 3′ |
| HSL | 5′ agacaccagccaacggatac 3′ | 5′ catcaccctcgaagaagagca 3′ |
| DGAT | 5′ tcctgaattggtgtgtggtg 3′ | 5′ ggcgcttctcaatctgaaat 3′ |
| LCAD | 5′ tcaacagcagttacttgg 3′ | 5′ gacaatatctgagtggag 3′ |
| LPL | 5′ actcatctccgccatgcc 3′ | 5′ ccagctttctcctagcaagg 3′ |
| ACADM | 5′ ggggaggatgacggagcagc 3′ | 5′ cgggtactttaggatctggg 3′ |
| Notch1 | 5′ ccagcatggccagctctgg 3′ | 5′ catccagatctgtggccctgtt 3′ |
| Jag1 | 5′ gtccacggcacctgcaatg 3′ | 5′ caaggtttggcctcgcact 3′ |
| RBPJ | 5′ tggcactgttcaatcgcctt 3′ | 5′ aatcttgggagtgccatgcca 3′ |
| Dll1 | 5′ actccttcagcctgcctga 3′ | 5′ tatcggatgcactcatcgc 3′ |
| Dll3 | 5′ ctggtgtcttcgagctaca 3′ | 5′ acacgtgctagcaggttcc 3′ |
| Hes1 | 5′ aaagacggcctctgagcaca 3′ | 5′ tcatggcgttgatctgggtca 3′ |
| Hes5 | 5′ aagtaccgtggcggtggagatgc 3′ | 5′ cgctggaagtggtaaagcagctt 3′ |
| PEPCK | 5′ agcctgctccagctttga 3′ | 5′ ccctagcctgttctctgtgc 3′ |
| G6Pase | 5′ tgctgtgtctggtaggcaac 3′ | 5′ agaatcctgggtctccttgc 3′ |
| Pdk4 | 5′ gatcctaaccaccgccagcc 3′ | 5′ gcaaaggacgttctttcacag 3′ |
| Pdha1 | 5′ gccgagtgctggttgcttccc 3′ | 5′ gtctgcatcatcctgtagtacttgagcc 3′ |
| Gys1 | 5′ cggttgtcggacttgctagattgg 3′ | 5′ cataggtgaagtggtctggaaaggc 3′ |
| Gys2 | 5′ ccttggggtgtttccatcgtac 3′ | 5′ cggagaggtttgtagtcacactgg 3′ |
| Pik3r1 | 5′ cccactactgtagccaacaacagc 3′ | 5′ gagtgtaatcgccgtgcattttag 3′ |
| IL-6 | 5′-gtactccagaagaccagagg-3′ | 5′-tgctggtgacaaccacggcc-3′ |
| IL-10 | 5′ ctggctcagcactgctat 3′ | 5′ attcatggccttgtagacac 3′ |
| TNFα | 5′ ttgacctcagcgctgagttg 3′ | 5′ cctgtagcccacgtcgtagc 3′ |
| SOCS1 | 5′ gtggttgtggagggtgagat 3′ | 5′ cccagacacaagctgctaca 3′ |
| SOCS3 | 5′ taggaggcgcagccccaagg 3′ | 5′ gcggcgggaaacttgctgtg 3′ |
| STAT3 | 5′ cgacccaggtgctgccccgta 3′ | 5′ atgggggaggtagcacactccga 3′ |
| SIRT1 | 5′ gcaacagcatcttgcctgat 3′ | 5′ gtgctactggtctcactt 3′ |
| PGC-1α | 5′ gactcagtgtcaccaccgaaa-3′ | 5′ tgaacgagagcgcatcctt 3′ |
| TFAM | 5′ ggaatgtggagcgtgctaaaa 3′ | 5′-tgctggaaaaacacttcggaata 3′ |
| UCP1 | 5′ cctgcctctctcggaaacaa 3′ | 5′-tgtaggctgcccaatgaaca 3′ |
| Complex I (20kDa) | 5′ ccagctgcgcagagttcatc 3′ | 5′ gagagagcttggggaccacg 3′ |
| Complex II (Ip) | 5′ tctaccgctgccacaccatc 3′ | 5′ aagccaatgctcgcttctcc 3′ |
| Complex III (Core II) | 5′ ccattggaaatgcagaggca 3′ | 5′ ggctggtgacttcctttggc 3′ |
| Complex IV (Cox2) | 5′ tcatgagcagtcccctccct 3′ | 5′ gccatagaataaccctggtcgg 3′ |
| Complex V (F1α) | 5′ atctatgcgggtgtacgggg 3′ | 5′ agggactggtgctggctgat 3′ |
| p22phox | 5′ tggcctgattctcatcactgg 3′ | 5′ gggacaactccacagaaactc 3′ |
| p47phox | 5′ acatcacaggccccatcatccttc 3′ | 5′-atggattgtcctttgtgcc 3′ |
