The Immunogenicity in Mice of HCV Core Delivered as DNA Is Modulated by Its Capacity to Induce Oxidative Stress and Oxidative Stress Response

 , , ,
, , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid DNA
2.2. Recombinant Proteins and Peptides
2.3. Eukaryotic Expression and Stability of HCV Core Protein Variants
2.4. Western Blot Analysis
2.5. Protein Stabilization Assays
2.6. Confocal Microscopy
2.7. Measurement of Production of the Reactive Oxygen Species
2.8. Reverse Transcription with Quantitative PCR (RT-qPCR)
2.9. Animals
2.10. Mouse Immunization Experiments
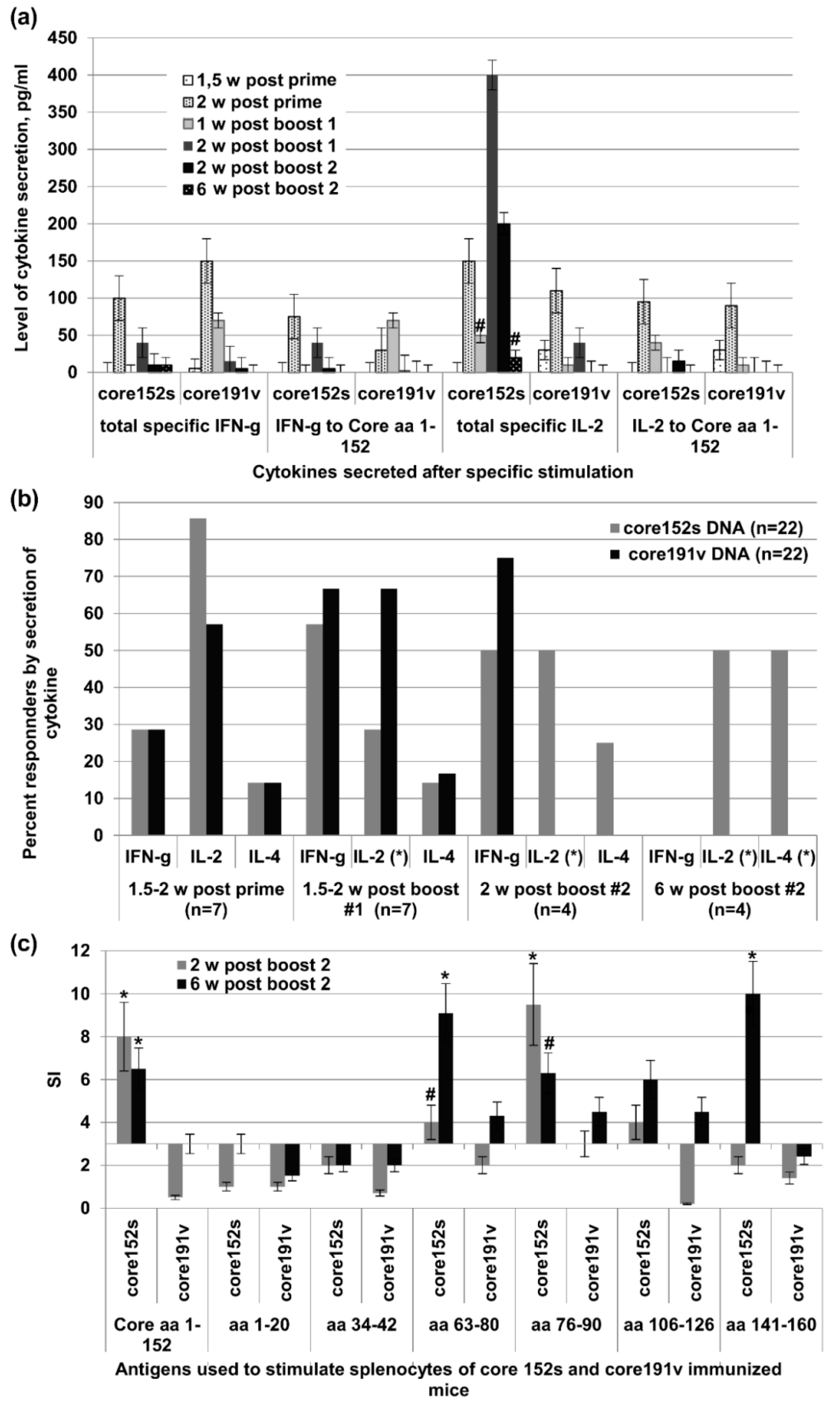
2.11. Preparation of Murine Splenocytes and Evaluation of Cytokine Secretion by Sandwich ELISA and IFN-γ/IL-2 Fluorospot Tests
2.12. Antibody ELISA
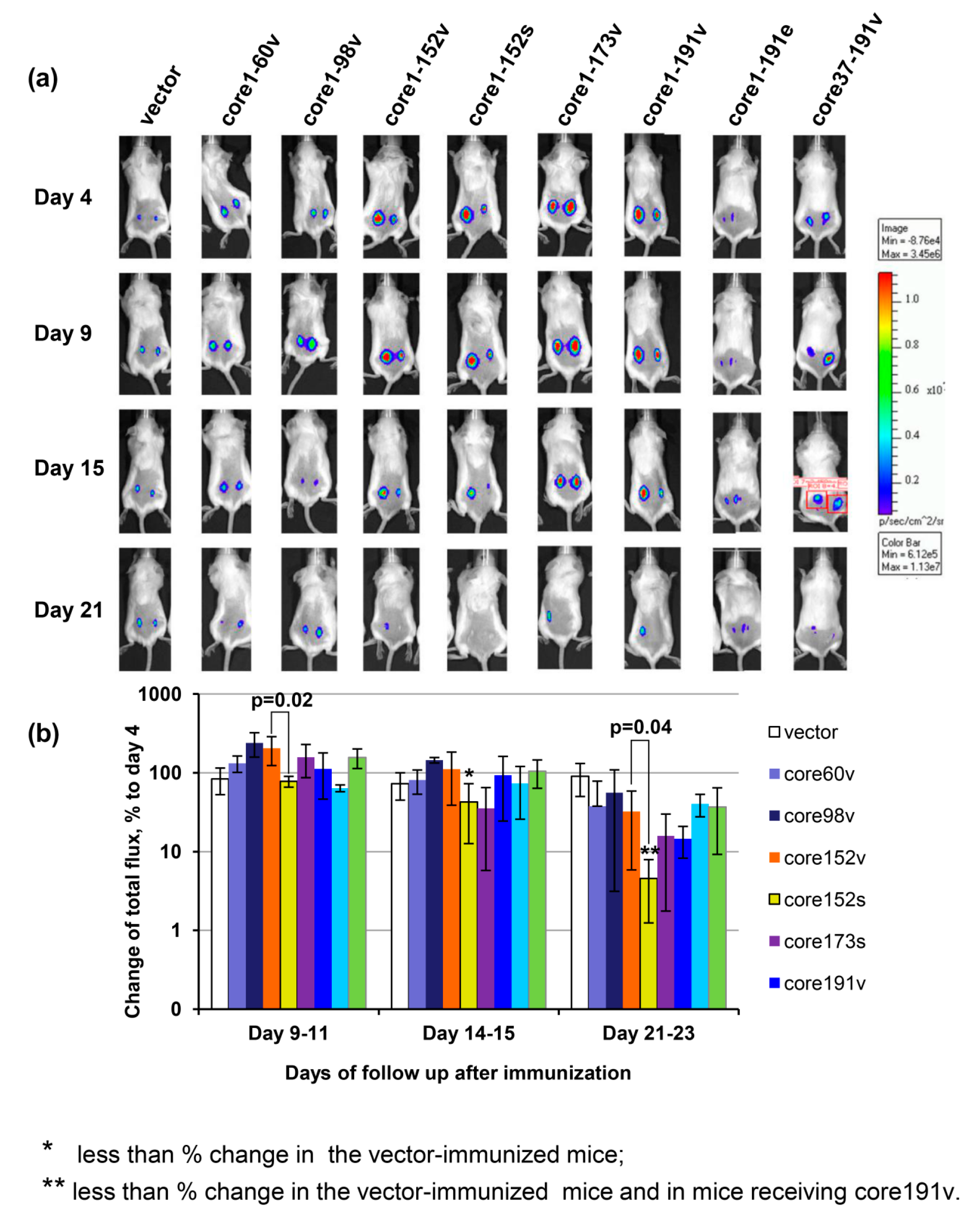
2.13. In Vivo Imaging of Luciferase Gene Expression
2.14. Statistical Analysis
3. Results
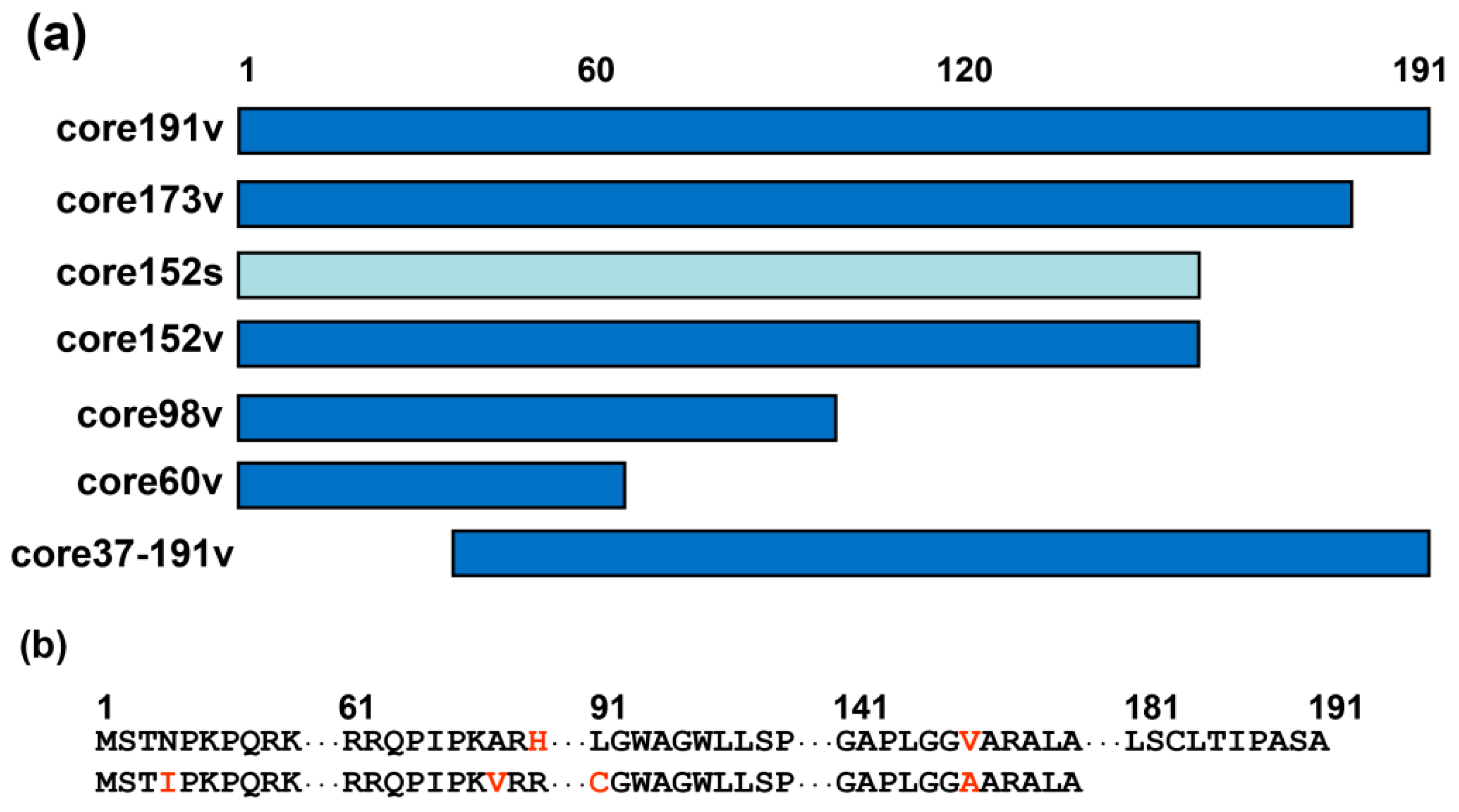
3.1. Design of DNA Immunogens Encoding the Full-Length and Truncated Variants of HCV Core
3.2. Eukaryotic Expression of HCV Core Variants
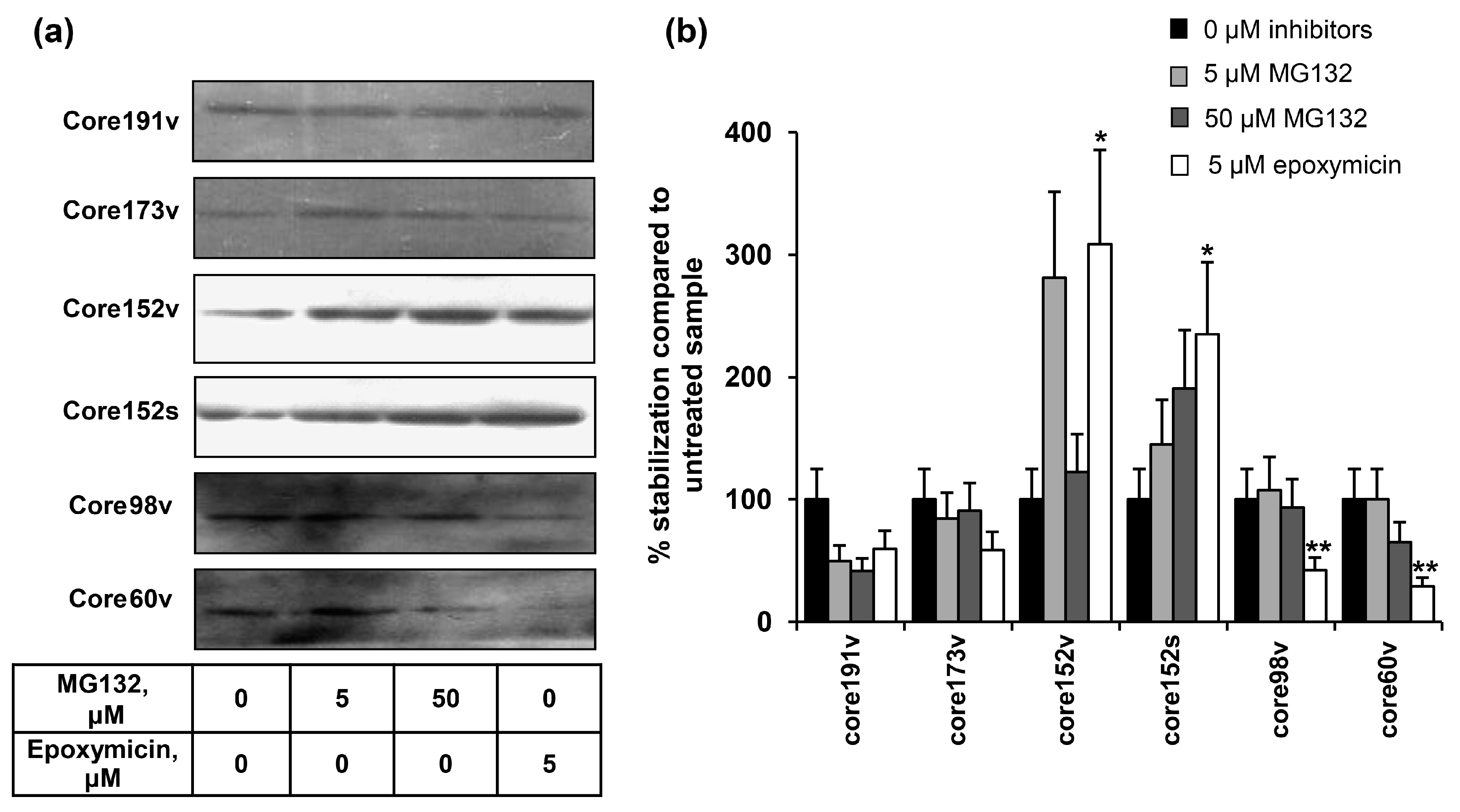
3.3. Proteasomal Degradation of HCV Core Variants in BHK-21 Cells
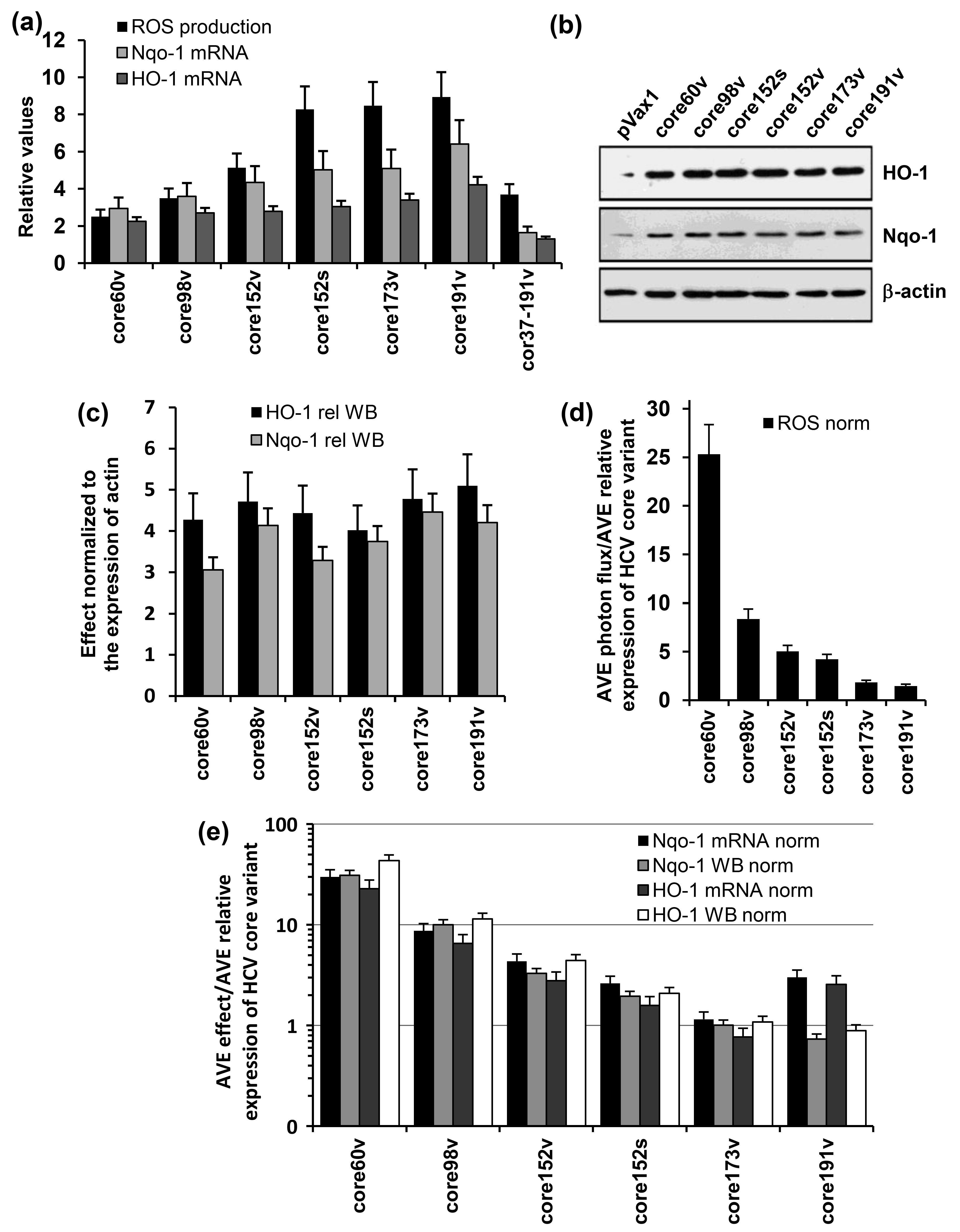
3.4. Both the Full-Length and the C-Terminally Truncated Versions of HCV Core Induce the Production of Reactive Oxygen Species (ROS) and an Oxidative Stress Response
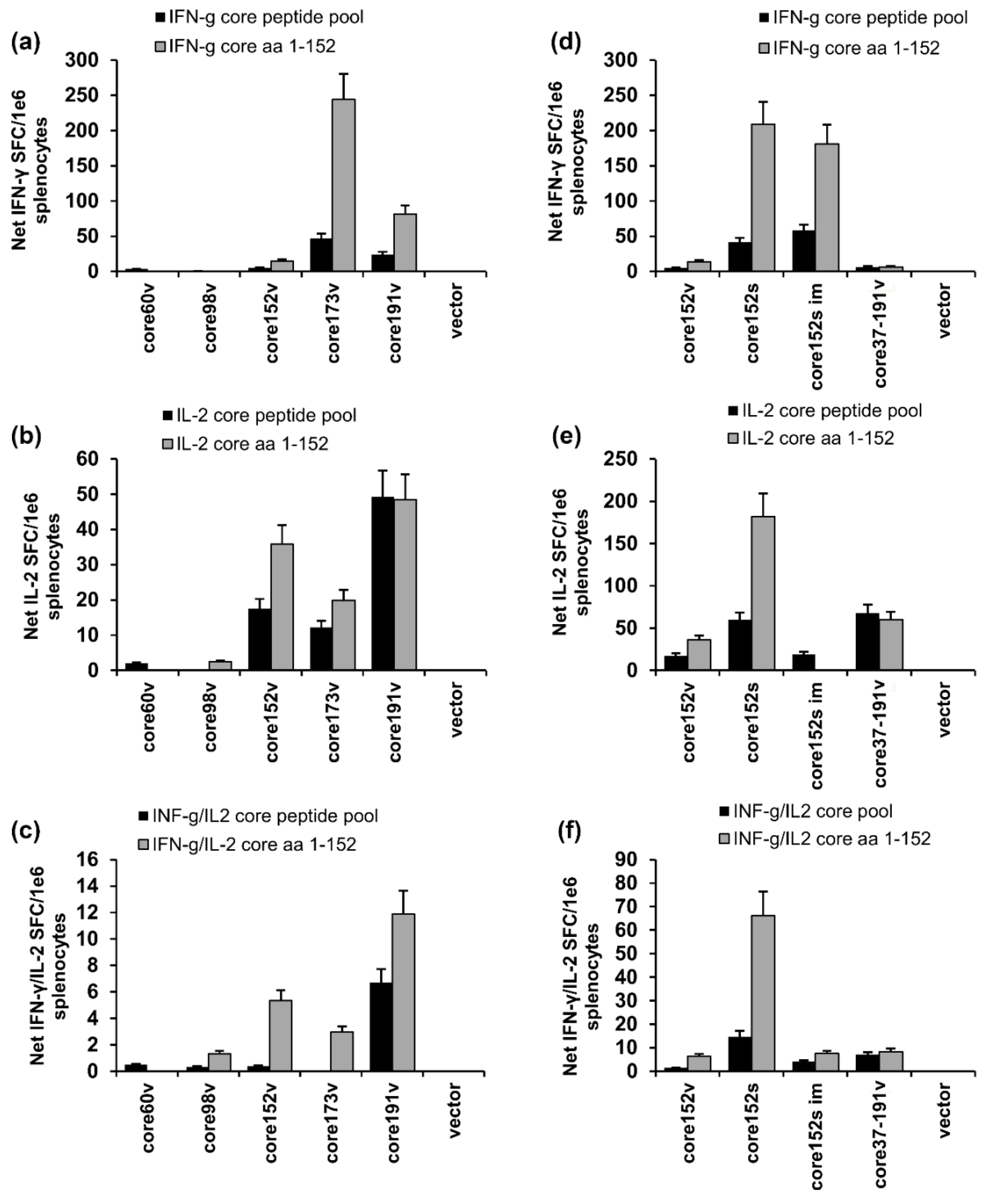
3.5. Immunogenic Performance of the Genes Encoding HCV Core Variants in BALB/c Mice
3.6. The Immunogenicity of HCV Core Gene Variants Does Not Depend on the Mouse Strain or Immunization Route
3.7. In Vivo Assessment of Effector Immune Response
3.8. Immunogenicity of HCV Core Gene Variants in Mice Is Associated with Their Ability to Induce Oxidative Stress and Oxidative Stress Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Christie, J.M.; Chapel, H.; Chapman, R.W.; Rosenberg, W.M. Immune selection and genetic sequence variation in core and envelope regions of hepatitis c virus. Hepatology 1999, 30, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Ina, Y.; Mizokami, M.; Ohba, K.; Gojobori, T. Reduction of synonymous substitutions in the core protein gene of hepatitis c virus. J. Mol. Evol. 1994, 38, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Rispeter, K.; Lu, M.; Behrens, S.E.; Fumiko, C.; Yoshida, T.; Roggendorf, M. Hepatitis c virus variability: Sequence analysis of an isolate after 10 years of chronic infection. Virus Genes 2000, 21, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wagoner, J.; Negash, A.; Austin, M.; McLauchlan, J.; Hahn, Y.S.; Rosen, H.R.; Polyak, S.J. Functional characterization of core genes from patients with acute hepatitis c virus infection. J. Infect. Dis. 2010, 201, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, E.; Sominskaya, I.; Skrastina, D.; Egorova, I.; Starodubova, E.; Kushners, E.; Mihailova, M.; Petrakova, N.; Bruvere, R.; Kozlovskaya, T.; et al. Enhancement of the expression of hcv core gene does not enhance core-specific immune response in DNA immunization: Advantages of the heterologous DNA prime, protein boost immunization regimen. Genet. Vaccines Ther. 2009, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Elmowalid, G.A.; Qiao, M.; Jeong, S.H.; Borg, B.B.; Baumert, T.F.; Sapp, R.K.; Hu, Z.; Murthy, K.; Liang, T.J. Immunization with hepatitis c virus-like particles results in control of hepatitis c virus infection in chimpanzees. Proc. Nat. Acad. Sci. USA 2007, 104, 8427–8432. [Google Scholar] [CrossRef] [PubMed]
- Rollier, C.S.; Paranhos-Baccala, G.; Verschoor, E.J.; Verstrepen, B.E.; Drexhage, J.A.; Fagrouch, Z.; Berland, J.L.; Komurian-Pradel, F.; Duverger, B.; Himoudi, N.; et al. Vaccine-induced early control of hepatitis c virus infection in chimpanzees fails to impact on hepatic pd-1 and chronicity. Hepatology 2007, 45, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.W.; Hu, Y.W.; Tricoche, N.; Pfahler, W.; Shata, M.T.; Dreux, M.; Cosset, F.L.; Folgori, A.; Lee, D.H.; Brotman, B.; et al. Evidence for protection against chronic hepatitis c virus infection in chimpanzees by immunization with replicating recombinant vaccinia virus. J. Virol. 2008, 82, 10896–10905. [Google Scholar] [CrossRef] [PubMed]
- Drane, D.; Maraskovsky, E.; Gibson, R.; Mitchell, S.; Barnden, M.; Moskwa, A.; Shaw, D.; Gervase, B.; Coates, S.; Houghton, M.; et al. Priming of cd4+ and cd8+ t cell responses using a hcv core iscomatrix vaccine: A phase I study in healthy volunteers. Hum. Vaccin. 2009, 5, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Geissler, M.; Gesien, A.; Wands, J.R. Inhibitory effects of chronic ethanol consumption on cellular immune responses to hepatitis c virus core protein are reversed by genetic immunizations augmented with cytokine-expressing plasmids. J. Immunol. 1997, 159, 5107–5113. [Google Scholar] [PubMed]
- Inchauspe, G.; Major, M.E.; Nakano, I.; Vitvitski, L.; Trepo, C. DNA vaccination for the induction of immune responses against hepatitis c virus proteins. Vaccine 1997, 15, 853–856. [Google Scholar] [CrossRef]
- Lagging, L.M.; Meyer, K.; Hoft, D.; Houghton, M.; Belshe, R.B.; Ray, R. Immune responses to plasmid DNA encoding the hepatitis c virus core protein. J. Virol. 1995, 69, 5859–5863. [Google Scholar] [PubMed]
- Brinster, C.; Inchauspe, G. DNA vaccines for hepatitis c virus. Intervirology 2001, 44, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Sominskaya, I.; Jansons, J.; Dovbenko, A.; Petrakova, N.; Lieknina, I.; Mihailova, M.; Latyshev, O.; Eliseeva, O.; Stahovska, I.; Akopjana, I.; et al. Comparative immunogenicity in rabbits of the polypeptides encoded by the 5’ terminus of hepatitis c virus RNA. J. Immunol. Res. 2015, 2015, 762426. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.L.; Ren, H.; Zhao, P.; Zhu, S.Y.; Cao, J.; Chen, Z.H.; Zhao, L.J.; Pan, W.; Feitelson, M.A.; Qui, Z.T. Efficient induction of mouse immune responses to hepatitis c virus by viral core protein-carrying attenuated salmonella typhimurium. Viral Immunol. 