Cellular Proteostasis During Influenza A Virus Infection—Friend or Foe?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
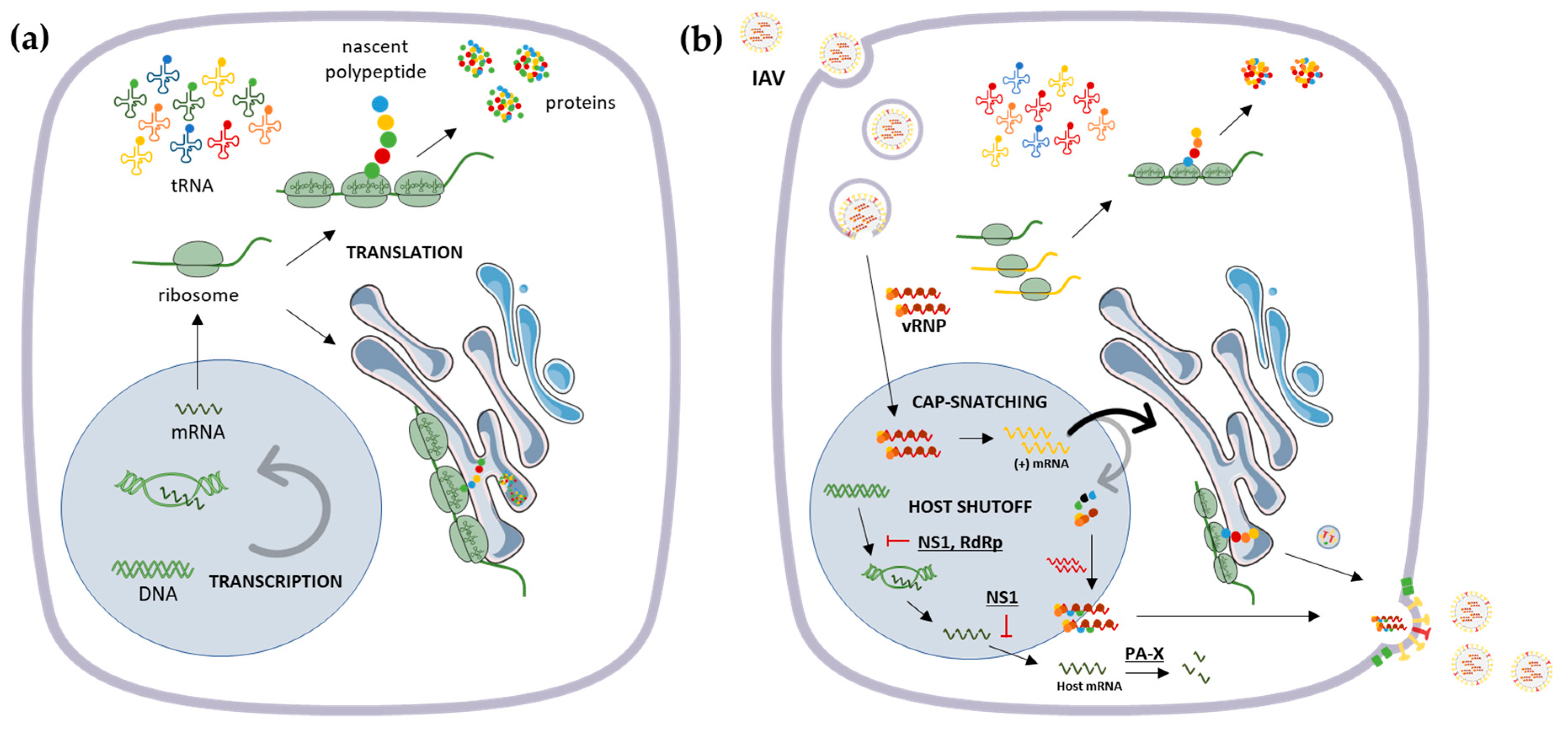
2. Influenza Virus Genome and Host Translational Machinery
3. Protein Quality Control Mechanisms and Their Interplay with Influenza A Virus
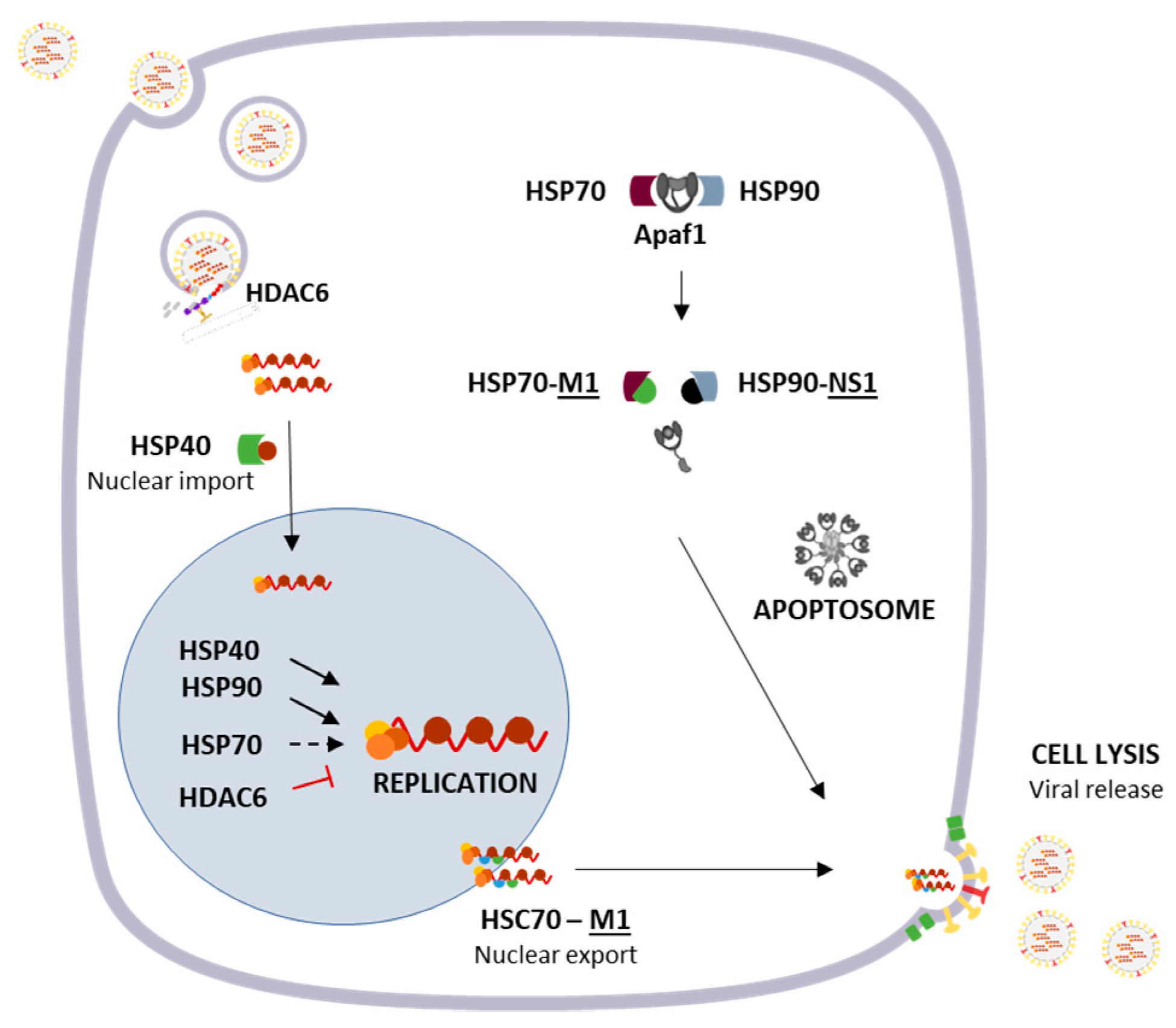
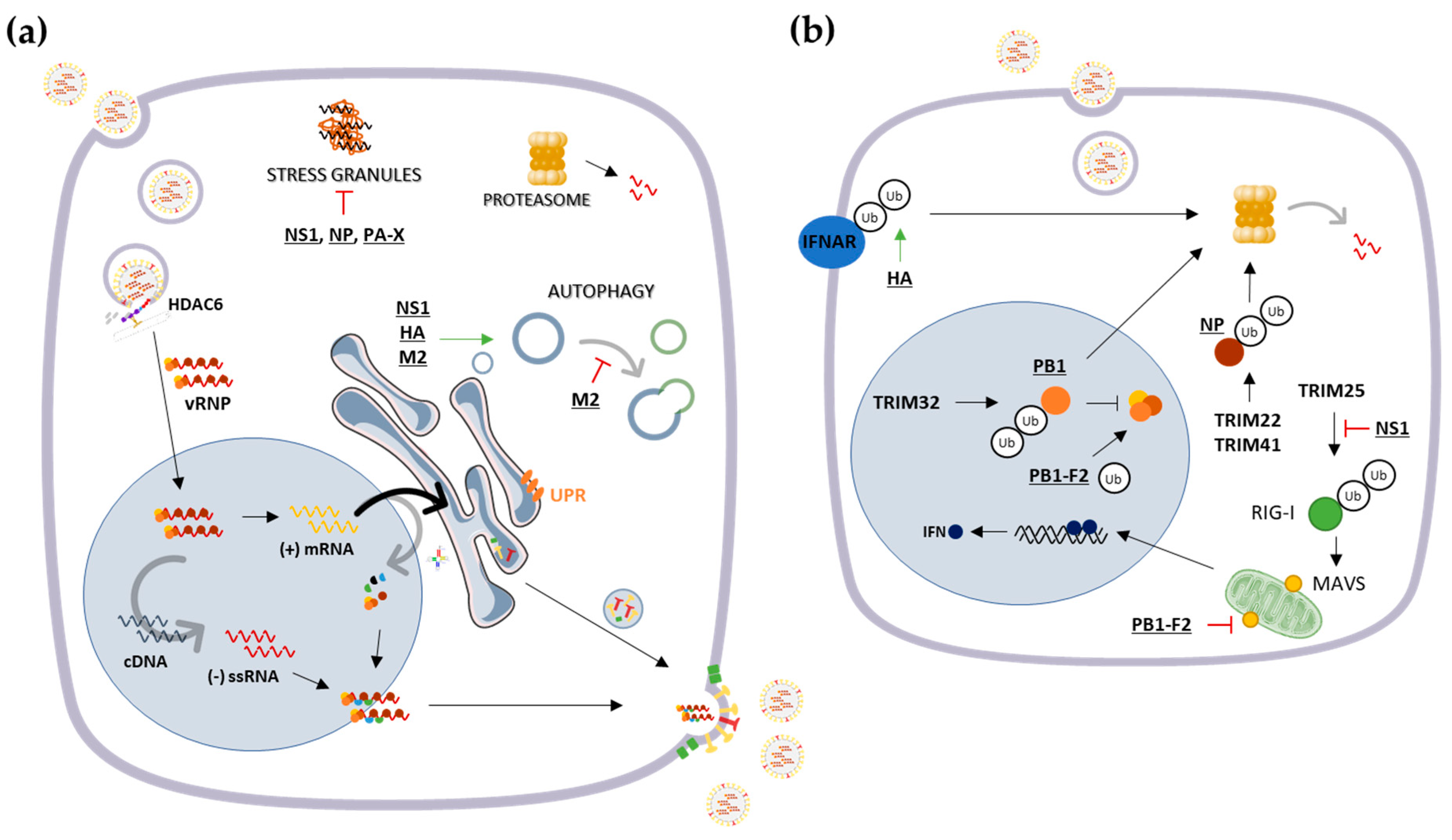
3.1. Cytosolic Responses: Protein Refolding, Degradation and Sequestration
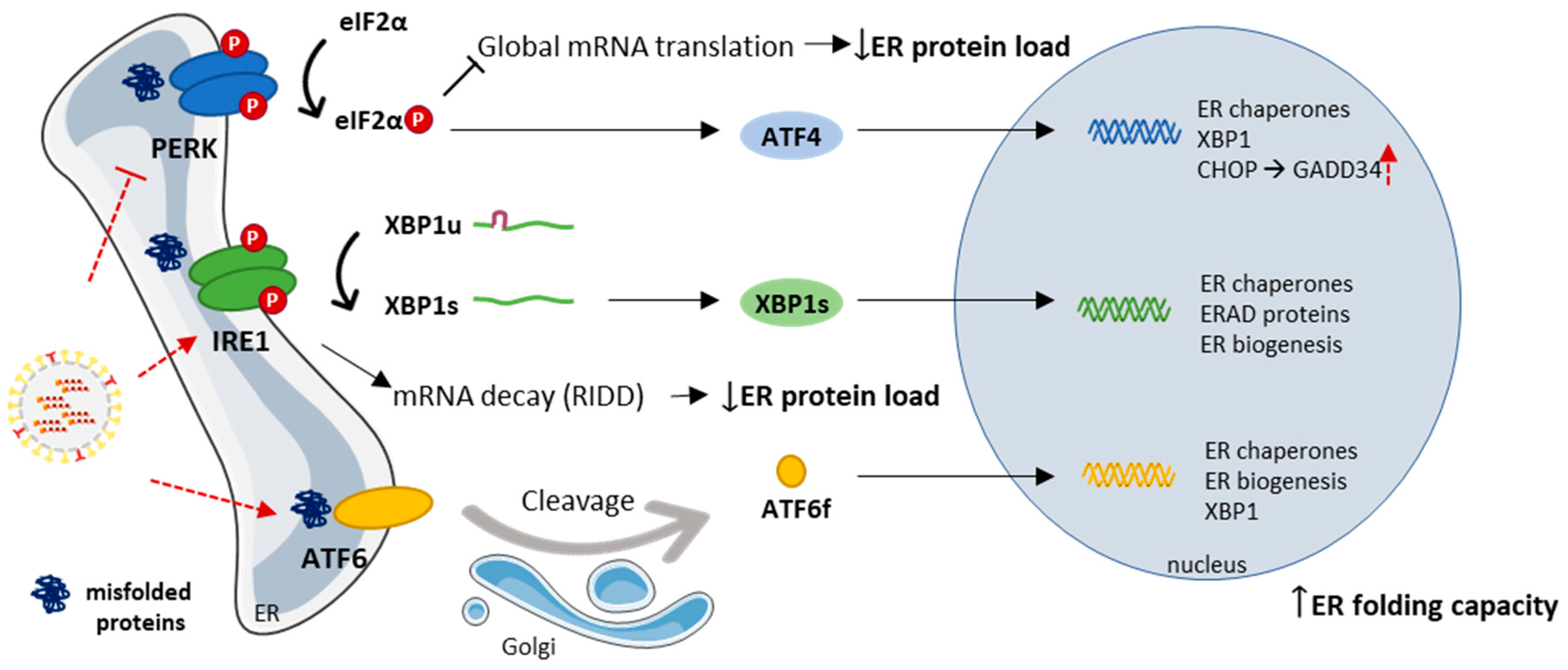
3.2. The Endoplasmic Reticulum and the Unfolded Protein Response
4. Concluding Remarks
Funding
Conflicts of Interest
References
- Ciryam, P.; Kundra, R.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol. Sci. 2015, 36, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.H.; Hartl, U.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Dillin, A. The UPR ER: Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular Strategies of Protein Quality Control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the Aging Proteome in Health and Disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 33–38. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Jean Beltran, P.M.; Cook, K.C.; Cristea, I.M. Exploring and Exploiting Proteome Organization during Viral Infection. J. Virol. 2017, 91, e00268-17. [Google Scholar] [CrossRef]
- Heaton, N.S.; Moshkina, N.; Fenouil, R.; Gardner, T.J.; Aguirre, S.; Shah, P.S.; Zhao, N.; Manganaro, L.; Hultquist, J.; Noel, J.; et al. Limiting influenza virus, HIV and dengue virus infection by targeting viral proteostasis. Immunity 2016, 44, 46–58. [Google Scholar] [CrossRef]
- Ravindran, M.S. Molecular chaperones: From proteostasis to pathogenesis. FEBS J. 2018, 285, 3353–3361. [Google Scholar] [CrossRef]
- Wileman, T. Aggresomes and Pericentriolar Sites of Virus Assembly: Cellular Defense or Viral Design? Annu. Rev. Microbiol. 2007, 61, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.; Moffat, K.; Brooks, E.; Wileman, T. A Guide to Viral Inclusions, Membrane Rearrangements, Factories, and Viroplasm Produced During Virus Replication. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2007; Volume 70, pp. 101–182. [Google Scholar]
- Schmid, M.; Speiseder, T.; Dobner, T.; Gonzalez, R.A. DNA Virus Replication Compartments. J. Virol. 2014, 88, 1404–1420. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Wileman, T. Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr. Opin. Virol. 2011, 1, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Moshe, A.; Gorovits, R. Virus-Induced Aggregates in Infected Cells. Viruses 2012, 4, 2218–2232. [Google Scholar] [CrossRef]
- Tekin, S.; Keske, S.; Alan, S.; Batirel, A.; Karakoc, C.; Tasdelen-Fisgin, N.; Simsek-Yavuz, S.; Işler, B.; Aydin, M.; Kapmaz, M.; et al. Predictors of Fatality in Influenza A Virus Subtypes Infection among inpatients in 2015-2016 season. Int. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Trucchi, C.; Paganino, C.; Orsi, A.; De Florentiis, D.; Ansaldi, F. Influenza vaccination in the elderly: Why are the overall benefits still hotly debated? J. Prev. Med. Hyg. 2015, 56, E37–E43. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.L. Evolution of influenza a virus by mutation and re-assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef]
