The Protein Tyrosine Phosphatase H1 PTPH1 Supports Proliferation of Keratinocytes and is a Target of the Human Papillomavirus Type 8 E6 Oncogene

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids, siRNA
2.3. RNA-Isolation, RT-PCRs
2.4. Antibodies
2.5. Western Blots, Co-Immunoprecipitation (Co-IP) and GST-Pull Down Assays
2.6. BrdU- and Proliferation Assays
2.7. Immunostainings
2.8. PTP-Assay
2.9. Statistics
2.10. Ethic Statement
3. Results
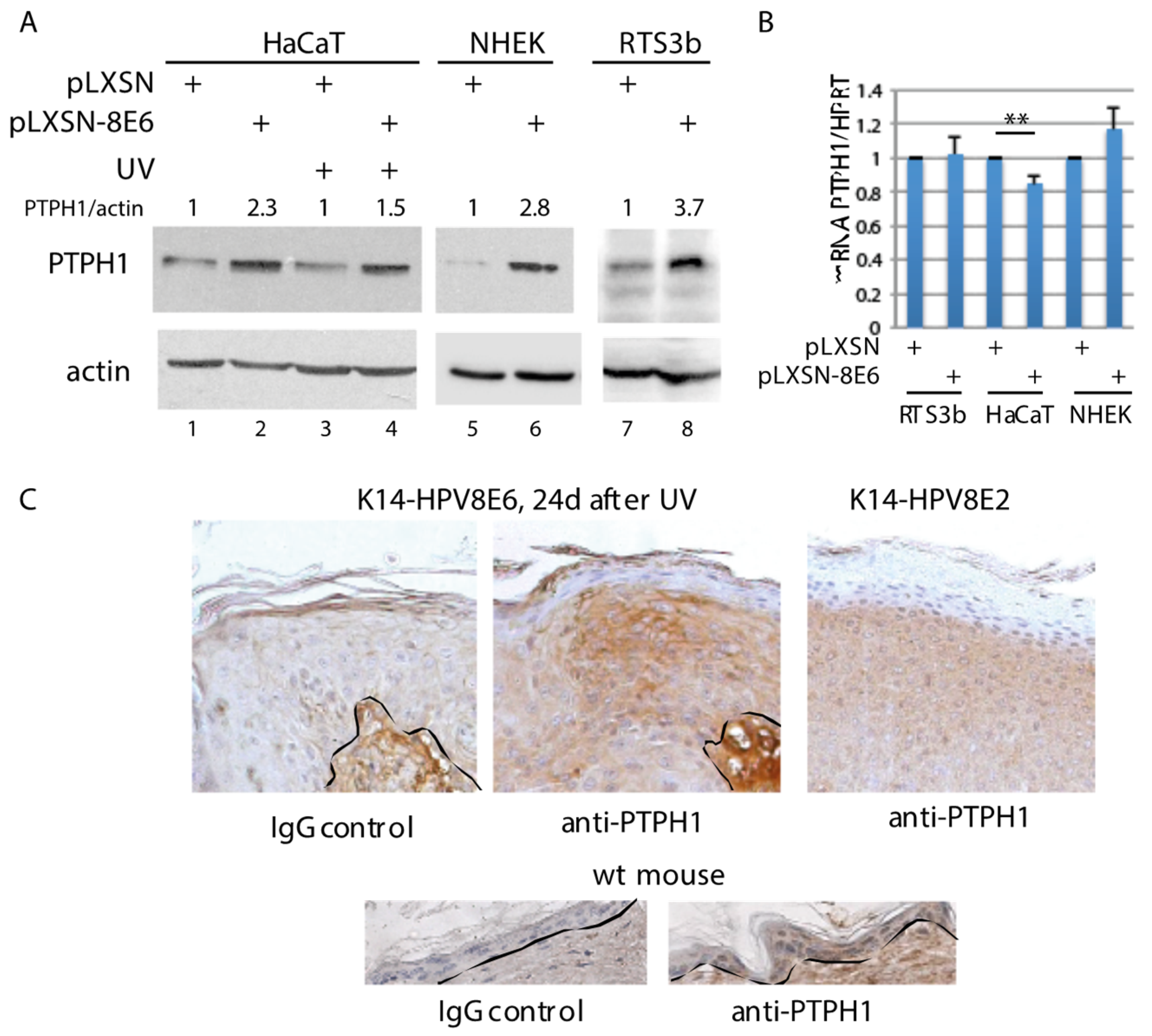
3.1. Increased PTPH1 Level in HPV8E6 Expressing Keratinocytes
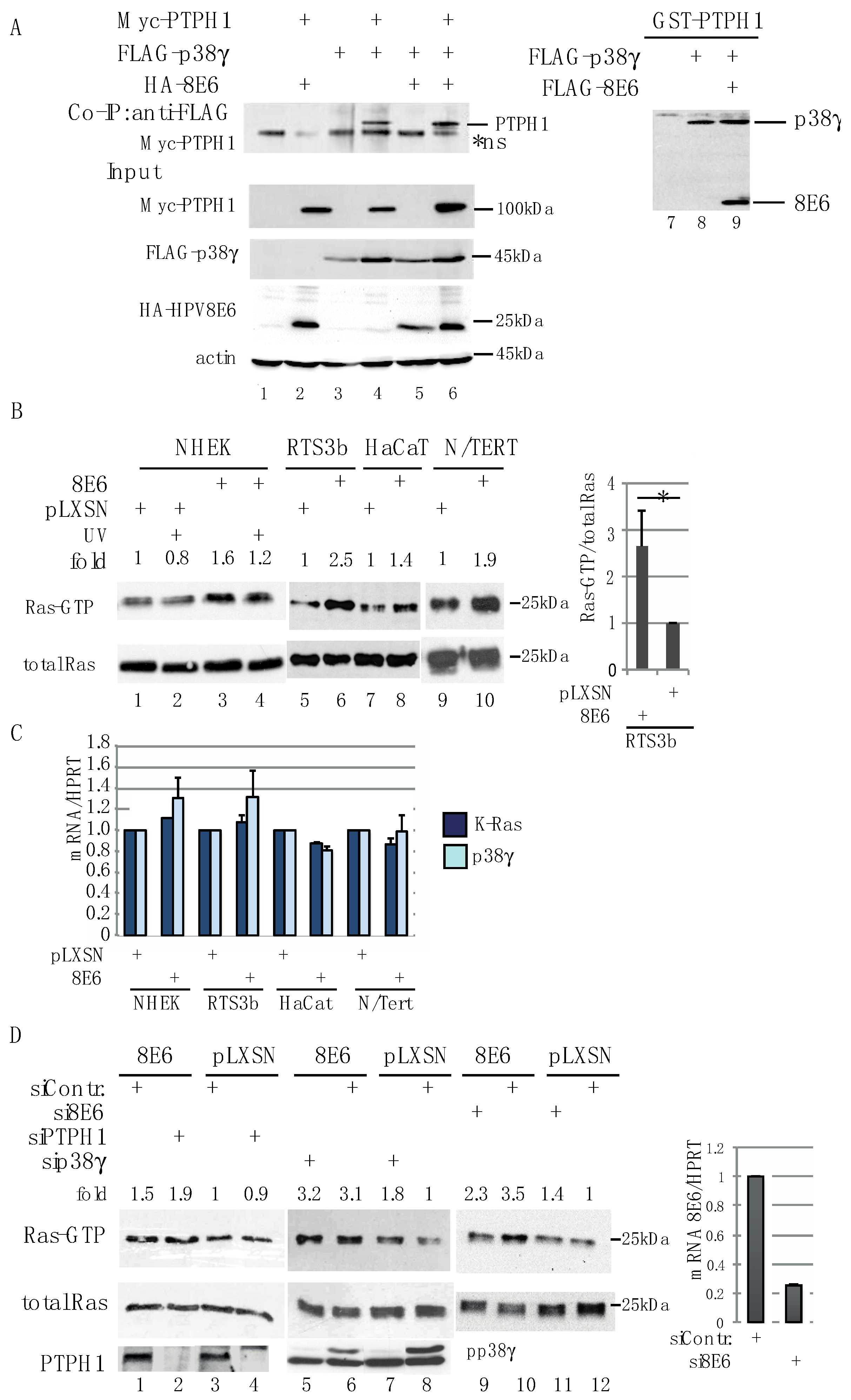
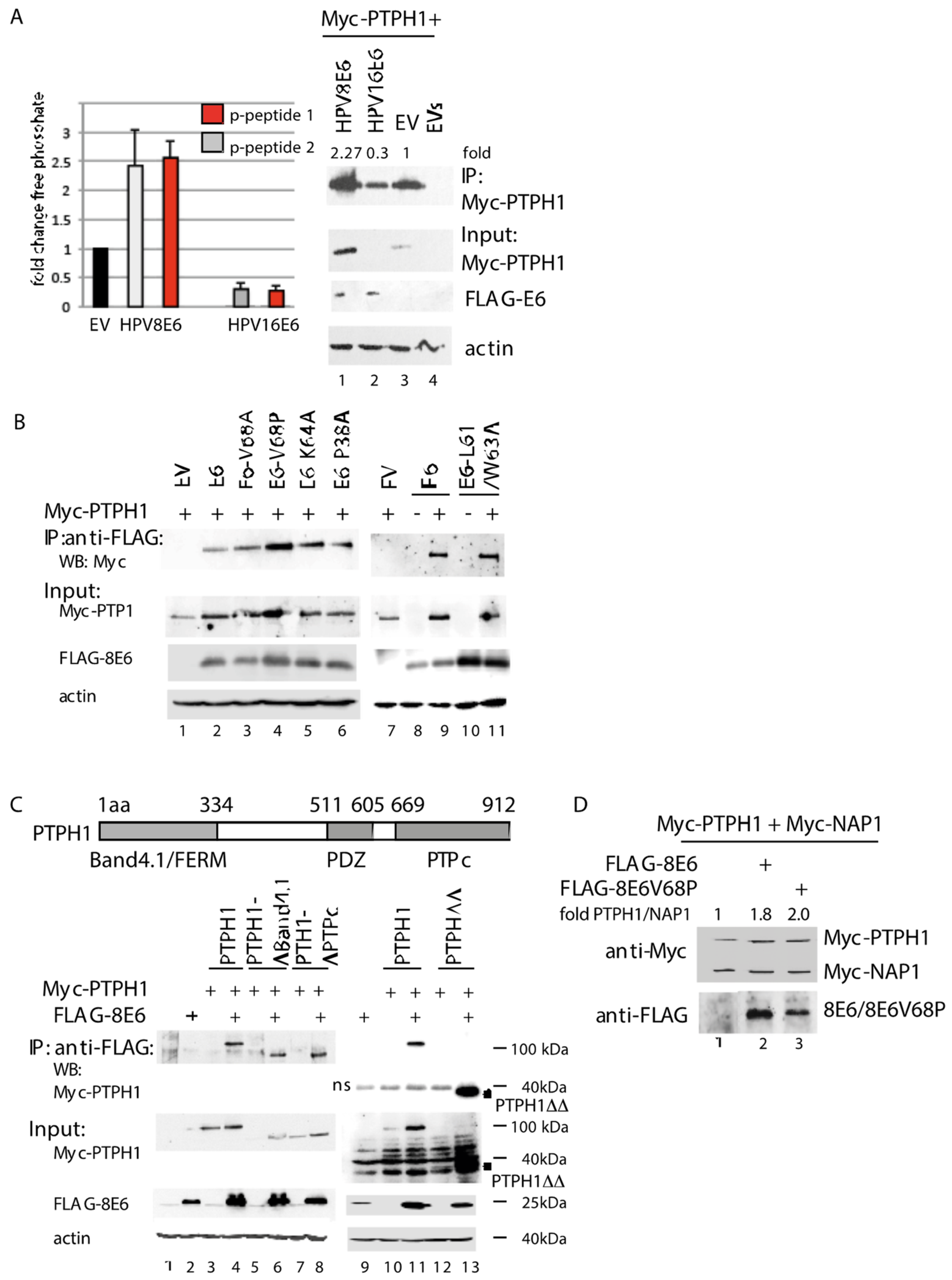
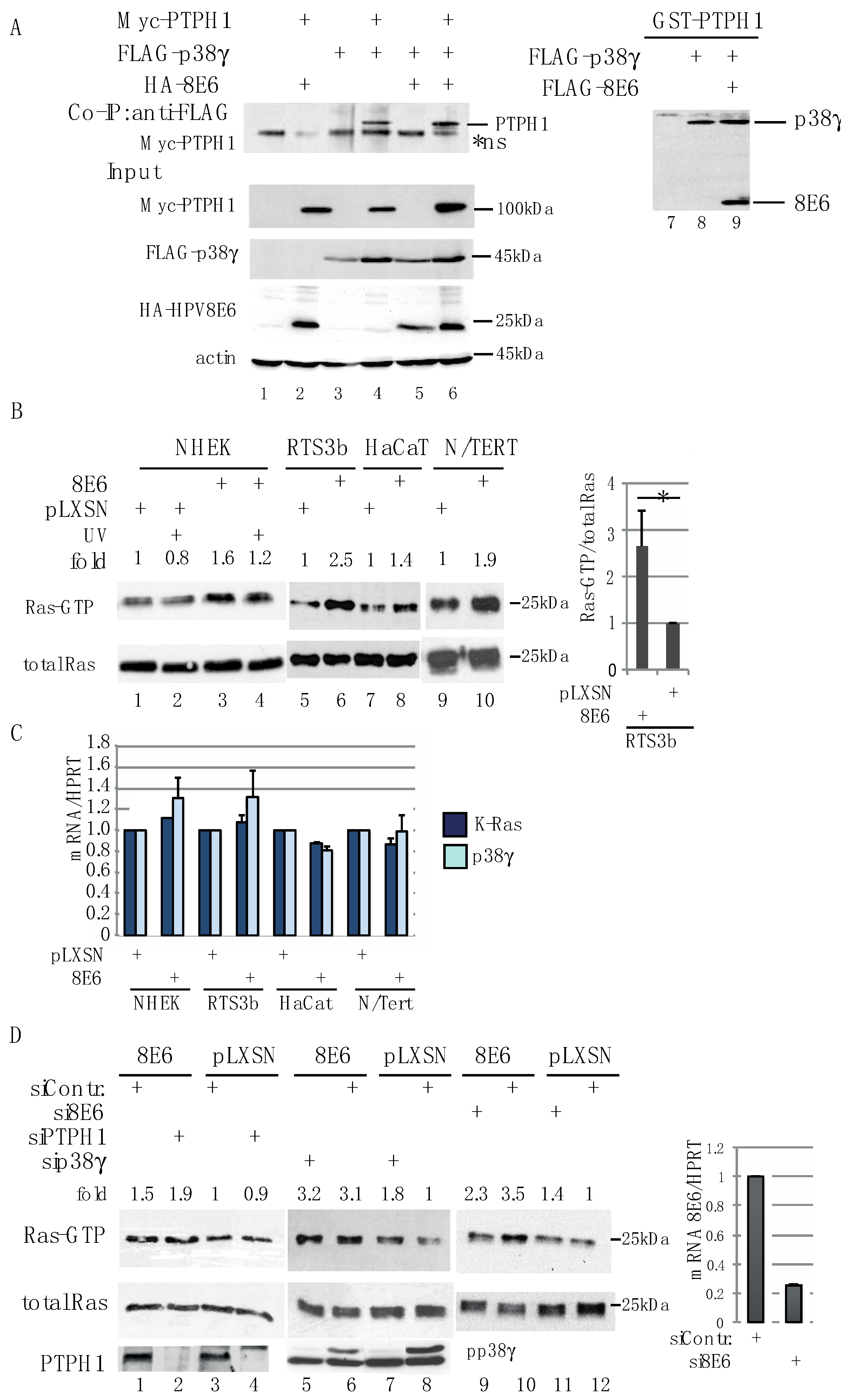
3.2. HPV8E6 Directly Targets PTPH1 and Does Not Interfere with the Phosphatase Activity of PTPH1
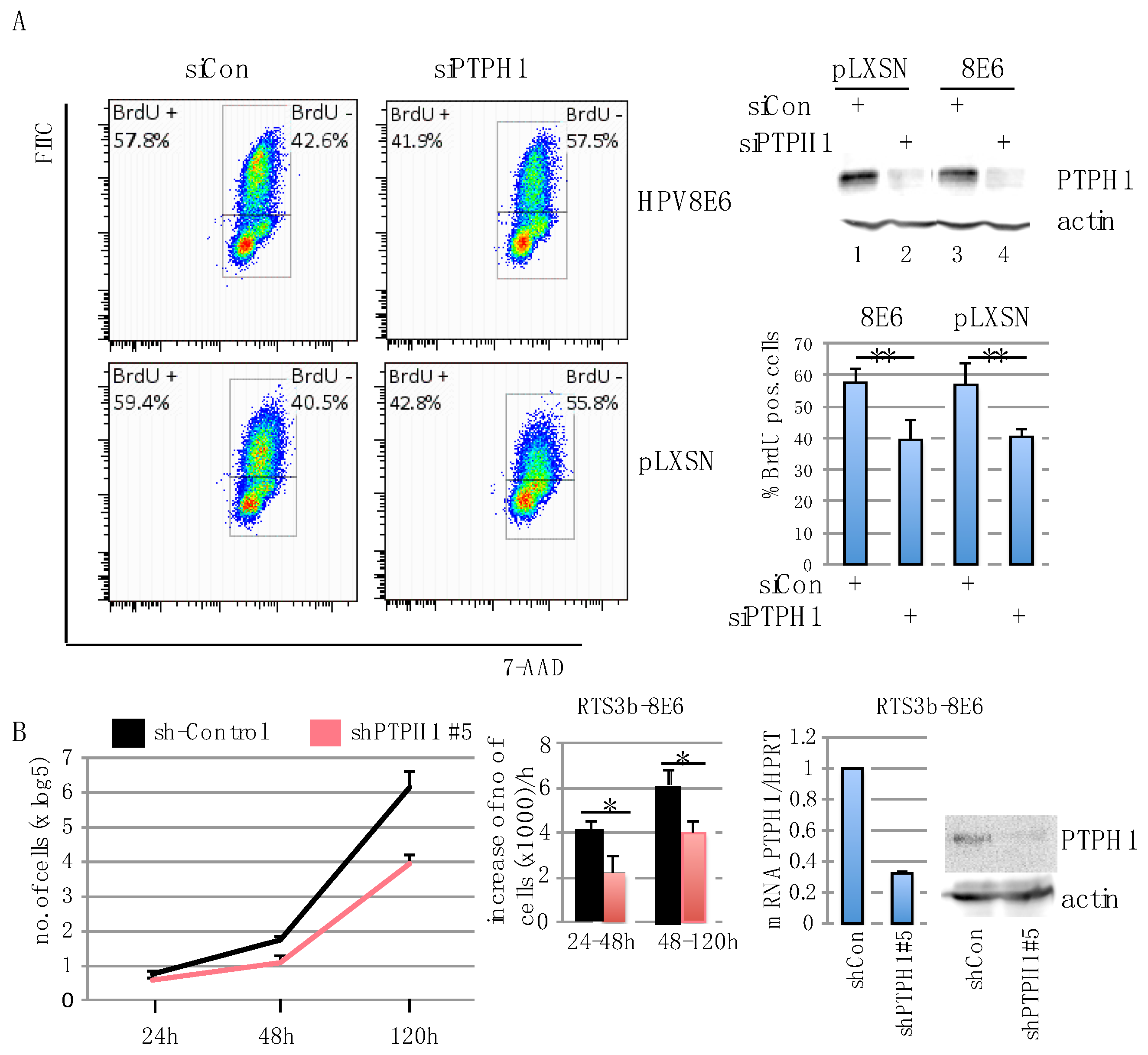
3.3. PTPH1 Supports Cell Growth and Proliferation in HPV8E6 Expressing Keratinocytes
3.4. HPV8E6 Leads to Higher Level of GTP-Ras
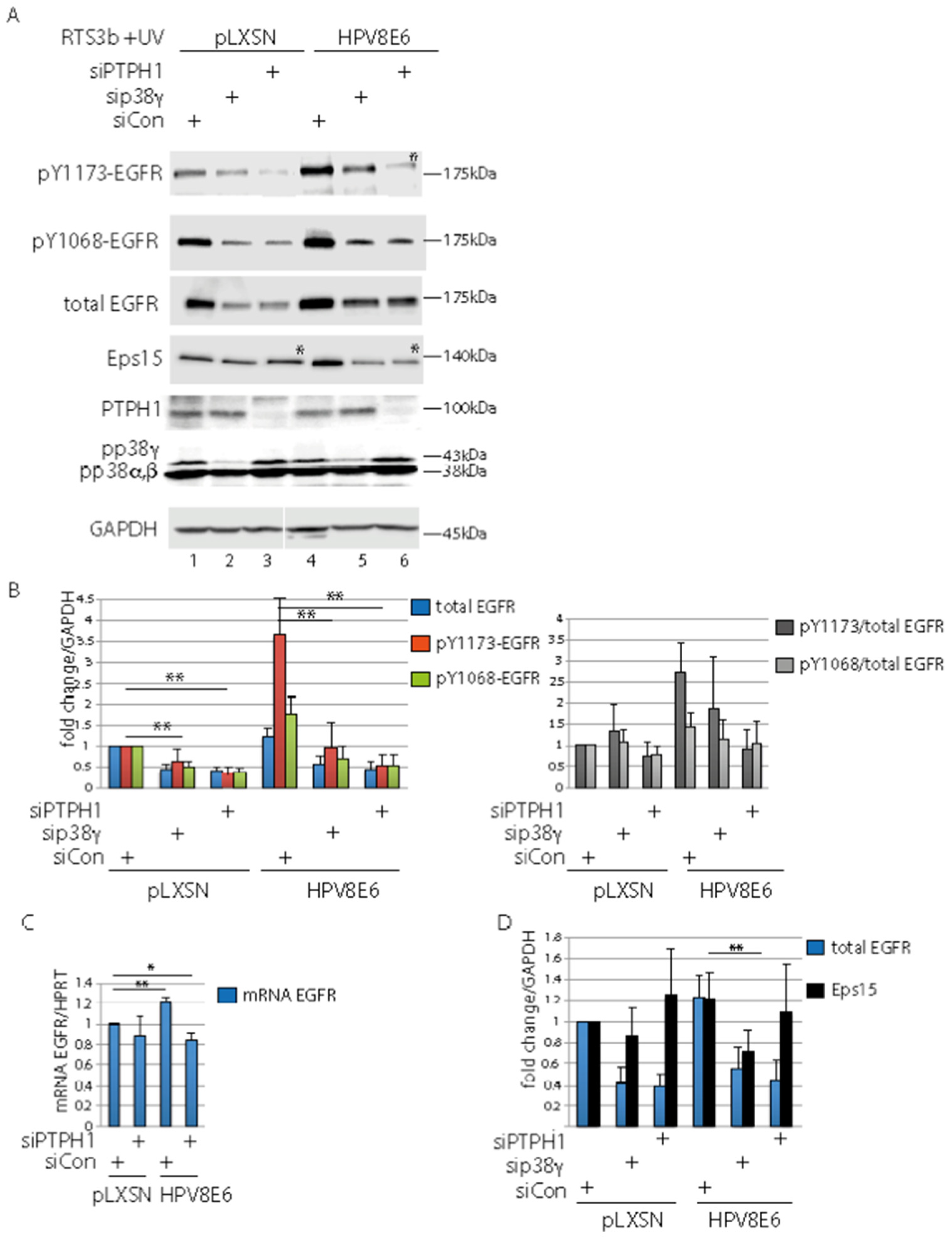
3.5. PTPH1 and p38γ Are Involved in Control of the UV-Activated EGFR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef]
- Bouwes Bavinck, J.N.; Feltkamp, M.C.W.; Green, A.C.; Fiocco, M.; Euvrard, S.; Harwood, C.A.; Nasir, S.; Thomson, J.; Proby, C.M.; Naldi, L.; et al. Human papillomavirus and posttransplantation cutaneous squamous cell carcinoma: A multicenter, prospective cohort study. Am. J. Transplant. 2018, 18, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Neale, R.E.; Weissenborn, S.; Abeni, D.; Bavinck, J.N.; Euvrard, S.; Feltkamp, M.C.; Green, A.C.; Harwood, C.; de Koning, M.; Naldi, L.; et al. Human papillomavirus load in eyebrow hair follicles and risk of cutaneous squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, S.; Neale, R.E.; Waterboer, T.; Abeni, D.; Bavinck, J.N.; Green, A.C.; Harwood, C.A.; Euvrard, S.; Feltkamp, M.C.; de Koning, M.N.; et al. β-papillomavirus DNA loads in hair follicles of immunocompetent people and organ transplant recipients. Med. Microbiol. Immunol. 2012, 201, 117–125. [Google Scholar] [CrossRef]