2007, 20, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chang, Y.; Wu, C.; Han, Q.; Pei, R.; Lu, M.; Chen, X. The wild-type hepatitis c virus core inhibits initiation of antigen-specific t- and b-cell immune responses in balb/c mice. Clin. Vaccine Immunol. 2010, 17, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Soguero, C.; Joo, M.; Chianese-Bullock, K.A.; Nguyen, D.T.; Tung, K.; Hahn, Y.S. Hepatitis c virus core protein leads to immune suppression and liver damage in a transgenic murine model. J. Virol. 2002, 76, 9345–9354. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Hahn, Y.S. Animal models for immune defects caused by hepatitis c virus. Mol. Med. Today 2000, 6, 176–177. [Google Scholar] [CrossRef]
- Large, M.K.; Kittlesen, D.J.; Hahn, Y.S. Suppression of host immune response by the core protein of hepatitis c virus: Possible implications for hepatitis c virus persistence. J. Immunol. 1999, 162, 931–938. [Google Scholar] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. Hcv and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M. Viral hepatitis and liver cancer: The case of hepatitis c. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.G.; Petrakova, N.V.; Mokhonov, V.V.; Pokrovskaya, K.; Suzdaltzeva, Y.G.; Krivonos, A.V.; Zaberezhny, A.D.; Garaev, M.M.; Smirnov, V.D.; Nordenfelt, E. DNA immunization efficiently targets conserved functional domains of protease and atpase/helicase of nonstructural 3 protein (ns3) of human hepatitis c virus. Immunol. Lett. 2003, 88, 1–13. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Stellacci, E.; Marcantonio, C.; Muto, V.; Equestre, M.; Marsili, G.; Rapicetta, M.; Battistini, A. Repression of interferon regulatory factor 1 by hepatitis c virus core protein results in inhibition of antiviral and immunomodulatory genes. J. Virol. 2007, 81, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.E.; Mitchell, A.; Brownell, J.; Miklin, D.J.; Golden-Mason, L.; Polyak, S.J.; Gale, M.J., Jr.; Rosen, H.R. Hepatitis c virus core protein inhibits interferon production by a human plasmacytoid dendritic cell line and dysregulates interferon regulatory factor-7 and signal transducer and activator of transcription (stat) 1 protein expression. PLoS ONE 2014, 9, e95627. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, S.; Ota, S.; Dimberg, L.Y.; Masucci, M.G.; Bergqvist, A. Hepatitis c virus core protein induces an anergic state characterized by decreased interleukin-2 production and perturbation of mitogen-activated protein kinase responses. J. Virol. 2005, 79, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Petrushanko, I.Y.; Ivanova, O.N.; Karpenko, I.L.; Alekseeva, E.; Sominskaya, I.; Makarov, A.A.; Bartosch, B.; Kochetkov, S.N.; et al. Hcv core protein uses multiple mechanisms to induce oxidative stress in human hepatoma huh7 cells. Viruses 2015, 7, 2745–2770. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Kato, N.; Otsuka, M.; Goto, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Hepatitis c virus core protein upregulates transforming growth factor-beta 1 transcription. J. Med. Virol. 