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159. [Google Scholar]
- Levene, R.E.; Gaglia, M.M. Host Shutoff in Influenza A Virus: Many Means to an End. Viruses 2018, 10, 475. [Google Scholar] [CrossRef]
- Rivas, H.; Schmaling, S.; Gaglia, M.; Rivas, H.G.; Schmaling, S.K.; Gaglia, M.M. Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses 2016, 8, 102. [Google Scholar] [CrossRef]
- Cao, S.; Dhungel, P.; Yang, Z. Going against the Tide: Selective Cellular Protein Synthesis during Virally Induced Host Shutoff. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3’-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Bercovich-Kinori, A.; Tai, J.; Gelbart, I.A.; Shitrit, A.; Ben-Moshe, S.; Drori, Y.; Itzkovitz, S.; Mandelboim, M.; Stern-Ginossar, N. A systematic view on influenza induced host shutoff. Elife 2016, 5, 1–20. [Google Scholar] [CrossRef]
- Wang, W.; Krug, R.M. The RNA-Binding and Effector Domains of the Viral NS1 Protein Are Conserved to Different Extents among Influenza A and B Viruses. Virology 1996, 223, 41–50. [Google Scholar] [CrossRef]
- de la Luna, S.; Fortes, P.; Beloso, A.; Ortín, J. Influenza virus NS1 protein enhances the rate of translation initiation of viral mRNAs. J. Virol. 1995, 69, 2427–2433. [Google Scholar]
- Aragón, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortín, J.; Nieto, A. Eukaryotic Translation Initiation Factor 4GI Is a Cellular Target for NS1 Protein, a Translational Activator of Influenza Virus. Mol. Cell. Biol. 2000, 20, 6259–6268. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.; Basler, C.F.; Parisien, J.-P.; Horvath, C.M.; Bourmakina, S.; Zheng, H.; Muster, T.; Palese, P.; García-Sastre, A. Effects of influenza A virus NS1 protein on protein expression: The NS1 protein enhances translation and is not required for shutoff of host protein synthesis. J. Virol. 2002, 76, 1206–1212. [Google Scholar] [CrossRef]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the Influenza Virus NS1 Protein to Double-Stranded RNA Inhibits the Activation of the Protein Kinase That Phosphorylates the eIF-2 Translation Initiation Factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef]
- Tan, S.-L.; Katze, M.G. Biochemical and Genetic Evidence for Complex Formation Between the Influenza A Virus NS1 Protein and the Interferon-induced PKR Protein Kinase. J. Interfaces Cytokine Res. 1998, 18, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Schierhorn, K.L.; Jolmes, F.; Bespalowa, J.; Saenger, S.; Peteranderl, C.; Dzieciolowski, J.; Mielke, M.; Budt, M.; Pleschka, S.; Herrmann, A.; et al. Influenza A Virus Virulence Depends on Two Amino Acids in the N-Terminal Domain of Its NS1 Protein To Facilitate Inhibition of the RNA-Dependent Protein Kinase PKR. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Zürcher, T.; Marión, R.M.; Ortín, J. Protein Synthesis Shut-Off Induced by Influenza Virus Infection Is Independent of PKR Activity. J. Virol. 2000, 74, 8781–8784. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2’-5’ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.-M.; Gale, M.; Núñez, G.; Silverman, R.H. RNase L Activates the NLRP3 Inflammasome during Viral Infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [CrossRef]
- Koppstein, D.; Ashour, J.; Bartel, D.P. Sequencing the cap-snatching repertoire of H1N1 influenza provides insight into the mechanism of viral transcription initiation. Nucleic Acids Res. 2015, 43, 5052–5064. [Google Scholar] [CrossRef]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza A Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Fan, J.; He, R.; Luo, M.; Zheng, X. The Crystal Structure of the PB2 Cap-binding Domain of Influenza B Virus Reveals a Novel Cap Recognition Mechanism. J. Biol. Chem. 2015, 290, 9141–9149. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef]
- Hayashi, T.; Chaimayo, C.; McGuinness, J.; Takimoto, T. Critical Role of the PA-X C-Terminal Domain of Influenza A Virus in Its Subcellular Localization and Shutoff Activity. J. Virol. 2016, 90, 7131–7141. [Google Scholar] [CrossRef] [PubMed]
- Mohr, I. Closing in on the causes of host shutoff. Elife 2016, 5, e20755. [Google Scholar] [CrossRef]
- Vester, D.; Rapp, E.; Gade, D.; Genzel, Y.; Reichl, U. Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics 2009, 9, 3316–3327. [Google Scholar] [CrossRef]
- Kroeker, A.L.; Ezzati, P.; Coombs, K.M.; Halayko, A.J. Influenza A infection of primary human airway epithelial cells up-regulates proteins related to purine metabolism and ubiquitin-related signaling. J. Proteome Res. 2013, 12, 3139–3151. [Google Scholar] [CrossRef] [PubMed]
- Söderholm, S.; Kainov, D.E.; Öhman, T.; Denisova, O.V.; Schepens, B.; Kulesskiy, E.; Imanishi, S.Y.; Corthals, G.; Hintsanen, P.; Aittokallio, T.; et al. Phosphoproteomics to Characterize Host Response During Influenza A Virus Infection of Human Macrophages. Mol. Cell. Proteomics 2016, 15, 3203–3219. [Google Scholar] [CrossRef] [PubMed]
- Lietzén, N.; Öhman, T.; Rintahaka, J.; Julkunen, I.; Aittokallio, T.; Matikainen, S.; Nyman, T.A. Quantitative Subcellular Proteome and Secretome Profiling of Influenza A Virus-Infected Human Primary Macrophages. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Albers, S.; Czech, A. Exploiting tRNAs to Boost Virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef]
- Pavon-Eternod, M.; David, A.; Dittmar, K.; Berglund, P.; Pan, T.; Bennink, J.R.; Yewdell, J.W. Vaccinia and influenza A viruses select rather than adjust tRNAs to optimize translation. Nucleic Acids Res. 2013, 41, 1914–1921. [Google Scholar] [CrossRef]