- Missero, C.; Antonini, D. Crosstalk among p53 family members in cutaneous carcinoma. Exp. Dermatol. 2014, 23, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Schaper, I.D.; Marcuzzi, G.P.; Weissenborn, S.J.; Kasper, H.U.; Dries, V.; Smyth, N.; Fuchs, P.G.; Pfister, H. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005, 65, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, R.; Marcuzzi, G.P.; Akgül, B.; Kasper, H.U.; Schulze, F.; Haase, I.; Wickenhauser, C.; Pfister, H. The human papillomavirus type 8 E2 protein induces skin tumors in transgenic mice. J. Investig. Dermatol. 2008, 128, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Mueller-Decker, K.; Kloz, U.; Aengeneyndt, B.; Kopp-Schneider, A.; Grone, H.J.; Gheit, T.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. E6 and E7 from β HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011, 7, e1002125. [Google Scholar] [CrossRef]
- Viarisio, D.; Decker, K.M.; Aengeneyndt, B.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. Human papillomavirus type 38 E6 and E7 act as tumour promoters during chemically induced skin carcinogenesis. J. Gen. Virol. 2013, 94, 749–752. [Google Scholar] [CrossRef]
- Heuser, S.; Hufbauer, M.; Steiger, J.; Marshall, J.; Sterner-Kock, A.; Mauch, C.; Zigrino, P.; Akgül, B. The fibronectin/α3β1 integrin axis serves as molecular basis for keratinocyte invasion induced by βHPV. Oncogene 2016, 35, 4529–4539. [Google Scholar] [CrossRef]
- Marcuzzi, G.P.; Hufbauer, M.; Kasper, H.U.; Weissenborn, S.J.; Smola, S.; Pfister, H. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 2009, 90, 2855–2864. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Cooke, J.; van der Horst, G.T.; Pfister, H.; Storey, A.; Akgül, B. Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol. Cancer 2015, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Underbrink, M.P.; Howie, H.L.; Bedard, K.M.; Koop, J.I.; Galloway, D.A. E6 proteins from multiple human βpapillomavirus types degrade Bak and protect keratinocytes from apoptosis after UVB irradiation. J. Virol. 