2004, 72, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kajdaniuk, D.; Marek, B.; Borgiel-Marek, H.; Kos-Kudla, B. Transforming growth factor beta1 (tgfbeta1) in physiology and pathology. Endokrynol. Pol. 2013, 64, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M. Transforming growth factor-beta: Innately bipolar. Curr. Opin. Immunol. 2007, 19, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.T.; Martin, S.L.; Xia, L.; Gorham, J.D. Tgf-beta 1 uses distinct mechanisms to inhibit ifn-gamma expression in cd4+ t cells at priming and at recall: Differential involvement of stat4 and t-bet. J. Immunol. 2005, 174, 5950–5958. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ueno, Y.; Wakui, Y.; Ninomiya, M.; Kakazu, E.; Inoue, J.; Kobayashi, K.; Obara, N.; Shimosegawa, T. Rapid reduction of hepatitis c virus-core protein in the peripheral blood improve the immunological response in chronic hepatitis c patients. Hepatol. Res. 2011, 41, 1153–1168. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Hahn, Y.S.; Strong, M.; Cheng, L.; Rosen, H.R. Extracellular hcv-core protein induces an immature regulatory phenotype in nk cells: Implications for outcome of acute infection. PLoS ONE 2014, 9, e103219. [Google Scholar] [CrossRef] [PubMed]
- De Lucas, S.; Bartolome, J.; Carreno, V. Hepatitis c virus core protein down-regulates transcription of interferon-induced antiviral genes. J. Infect. Dis. 2005, 191, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Kim, S.S.; Yeung, E.; Kamegaya, Y.; Blackard, J.T.; Kim, K.A.; Holtzman, M.J.; Chung, R.T. Hepatitis c virus core protein blocks interferon signaling by interaction with the stat1 sh2 domain. J. Virol. 2006, 80, 9226–9235. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the nf-kappab pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. Nf-kappa b and rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- You, L.R.; Chen, C.M.; Lee, Y.H. Hepatitis c virus core protein enhances nf-kappab signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J. Virol. 1999, 73, 1672–1681. [Google Scholar] [PubMed]
- Tai, D.I.; Tsai, S.L.; Chen, Y.M.; Chuang, Y.L.; Peng, C.Y.; Sheen, I.S.; Yeh, C.T.; Chang, K.S.; Huang, S.N.; Kuo, G.C.; et al. Activation of nuclear factor kappab in hepatitis c virus infection: Implications for pathogenesis and hepatocarcinogenesis. Hepatology 2000, 31, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Hahn, Y.S.; Kwon, M.; Sadikot, R.T.; Blackwell, T.S.; Christman, J.W. Hepatitis c virus core protein suppresses nf-kappab activation and cyclooxygenase-2 expression by direct interaction with ikappab kinase beta. J. Virol. 2005, 79, 7648–7657. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kang, W.; Ryu, S.W.; Kim, W.I.; Chang, D.Y.; Lee, D.H.; Park, D.Y.; Choi, Y.H.; Choi, K.; Shin, E.C.; et al. Hepatitis c virus infection enhances tnfalpha-induced cell death via suppression of nf-kappab. Hepatology 2012, 56, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Ray, R. Hepatitis c virus core protein: Intriguing properties and functional relevance. FEMS Microbiol. Lett. 2001, 202, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of nf-kappab in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lajonchere, L.; Shoukry, N.H.; Gra, B.; Amador-Canizares, Y.; Helle, F.; Bedard, N.; Guerra, I.; Drouin, C.; Dubuisson, J.; Gonzalez-Horta, E.E.; et al. Immunogenicity of cigb-230, a therapeutic DNA vaccine preparation, in hcv-chronically infected individuals in a phase I clinical trial. J. Viral Hepat. 