- Smith, B.L.; Chen, G.; Wilke, C.O.; Krug, R.M. Avian Influenza Virus PB1 Gene in H3N2 Viruses Evolved in Humans To Reduce Interferon Inhibition by Skewing Codon Usage toward Interferon-Altered tRNA Pools. MBio 2018, 9, e01222-18. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.-Y.; Lu, J.-S.; Xia, B.-H.; Yang, Z.-X.; Zhu, X.-D.; Zhou, X.-W.; Huang, P.-T. The highly pathogenic H5N1 influenza A virus down-regulated several cellular MicroRNAs which target viral genome. J. Cell. Mol. Med. 2017, 21, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, J.; Song, L.; Wu, J.; Huang, W. Influenza A virus-induced downregulation of miR-26a contributes to reduced IFNα/β production. Virol. Sin. 2017, 32, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, J.; Wei, S.; Li, C.; Zhou, K.; Hu, J.; Ye, X.; Yan, J.; Liu, W.; Gao, G.F.; et al. Endogenous Cellular MicroRNAs Mediate Antiviral Defense against Influenza A Virus. Mol. Ther. Nucleic Acids 2018, 10, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, X.; Wu, Z.; Huang, K.; Sun, X.; Chen, H.; Jin, M. The Downregulation of MicroRNA hsa-miR-340-5p in IAV-Infected A549 Cells Suppresses Viral Replication by Targeting RIG-I and OAS2. Mol. Ther. Nucleic Acids 2019, 14, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Dianat-Moghadam, H.; Sofiani, V.H.; Karimzadeh, M.; Zargar, M.; Moghoofei, M.; Biglari, H.; Ghorbani, S.; Nahand, J.S.; Mirzaei, H. MiRNA-based strategy for modulation of influenza A virus infection. Epigenomics 2018, 10, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Kuciak, M.; Gabus, C.; Ivanyi-Nagy, R.; Semrad, K.; Storchak, R.; Chaloin, O.; Muller, S.; Mély, Y.; Darlix, J.L. The HIV-1 transcriptional activator Tat has potent nucleic acid chaperoning activities in vitro. Nucleic Acids Res. 2008, 36, 3389–3400. [Google Scholar] [CrossRef]
- Batra, J.; Tripathi, S.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; Lal, S.K. Human Heat shock protein 40 (Hsp40/DnaJB1) promotes influenza A virus replication by assisting nuclear import of viral ribonucleoproteins. Sci. Rep. 2016, 6, 19063. [Google Scholar] [CrossRef]
- Cao, M.; Wei, C.; Zhao, L.; Wang, J.; Jia, Q.; Wang, X.; Jin, Q.; Deng, T. DnaJA1/Hsp40 Is Co-Opted by Influenza A Virus To Enhance Its Viral RNA Polymerase Activity. J. Virol. 2014, 88, 14078–14089. [Google Scholar] [CrossRef]
- Li, G.; Zhang, J.; Tong, X.; Liu, W.; Ye, X. Heat shock protein 70 inhibits the activity of influenza a virus ribonucleoprotein and blocks the replication of virus In Vitro and In Vivo. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Manzoor, R.; Kuroda, K.; Yoshida, R.; Tsuda, Y.; Fujikura, D.; Miyamoto, H.; Kajihara, M.; Kida, H.; Takada, A. Heat shock protein 70 modulates influenza A virus polymerase activity. J. Biol. Chem. 2014, 289, 7599–7614. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Iguchi, A.; Gomyou, R.; Ono, Y. Influenza Virus Inhibits Cleavage of the HSP70 Pre-mRNAs at the Polyadenylation Site. Virology 1999, 254, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Momose, F.; Kawaguchi, A.; Nagata, K. Involvement of Hsp90 in Assembly and Nuclear Import of Influenza Virus RNA Polymerase Subunits. J. Virol. 