| NOX2 | 5′ actccttgggtcagcactgg 3′ | 5′ gttcctgtccagttgtcttcg 3′ |
| NOX4 | 5′ tgaactacagtgaagatttccttgaac 3′ | 5′ gacacccgtcagaccaggaa 3′ |
| Oatp1 | 5′ gtcttacgagtgtgctccagat 3′ | 5′ ggaatactgcctctgaagtggatt 3′ |
| Ntcp | 5′ caccatggagttcagcaaga 3′ | 5′ agcactgaggggcatgatac 3′ |
| Mrp2 | 5′ gcttcccatggtgatctctt 3′ | 5′ atcatcgcttcccaggtact 3′ |
| Mrp4 | 5′ ttagatgggcctctggttct ’ | 5′ gcccacaattccaaccttt 3′ |
| Sult2a1 | 5′ ggaaggaccacgactcataac 3′ | 5′ gattcttcacaaggtttgtgttacc 3′ |
| ABCA1 | 5′ tggacatcctgaagccag 3′ | 5′ ttcttcccacatgccct 3′ |
| ABCG1 | 5′ gctgggaagtccacactc 3′ | 5′ gatacggcacgagattgg 3′ |
| HMG Co S | 5′ tatgatggtgtagatgctgggaagtatacc 3′ | 5′ taagttcttctgtgcttttcatccac 3′ |
| HMG Co R | 5′ gggacggtgacacttaccatctgtatgatg 3′ | 5′ atcatcttggagagataaaactgcca 3′ |
| Tmem26 | 5′ accctgtcatcccacagag 3′ | 5′ tgtttggtggagtcctaaggtc 3′ |
| CD137 | 5′ cgtgcagaactcctgtgataac 3′ | 5′ gtccacctatgctggagaagg 3′ |
| Prdm16 | 5′ cagcacggtgaagccattc | 5′ gcgtgcatccgcttgtg 3′ |
| GAPDH | 5′ tcaccaccatggagaaggc 3′ | 5′ gctaagcagttggtggtgca 3′ |
| Items | Normal | ob/ob | ob/ob + UDCA 50 |
|---|---|---|---|
| Body weight (g) | 27.57 ± 0.74 | 63.68 ± 3.15 ** | 55.97 ± 2.09 # |
| Liver weight/Body weight (%) | 4.69 ± 0.29 | 5.42 ± 0.24 * | 4.53 ± 0.17 # |
| Plasma ALT (IU/dL) | 26.87 ± 7.62 | 111.47 ± 21.40 ** | 56.93 ± 12.34 ## |
| Plasma TBA (μ mol/L) | 27.46 ± 11.36 | 61.52 ± 3.84 ** | 57.95 ± 2.77 |
| Plasma TG (mg/dL) | 85.26 ± 11.12 | 157.62 ± 5.30 ** | 123.15 ± 8.44 ## |
| Plasma FFA (mmol/L) | 1.22 ± 0.02 | 1.29 ± 0.01 ** | 1.13 ± 0.04 ## |
| Plasma Cholesterol (mmol/L) | 58.15 ± 2.93 | 111.53 ± 12.55 ** | 76.96 ± 5.77 ## |
| Liver TBA (mmol/g liver) | 0.85 ± 0.04 | 1.02 ± 0.06 ** | 1.06 ± 0.11 |
| Liver TG (mmol/g liver) | 48.18 ± 2.18 | 111.21 ± 1.39 ** | 102.02 ± 3.32 ## |
| Live FFA (mmol/g liver) | 36.61 ± 0.06 | 69.71 ± 0.78 ** | 56.29 ± 1.17 ## |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-S.; Liu, H.-M.; Lee, T.-Y. Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice. Cells 2019, 8, 253. https://doi.org/10.3390/cells8030253
Chen Y-S, Liu H-M, Lee T-Y. Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice. Cells. 2019; 8(3):253. https://doi.org/10.3390/cells8030253
Chicago/Turabian StyleChen, Yu-Sheng, Hsuan-Miao Liu, and Tzung-Yan Lee. 2019. "Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice" Cells 8, no. 3: 253. https://doi.org/10.3390/cells8030253
APA StyleChen, Y.-S., Liu, H.-M., & Lee, T.-Y. (2019). Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice. Cells, 8(3), 253. https://doi.org/10.3390/cells8030253