2009, 16, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Noriega, D.; Alvarez-Lajonchere, L.; Brown, E.; Santana, F.L.; Dubuisson, J.; Wychowski, C.; Guerra, I.; Martinez-Donato, G.; Perez, A.; Amador-Canizares, Y.; et al. A chimeric protein encompassing hepatitis c virus epitopes is able to elicit both humoral and cell-mediated immune responses in mice. Biotechnol. Appl. Biochem. 2014, 61, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Amador-Canizares, Y.; Martinez-Donato, G.; Alvarez-Lajonchere, L.; Vasallo, C.; Dausa, M.; Aguilar-Noriega, D.; Valenzuela, C.; Raices, I.; Dubuisson, J.; Wychowski, C.; et al. Hcv-specific immune responses induced by cigb-230 in combination with ifn-alpha plus ribavirin. World J. Gastroenterol. 2014, 20, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Donato, G.; Amador-Canizares, Y.; Alvarez-Lajonchere, L.; Guerra, I.; Perez, A.; Dubuisson, J.; Wychowsk, C.; Musacchio, A.; Aguilar, D.; Duenas-Carrera, S. Neutralizing antibodies and broad, functional t cell immune response following immunization with hepatitis c virus proteins-based vaccine formulation. Vaccine 2014, 32, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Wang, Y.; Chang, J.; Bian, T.; Tan, W.; Sun, M.; Li, W.; Yang, H.; Chen, J.; Zhang, X.; et al. Hepatitis b virus precore protein augments genetic immunizations of the truncated hepatitis c virus core in balb/c mice. Hepatology 2008, 47, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.B.; Yue, Q.H.; Ouyang, H.F.; Chen, Y.Z.; Xu, X.Q.; Yin, W.; Wei, S.H.; Zhang, X.Q.; Mu, S.J. Immune response to fused core protein of hepatitis c virus and truncated tetanus toxin peptides in mice. Arch. Virol. 2010, 155, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, A.; Ghaemi, A.; Tabarraei, A.; Moradi, A.; Gorji, A.; Semnani, S.; Soleimanjahi, H.; Adli, A.H.; Hosseini, S.Y.; Vakili, M.A. Enhanced cell immune responses to hepatitis c virus core by novel heterologous DNA prime/lambda nanoparticles boost in mice. Virus Genes 2014, 49, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis c virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Gnoni, A.; Casadei Gardini, A.; Pisconti, S.; Licchetta, A.; Scartozzi, M.; Memeo, R.; Palmieri, V.O.; Aprile, G.; Santini, D.; et al. Immunotherapeutic approaches for hepatocellular carcinoma. Oncotarget 2017, 8, 33897–33910. [Google Scholar] [CrossRef] [PubMed]