2007, 81, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Momose, F.; Naito, T.; Yano, K.; Sugimoto, S.; Morikawa, Y.; Nagata, K. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J. Biol. Chem. 2002, 277, 45306–45314. [Google Scholar] [CrossRef] [PubMed]
- Chase, G.; Deng, T.; Fodor, E.; Leung, B.W.; Mayer, D.; Schwemmle, M.; Brownlee, G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 2008, 377, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Liu, D.; Mi, S.; Zhang, J.; Ye, Q.; Wang, M.; Gao, G.F.; Yan, J. Interaction of Hsp40 with influenza virus M2 protein: Implications for PKR signaling pathway. Protein Cell 2010, 1, 944–955. [Google Scholar] [CrossRef]
- Sharma, K.; Tripathi, S.; Ranjan, P.; Kumar, P.; Garten, R.; Deyde, V.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; et al. Influenza a virus nucleoprotein exploits Hsp40 to inhibit PKR activation. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The Cellular Protein P58IPK Regulates Influenza Virus mRNA Translation and Replication through a PKR-Mediated Mechanism. J. Virol. 2007, 81, 2221–2230. [Google Scholar] [CrossRef]
- Watanabe, K.; Fuse, T.; Asano, I.; Tsukahara, F.; Maru, Y.; Nagata, K.; Kitazato, K.; Kobayashi, N. Identification of Hsc70 as an influenza virus matrix protein (M1) binding factor involved in the virus life cycle. FEBS Lett. 2006, 580, 5785–5790. [Google Scholar] [CrossRef]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix (M1) protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Zhou, X.; Yang, Z.; Liu, X.; Cao, Z.; Song, H.; He, Y.; Huang, P. The NS1 protein of influenza a virus interacts with heat shock protein Hsp90 in human alveolar basal epithelial cells: Implication for virus-induced apoptosis. Virol. J. 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Luo, H. Interplay between the virus and the ubiquitin–proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, A.; Yamauchi, Y. Ubiquitin in influenza virus entry and innate immunity. Viruses 2016, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, I.; de Vries, E.; Tscherne, D.M.; Garcia-Sastre, A.; Rottier, P.J.M.; de Haan, C.A.M. Inhibition of the Ubiquitin-Proteasome System Affects Influenza A Virus Infection at a Postfusion Step. J. Virol. 2010, 84, 9625–9631. [Google Scholar] [CrossRef] [PubMed]
- Khor, R.; McElroy, L.J.; Whittaker, G.R. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 2003, 4, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 Inhibits Influenza A Virus Infection by Targeting the Viral Nucleoprotein for Degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef]