2008, 82, 10408–10417. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Storey, A. E6 proteins from diverse cutaneous HPV types inhibit apoptosis in response to UV damage. Oncogene 2000, 19, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Gasior, S.L.; Faber, Z.J.; Howie, H.L.; Deininger, P.L.; Galloway, D.A. HPV 5 and 8 E6 expression reduces ATM protein levels and attenuates LINE-1 retrotransposition. Virology 2013, 443, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.J.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous β-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–E1480. [Google Scholar] [CrossRef]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 Proteins Target the NOTCH and TGF-β Tumor Suppressors to Inhibit Differentiation and Sustain Keratinocyte Proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef]
- Bedard, K.M.; Underbrink, M.P.; Howie, H.L.; Galloway, D.A. The E6 oncoproteins from human βpapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J. Virol. 2008, 82, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Taute, S.; Pfister, H.J.; Steger, G. Induction of Tyrosine Phosphorylation of UV-Activated EGFR by the β-Human Papillomavirus Type 8 E6 Leads to Papillomatosis. Front. Microbiol. 2017, 8, 2197. [Google Scholar] [CrossRef] [PubMed]
- Topffer, S.; Muller-Schiffmann, A.; Matentzoglu, K.; Scheffner, M.; Steger, G. Protein tyrosine phosphatase H1 is a target of the E6 oncoprotein of high-risk genital human papillomaviruses. J. Gen. Virol. 2007, 88, 2956–2965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-H.; Eckberg, W.R.; Yang, Q.; Samatar, A.A.; Tonks, N.K. Biochemical characterization of human band 4.1-related protein-tyrosine phosphatase, PTPH1. J. Biol. Chem. 1995, 270, 20067–20072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Lai, P.L.; Chou, Y.T.; Chi, A.P.; Mi, Y.Z.; Khoo, K.H.; Chang, G.D.; Wu, C.W.; Meng, T.C.; Chen, G.C. Protein tyrosine phosphatase PTPN3 inhibits lung cancer cell proliferation and migration by promoting EGFR endocytic degradation. Oncogene 2015, 34, 3791–3803. [Google Scholar] [CrossRef]