- Virlogeux, V.; Pradat, P.; Hartig-Lavie, K.; Bailly, F.; Maynard, M.; Ouziel, G.; Poinsot, D.; Lebosse, F.; Ecochard, M.; Radenne, S.; et al. Direct-acting antiviral therapy decreases hepatocellular carcinoma recurrence rate in cirrhotic patients with chronic hepatitis C. Liver Int. 2017, 37, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Colombo, M. Hepatocellular carcinoma and direct acting antiviral treatments: Controversy after the revolution. J. Hepatol. 2016, 65, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Nahon, P.; Layese, R.; Bourcier, V.; Cagnot, C.; Marcellin, P.; Guyader, D.; Pol, S.; Larrey, D.; De Ledinghen, V.; Ouzan, D.; et al. Incidence of hepatocellular carcinoma after direct antiviral therapy for hcv in patients with cirrhosis included in surveillance programs. Gastroenterology 2018, 155, 1436–1450. [Google Scholar] [CrossRef] [PubMed]
- Aleman, S.; Rahbin, N.; Weiland, O.; Davidsdottir, L.; Hedenstierna, M.; Rose, N.; Verbaan, H.; Stal, P.; Carlsson, T.; Norrgren, H.; et al. A risk for hepatocellular carcinoma persists long-term after sustained virologic response in patients with hepatitis c-associated liver cirrhosis. Clin. Infect. Dis. 2013, 57, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Beste, L.A.; Green, P.K.; Berry, K.; Kogut, M.J.; Allison, S.K.; Ioannou, G.N. Effectiveness of hepatitis c antiviral treatment in a USA cohort of veteran patients with hepatocellular carcinoma. J. Hepatol. 2017, 67, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Prenner, S.B.; VanWagner, L.B.; Flamm, S.L.; Salem, R.; Lewandowski, R.J.; Kulik, L. Hepatocellular carcinoma decreases the chance of successful hepatitis c virus therapy with direct-acting antivirals. J. Hepatol. 2017, 66, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Khudyakov, Y.E.; Fields, H.A.; Favorov, M.O.; Khudyakova, N.S.; Bonafonte, M.T.; Holloway, B. Synthetic gene for the hepatitis c virus nucleocapsid protein. Nucl. Acids Res. 1993, 21, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.G.; Iakimtchouk, K.; Petrakova, N.V.; Yermalovich, M.A.; Zuber, A.K.; Kashuba, V.I.; Belikov, S.V.; Andersson, S.; Kochetkov, S.N.; Klinman, D.M.; et al. Gene immunization may induce secondary antibodies reacting with DNA. Vaccine 2004, 22, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.G.; Petrakova, N.V.; Kashuba, E.V.; Suzdaltzeva, Y.G.; Belikov, S.V.; Mokhonov, V.V.; Prilipov, A.G.; Matskova, L.; Smirnova, I.S.; Jolivet-Reynaud, C.; et al. Immunization with hepatitis c virus core gene triggers potent t-cell response, but affects cd4+ t-cells. Vaccine 2004, 22, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Rispeter, K.; Lu, M.; Lechner, S.; Zibert, A.; Roggendorf, M. Cloning and characterization of a complete open reading frame of the hepatitis c virus genome in only two cDNA fragments. J. Gen. Virol. 1997, 78, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Mihailova, M.; Fiedler, M.; Boos, M.; Petrovskis, I.; Sominskaya, I.; Roggendorf, M.; Viazov, S.; Pumpens, P. Preparation of hepatitis c virus structural and non-structural protein fragments and studies of their immunogenicity. Protein Expr. Purif. 2006, 50, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis c virus proteins activate nrf2/are pathway by distinct ros-dependent and independent mechanisms in huh7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [PubMed]