- Patil, G.; Zhao, M.; Song, K.; Hao, W.; Bouchereau, D.; Wang, L.; Li, S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Influenza A Virus Infection. J. Virol. 2018, 92, e00905-18. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; Van Der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis E Sousa, C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-Specific Inhibition of RIG-I Ubiquitination and IFN Induction by the Influenza A Virus NS1 Protein. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aparicio, M.T.; Ayllón, J.; Leo-Macias, A.; Wolff, T.; García-Sastre, A. Subcellular Localizations of RIG-I, TRIM25, and MAVS Complexes. J. Virol. 2017, 91, e01155-16. [Google Scholar] [CrossRef]
- Xia, C.; Vijayan, M.; Pritzl, C.J.; Fuchs, S.Y.; McDermott, A.B.; Hahm, B. Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1. J. Virol. 2015, 90, 2403–2417. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-C.; Yu, W.-Y.; Huang, S.-H.; Lai, M.M.C. Ubiquitination of the Cytoplasmic Domain of Influenza A Virus M2 Protein is Crucial for Production of Infectious Virus Particles. J. Virol. 2017, 92, e01972-17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Jeng, K.-S.; Lai, M.M.C. CNOT4-Mediated Ubiquitination of Influenza A Virus Nucleoprotein Promotes Viral RNA Replication. MBio 2017, 8, e00597-17. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.L.; Wu, C.Y.; Su, W.C.; Jeng, K.S.; Lai, M.M.C. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 2010, 29, 3879–3890. [Google Scholar] [CrossRef] [PubMed]
- Košík, I.; Práznovská, M.; Košíková, M.; Bobišová, Z.; Hollý, J.; Varečková, E.; Kostolanský, F.; Russ, G. The ubiquitination of the influenza a virus PB1-F2 protein is crucial for its biological function. PLoS ONE 2015, 10, e0118477. [Google Scholar] [CrossRef]
- Liu, G.; Zhong, M.; Guo, C.; Komatsu, M.; Xu, J.; Wang, Y.; Kitazato, K. Autophagy is involved in regulating influenza A virus RNA and protein synthesis associated with both modulation of Hsp90 induction and mTOR/p70S6K signaling pathway. Int. J. Biochem. Cell Biol. 2016, 72, 100–108. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef]
- Yeganeh, B.; Ghavami, S.; Rahim, M.N.; Klonisch, T.; Halayko, A.J.; Coombs, K.M. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Feizi, N.; Mehrbod, P.; Romani, B.; Soleimanjahi, H.; Bamdad, T.; Feizi, A.; Jazaeri, E.O.; Targhi, H.S.; Saleh, M.; Jamali, A.; et al. Autophagy induction regulates influenza virus replication in a time-dependent manner. J. Med. Microbiol. 2017, 66, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Zhao, J.; Ren, C.; Li, P.; Chen, H.; Jin, M.; Zhou, H. Autophagy Promotes Replication of Influenza A Virus In Vitro. J. Virol. 2019, 93, e01984-18. [Google Scholar] [CrossRef] [PubMed]