- Zhi, H.Y.; Hou, S.W.; Li, R.S.; Basir, Z.; Xiang, Q.; Szabo, A.; Chen, G. PTPH1 cooperates with vitamin D receptor to stimulate breast cancer growth through their mutual stabilization. Oncogene 2011, 30, 1706–1715. [Google Scholar] [CrossRef]
- Suresh, P.S.; Ma, S.; Migliaccio, A.; Chen, G. Protein-tyrosine phosphatase H1 increases breast cancer sensitivity to antiestrogens by dephosphorylating estrogen receptor at Tyr537. Mol. Cancer Ther. 2014, 13, 230–238. [Google Scholar] [CrossRef]
- Wang, Y.; Su, Y.; Ji, Z.; Lv, Z. High Expression of PTPN3 Predicts Progression and Unfavorable Prognosis of Glioblastoma. Med. Sci. Monit. 2018, 24, 7556–7562. [Google Scholar] [CrossRef]
- Gao, Q.; Zhao, Y.J.; Wang, X.Y.; Guo, W.J.; Gao, S.; Wei, L.; Shi, J.Y.; Shi, G.M.; Wang, Z.C.; Zhang, Y.N.; et al. Activating Mutations in PTPN3 Promote Cholangiocarcinoma Cell Proliferation and Migration and are Associated with Tumor Recurrence in Patients. Gastroenterology 2014, 146, 1397–1407. [Google Scholar] [CrossRef]
- Li, S.; Cao, J.; Zhang, W.; Zhang, F.; Ni, G.; Luo, Q.; Wang, M.; Tao, X.; Xia, H. Protein tyrosine phosphatase PTPN3 promotes drug resistance and stem cell-like characteristics in ovarian cancer. Sci. Rep. 2016, 6, 36873. [Google Scholar] [CrossRef]
- Shi, Z.H.; Li, X.G.; Jie, W.D.; Zhao, H.L.; Zeng, Y.; Liu, Y. PTPH1 promotes tumor growth and metastasis in human glioma. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3777–3787. [Google Scholar]
- Qi, X.M.; Wang, F.; Mortensen, M.; Wertz, R.; Chen, G. Targeting an oncogenic kinase/phosphatase signaling network for cancer therapy. Acta Pharm. Sin. B 2018, 8, 511–517. [Google Scholar] [CrossRef]
- El-Abaseri, T.B.; Putta, S.; Hansen, L.A. Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis 2006, 27, 225–231. [Google Scholar] [CrossRef]
- El-Abaseri, T.B.; Fuhrman, J.; Trempus, C.; Shendrik, I.; Tennant, R.W.; Hansen, L.A. Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 2005, 65, 3958–3965. [Google Scholar] [CrossRef]