- Starodubova, E.S.; Boberg, A.; Litvina, M.; Morozov, A.; Petrakova, N.V.; Timofeev, A.; Latyshev, O.; Tunitskaya, V.; Wahren, B.; Isaguliants, M.G.; et al. Hiv-1 reverse transcriptase artificially targeted for proteasomal degradation induces a mixed th1/th2-type immune response. Vaccine 2008, 26, 5170–5176. [Google Scholar] [CrossRef] [PubMed]
- Krotova, O.; Starodubova, E.; Petkov, S.; Kostic, L.; Agapkina, J.; Hallengard, D.; Viklund, A.; Latyshev, O.; Gelius, E.; Dillenbeck, T.; et al. Consensus hiv-1 fsu-a integrase gene variants electroporated into mice induce polyfunctional antigen-specific cd4+ and cd8+ t cells. PLoS ONE 2013, 8, e62720. [Google Scholar] [CrossRef] [PubMed]
- Latanova, A.; Petkov, S.; Kuzmenko, Y.; Kilpelainen, A.; Ivanov, A.; Smirnova, O.; Krotova, O.; Korolev, S.; Hinkula, J.; Karpov, V.; et al. Fusion to flaviviral leader peptide targets HIV-1 reverse transcriptase for secretion and reduces its enzymatic activity and ability to induce oxidative stress but has no major effects on its immunogenic performance in DNA-immunized mice. J. Immunol. Res. 2017, 2017, 7407136. [Google Scholar] [CrossRef] [PubMed]
- Petkov, S.P.; Heuts, F.; Krotova, O.A.; Kilpelainen, A.; Engstrom, G.; Starodubova, E.S.; Isaguliants, M.G. Evaluation of immunogen delivery by DNA immunization using non-invasive bioluminescence imaging. Hum. Vaccin. Immunother. 2013, 9, 2228–2236. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Hijikata, M.; Ootsuyama, Y.; Nakagawa, M.; Ohkoshi, S.; Sugimura, T.; Shimotohno, K. Molecular cloning of the human hepatitis c virus genome from Japanese patients with non-a, non-b hepatitis. Proc. Nat. Acad. Sci. USA 1990, 87, 9524–9528. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.D.; Wimmer, E.; Lahser, F.C. Poliovirus/hepatitis c virus (internal ribosomal entry site-core) chimeric viruses: Improved growth properties through modification of a proteolytic cleavage site and requirement for core RNA sequences but not for core-related polypeptides. J. Virol. 1999, 73, 1546–1554. [Google Scholar] [PubMed]
- Mokhonov, V.V.; Novikov, D.V.; Samokhvalov, E.I.; Shatalov, A.G.; Selivanov, N.A.; Prilipov, A.G.; L’Vov, D.K. Genome analysis of hepatitis c virus strain 274933RU isolated in Russian Federation. Vopr. Virusol. 2002, 47, 9–12. [Google Scholar] [PubMed]
- Masalova, O.V.; Atanadze, S.N.; Samokhvalov, E.I.; Petrakova, N.V.; Kalinina, T.I.; Smirnov, V.D.; Khudyakov, Y.E.; Fields, H.A.; Kushch, A.A. Detection of hepatitis c virus core protein circulating within different virus particle populations. J. Med. Virol. 1998, 55, 1–6. [Google Scholar] [CrossRef]