- Perot, B.P.; Boussier, J.; Yatim, N.; Rossman, J.S.; Ingersoll, M.A.; Albert, M.L. Autophagy diminishes the early interferon-β response to influenza A virus resulting in differential expression of interferon-stimulated genes. Cell Death Dis. 2018, 9, 539. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Klenk, H.D. Influenza A Virus Proteins NS1 and Hemagglutinin Along with M2 Are Involved in Stimulation of Autophagy in Infected Cells. J. Virol. 2013, 87, 13107–13114. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Hanzén, S.; Nyström, T. Restricted access: Spatial sequestration of damaged proteins during stress and aging. EMBO Rep. 2017, 18, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ko, S.; Xu, Y.; Fattah, E.A.; Xiang, Q.; Jagannath, C.; Ishii, T.; Komatsu, M.; Eissa, N.T. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 19687–19698. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Osterloh, P.; Hilf, N.; Rechtsteiner, G.; Hohfeld, J.; Rammensee, H.-G.; Schild, H. Dendritic Cell Aggresome-Like-Induced Structure Formation and Delayed Antigen Presentation Coincide in Influenza Virus-Infected Dendritic Cells. J. Immunol. 2005, 175, 891–898. [Google Scholar] [CrossRef]
- Vidic, J.; Richard, C.A.; Péchoux, C.; Da Costa, B.; Bertho, N.; Mazerat, S.; Delmas, B.; Chevalier, C. Amyloid assemblies of influenza a virus PB1-F2 protein damage membrane and induce cytotoxicity. J. Biol. Chem. 2016, 291, 739–751. [Google Scholar] [CrossRef]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.I.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon by Binding to MAVS and Decreasing Mitochondrial Membrane Potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E.; Santangelo, P.J. Human Respiratory Syncytial Virus Nucleoprotein and Inclusion Bodies Antagonize the Innate Immune Response Mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.J.; Wang, M.; Deng, M.; Shen, S.; Liu, W.; Cao, W.C.; Deng, F.; Wang, Y.Y.; Hu, Z.; Wang, H. Viral suppression of innate immunity via spatial isolation of TBK1/IKKε from mitochondrial antiviral platform. J. Mol. Cell Biol. 2014, 6, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qi, X.; Qu, B.; Zhang, Z.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Evasion of Antiviral Immunity through Sequestering of TBK1/IKK /IRF3 into Viral Inclusion Bodies. J. Virol. 2014, 88, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; Mclaurin, A.; Vance, J.M.; Ito, A.; Yao, T.; Kovacs, J.J.; Yao, T. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Banerjee, I.; Miyake, Y.; Philip Nobs, S.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, Y.; Chen, X.; Ruan, Z.; Ye, Y.; Chen, H.; Babiuk, L.A.; Jung, Y.-S.; Dai, J. HDAC6 Restricts Influenza A Virus by Deacetylation of the RNA Polymerase PA Subunit. J. Virol. 2019, 93, e01896-18. [Google Scholar] [CrossRef] [PubMed]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 2012, 26, 1629–1639. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; McCormick, C. Timing Is Everything: Coordinated Control of Host Shutoff by Influenza A Virus NS1 and PA-X Proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Emara, M.M.; Johnston, B.P.; Anderson, P.; Hatchette, T.F.; McCormick, C. Influenza A Virus Host Shutoff Disables Antiviral Stress-Induced Translation Arrest. PLoS Pathog. 2014, 10, e1004217. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kong, L.; Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 2015, 41, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Yamate, M.; Du, A.; Daidoji, T.; Okuno, Y.; Ikuta, K.; Nakaya, T. Maturation efficiency of viral glycoproteins in the ER impacts the production of influenza A virus. Virus Res. 2008, 136, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.; Kurowski, B.; Johnson, A.E.; Hebert, D.N. N-Linked Glycans Direct the Cotranslational Folding Pathway of Influenza Hemagglutinin. Mol. Cell 2003, 11, 79–90. [Google Scholar] [CrossRef]
- Diehl, J.A.; Fuchs, S.Y.; Koumenis, C. The Cell Biology of the Unfolded Protein Response. Gastroenterology 2011, 141, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Hrincius, E.R.; Liedmann, S.; Finkelstein, D.; Vogel, P.; Gansebom, S.; Samarasinghe, A.E.; You, D.; Cormier, S.A.; McCullers, J.A. Acute Lung Injury Results from Innate Sensing of Viruses by an ER Stress Pathway. Cell Rep. 2015, 11, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Frabutt, D.A.; Wang, B.; Riaz, S.; Schwartz, R.C.; Zheng, Y.-H. Innate sensing of influenza A virus hemagglutinin glycoproteins by the host endoplasmic reticulum (ER) stress pathway triggers a potent antiviral response via ER-associated protein degradation. J. Virol. 2018, 92, e01690-17. [Google Scholar] [CrossRef]
- Hassan, I.H.; Zhang, M.S.; Powers, L.S.; Shao, J.Q.; Baltrusaitis, J.; Rutkowski, D.T.; Legge, K.; Monick, M.M. Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 2012, 287, 4679–4689. [Google Scholar] [CrossRef]
- Roberson, E.C.; Tully, J.E.; Guala, A.S.; Reiss, J.N.; Godburn, K.E.; Pociask, D.A.; Alcorn, J.F.; Riches, D.W.H.; Dienz, O.; Janssen-Heininger, Y.M.W.; et al. Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-β release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 573–581. [Google Scholar] [CrossRef]
- Landeras-Bueno, S.; Fernández, Y.; Falcón, A.; Oliveros, J.C.; Ortín, J. Chemical Genomics Identifies the PERK-Mediated Unfolded Protein Stress Response as a Cellular Target for Influenza Virus Inhibition. MBio 2016, 7, e00085-16. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, M.; Ramos, B.; Soares, A.R.; Ribeiro, D. Cellular Proteostasis During Influenza A Virus Infection—Friend or Foe? Cells 2019, 8, 228. https://doi.org/10.3390/cells8030228
Marques M, Ramos B, Soares AR, Ribeiro D. Cellular Proteostasis During Influenza A Virus Infection—Friend or Foe? Cells. 2019; 8(3):228. https://doi.org/10.3390/cells8030228
Chicago/Turabian StyleMarques, Mariana, Bruno Ramos, Ana Raquel Soares, and Daniela Ribeiro. 2019. "Cellular Proteostasis During Influenza A Virus Infection—Friend or Foe?" Cells 8, no. 3: 228. https://doi.org/10.3390/cells8030228
APA StyleMarques, M., Ramos, B., Soares, A. R., & Ribeiro, D. (2019). Cellular Proteostasis During Influenza A Virus Infection—Friend or Foe? Cells, 8(3), 228. https://doi.org/10.3390/cells8030228