- Ma, S.; Yin, N.; Qi, X.; Pfister, S.L.; Zhang, M.J.; Ma, R.; Chen, G. Tyrosine dephosphorylation enhances the therapeutic target activity of epidermal growth factor receptor (EGFR) by disrupting its interaction with estrogen receptor (ER). Oncotarget 2015, 6, 13320–13333. [Google Scholar] [CrossRef]
- Dellambra, E. Oncogenic Ras: A double-edged sword for human epidermal stem and transient amplifying cells. Small GTPases 2016, 7, 147–155. [Google Scholar] [CrossRef]
- Drosten, M.; Lechuga, C.G.; Barbacid, M. Ras signaling is essential for skin development. Oncogene 2014, 33, 2857–2865. [Google Scholar] [CrossRef]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef]
- Jing, M.; Bohl, J.; Brimer, N.; Kinter, M.; Vande Pol, S.B. Degradation of tyrosine phosphatase PTPN3 (PTPH1) by association with oncogenic human papillomavirus E6 proteins. J. Virol. 2007, 81, 2231–2239. [Google Scholar] [CrossRef]
- Kranjec, C.; Banks, L. A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J. Virol. 2011, 85, 1757–1764. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Purdie, K.J.; Sexton, C.J.; Proby, C.M.; Glover, M.T.; Williams, A.T.; Stables, J.N.; Leigh, I.M. Malignant transformation of cutaneous lesions in renal allograft patients: A role for human papillomavirus. Cancer Res. 1993, 53, 5328–5333. [Google Scholar]
- Meyers, C.; Frattini, M.G.; Laimins, L.A. Tissue Culture Techniques for the Study of Human Papillomaviruses in Stratified Epithelia; Academic Press: Orlando, FL, USA, 1994; pp. 491–499. [Google Scholar]
- Leverrier, S.; Bergamaschi, D.; Ghali, L.; Ola, A.; Warnes, G.; Akgul, B.; Blight, K.; Garcia-Escudero, R.; Penna, A.; Eddaoudi, A.; et al. Role of HPV E6 proteins in preventing UVB-induced release of pro-apoptotic factors from the mitochondria. Apoptosis 2007, 12, 549–560. [Google Scholar] [CrossRef]
- Müller-Schiffmann, A.; Beckmann, J.; Steger, G. The E6 protein of the cutaneous human papillomavirus type 8 can stimulate the viral early and late promoters by distinct mechanisms. J. Virol. 2006, 80, 8718–8728. [Google Scholar] [CrossRef]
- Rehtanz, M.; Schmidt, H.-M.; Warthorst, U.; Steger, G. Direct interaction between nucleosome assembly protein-1 and the papillomavirus E2 proteins involved in activation of transcription. Mol. Cell. Biol. 2004, 24, 2153–2168. [Google Scholar] [CrossRef]
- Avitzour, M.; Diskin, R.; Raboy, B.; Askari, N.; Engelberg, D.; Livnah, O. Intrinsically active variants of all human p38 isoforms. FEBS J. 2007, 274, 963–975. [Google Scholar] [CrossRef]
- Hufbauer, M.; Lazic, D.; Akgul, B.; Brandsma, J.L.; Pfister, H.; Weissenborn, S.J. Enhanced human papillomavirus type 8 oncogene expression levels are crucial for skin tumorigenesis in transgenic mice. Virology 2010, 403, 128–136. [Google Scholar] [CrossRef]
- Zhang, S.-H.; Kobayashi, R.; Graves, P.R.; Piwnica-Worms, H.; Tonks, N.K. Serine phosphorylation-dependent association of the band 4.1-related protein-tyrosine phosphatase PTPH1 with 14-3-3ß protein. J. Biol. Chem. 1997, 272, 27281–27287. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Simmonds, M.; Storey, A. Identification of the regions of the HPV 5 E6 protein involved in Bak degradation and inhibition of apoptosis. Int. J. Cancer 2008, 123, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Galloway, D.A. β human papillomavirus E6 expression inhibits stabilization of p53 and increases tolerance of genomic instability. J. Virol. 2014, 88, 6112–6127. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Pohl, N.M.; Loesch, M.; Hou, S.; Li, R.; Qin, J.Z.; Cuenda, A.; Chen, G. p38α antagonizes p38gamma activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response. J. Biol. Chem. 2007, 282, 31398–31408. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Qi, X.; Mercola, D.; Han, J.; Chen, G. Essential role of p38γ in K-Ras transformation independent of phosphorylation. J. Biol. Chem. 2005, 280, 23910–23917. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.W.; Zhi, H.Y.; Pohl, N.; Loesch, M.; Qi, X.M.; Li, R.S.; Basir, Z.; Chen, G. PTPH1 dephosphorylates and cooperates with p38γ MAPK to increase ras oncogenesis through PDZ-mediated interaction. Cancer Res. 2010, 70, 2901–2910. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Suresh, P.S.; Qi, X.; Lepp, A.; Mirza, S.P.; Chen, G. p38γ Mitogen-activated protein kinase signals through phosphorylating its phosphatase PTPH1 in regulating ras protein oncogenesis and stress response. J. Biol. Chem. 2012, 287, 27895–27905. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Bos, J.L. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene 1997, 14, 623–625. [Google Scholar] [CrossRef]
- Confalonieri, S.; Salcini, A.E.; Puri, C.; Tacchetti, C.; Di Fiore, P.P. Tyrosine phosphorylation of Eps15 is required for ligand-regulated, but not constitutive, endocytosis. J. Cell Biol. 2000, 150, 905–912. [Google Scholar] [CrossRef]
- Haugen, L.H.; Skjeldal, F.M.; Bergeland, T.; Bakke, O. Endosomal binding kinetics of Eps15 and Hrs specifically regulate the degradation of RTKs. Sci. Rep. 2017, 7, 17962. [Google Scholar] [CrossRef]
- Howie, H.L.; Koop, J.I.; Weese, J.; Robinson, K.; Wipf, G.; Kim, L.; Galloway, D.A. β-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011, 7, e1002211. [Google Scholar] [CrossRef]
- Morandell, S.; Stasyk, T.; Skvortsov, S.; Ascher, S.; Huber, L.A. Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics 2008, 8, 4383–4401. [Google Scholar] [CrossRef]
- Martens, M.C.; Seebode, C.; Lehmann, J.; Emmert, S. Photocarcinogenesis and Skin Cancer Prevention Strategies: An Update. Anticancer Res. 2018, 38, 1153–1158. [Google Scholar] [CrossRef]
- Holderfield, M.; Lorenzana, E.; Weisburd, B.; Lomovasky, L.; Boussemart, L.; Lacroix, L.; Tomasic, G.; Favre, M.; Vagner, S.; Robert, C.; et al. Vemurafenib cooperates with HPV to promote initiation of cutaneous tumors. Cancer Res. 2014, 74, 2238–2245. [Google Scholar] [CrossRef]
- Hasche, D.; Vinzon, S.E.; Rosl, F. Cutaneous Papillomaviruses and Non-melanoma Skin Cancer: Causal Agents or Innocent Bystanders? Front. Microbiol. 2018, 9, 874. [Google Scholar] [CrossRef]
- Maurelli, R.; Tinaburri, L.; Gangi, F.; Bondanza, S.; Severi, A.L.; Scarponi, C.; Albanesi, C.; Mesiti, G.; Guerra, L.; Capogrossi, M.C.; et al. The role of oncogenic Ras in human skin tumorigenesis depends on the clonogenic potential of the founding keratinocytes. J. Cell Sci. 2016, 129, 1003–1017. [Google Scholar] [CrossRef]
- White, A.C.; Tran, K.; Khuu, J.; Dang, C.; Cui, Y.; Binder, S.W.; Lowry, W.E. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 7425–7430. [Google Scholar] [CrossRef]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar] [CrossRef]
- Lanfredini, S.; Olivero, C.; Borgogna, C.; Calati, F.; Powell, K.; Davies, K.J.; De Andrea, M.; Harries, S.; Tang, H.K.C.; Pfister, H.; et al. HPV8 Field Cancerization in a Transgenic Mouse Model Is due to Lrig1+ Keratinocyte Stem Cell Expansion. J. Investig. Dermatol. 2017, 137, 2208–2216. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taute, S.; Böhnke, P.; Sprissler, J.; Buchholz, S.; Hufbauer, M.; Akgül, B.; Steger, G. The Protein Tyrosine Phosphatase H1 PTPH1 Supports Proliferation of Keratinocytes and is a Target of the Human Papillomavirus Type 8 E6 Oncogene. Cells 2019, 8, 244. https://doi.org/10.3390/cells8030244
Taute S, Böhnke P, Sprissler J, Buchholz S, Hufbauer M, Akgül B, Steger G. The Protein Tyrosine Phosphatase H1 PTPH1 Supports Proliferation of Keratinocytes and is a Target of the Human Papillomavirus Type 8 E6 Oncogene. Cells. 2019; 8(3):244. https://doi.org/10.3390/cells8030244
Chicago/Turabian StyleTaute, Stefanie, Philipp Böhnke, Jasmin Sprissler, Stephanie Buchholz, Martin Hufbauer, Baki Akgül, and Gertrud Steger. 2019. "The Protein Tyrosine Phosphatase H1 PTPH1 Supports Proliferation of Keratinocytes and is a Target of the Human Papillomavirus Type 8 E6 Oncogene" Cells 8, no. 3: 244. https://doi.org/10.3390/cells8030244
APA StyleTaute, S., Böhnke, P., Sprissler, J., Buchholz, S., Hufbauer, M., Akgül, B., & Steger, G. (2019). The Protein Tyrosine Phosphatase H1 PTPH1 Supports Proliferation of Keratinocytes and is a Target of the Human Papillomavirus Type 8 E6 Oncogene. Cells, 8(3), 244. https://doi.org/10.3390/cells8030244