- Lo, S.Y.; Masiarz, F.; Hwang, S.B.; Lai, M.M.; Ou, J.H. Differential subcellular localization of hepatitis c virus core gene products. Virology 1995, 213, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Realdon, S.; Gerotto, M.; Dal Pero, F.; Marin, O.; Granato, A.; Basso, G.; Muraca, M.; Alberti, A. Proapoptotic effect of hepatitis c virus core protein in transiently transfected cells is enhanced by nuclear localization and is dependent on PKR activation. J. Hepatol. 2004, 40, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Ren, S.; Pietschmann, T.; Kartenbeck, J.; Kaehlcke, K.; Bartenschlager, R.; Yen, T.S.; Ott, M. Targeting of hepatitis c virus core protein to mitochondria through a novel c-terminal localization motif. J. Virol. 2004, 78, 7958–7968. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular determinants for subcellular localization of hepatitis c virus core protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, P.; Jibard, N.; Meng, X.; Schweizer-Groyer, G.; Fortin, D.; Rajkowski, K.; Kang, K.; Catelli, M.G.; Baulieu, E.E.; Cadepond, F. Quantification of the nucleocytoplasmic distribution of wild type and modified proteins using confocal microscopy: Interaction between 90-kda heat shock protein (hsp90 alpha) and glucocorticosteroid receptor (gr). Exp. Cell Res. 1998, 242, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Huber, R. Inhibitors of the eukaryotic 20s proteasome core particle: A structural approach. Biochim. Biophys. Acta 2004, 1695, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Todorova, B.; Adam, L.; Culina, S.; Boisgard, R.; Martinon, F.; Cosma, A.; Ustav, M.; Kortulewski, T.; Le Grand, R.; Chapon, C. Electroporation as a vaccine delivery system and a natural adjuvant to intradermal administration of plasmid DNA in macaques. Sci. Rep. 2017, 7, 4122. [Google Scholar] [CrossRef] [PubMed]
- Latanova, A.A.; Petkov, S.; Kilpelainen, A.; Jansons, J.; Latyshev, O.E.; Kuzmenko, Y.V.; Hinkula, J.; Abakumov, M.A.; Valuev-Elliston, V.T.; Gomelsky, M.; et al. Codon optimization and improved delivery/immunization regimen enhance the immune response against wild-type and drug-resistant hiv-1 reverse transcriptase, preserving its th2-polarity. Sci. Rep. 2018, 8, 8078. [Google Scholar] [CrossRef] [PubMed]
- Harase, I.; Moriyama, T.; Kaneko, T.; Kita, H.; Nomura, M.; Suzuki, G.; Ohnishi, H.; Muto, Y.; Yazaki, Y.; Imawari, M. Immune response to hepatitis c virus core protein in mice. Immun. Cell Biol. 1995, 73, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Petkov, S.; Starodubova, E.; Latanova, A.; Kilpelainen, A.; Latyshev, O.; Svirskis, S.; Wahren, B.; Chiodi, F.; Gordeychuk, I.; Isaguliants, M. DNA immunization site determines the level of gene expression and the magnitude, but not the type of the induced immune response. PLoS ONE 2018, 13, e0197902. [Google Scholar] [CrossRef] [PubMed]
- Starodubova, E.; Krotova, O.; Hallengard, D.; Kuzmenko, Y.; Engstrom, G.; Legzdina, D.; Latyshev, O.; Eliseeva, O.; Karin Maltais, A.; Tunitskaya, V.; et al. Cellular immunogenicity of novel gene immunogens in mice monitored by in vivo imaging. Mol. Imaging 2012, 11, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.A.; Singh, R.A.; Barry, M.A. Activation of refractory t cell responses against hepatitis c virus core protein by ablation of interfering hydrophobic domains. Mol. Ther. 2006, 13, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Hope, R.G.; McLauchlan, J. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis c virus core protein. J. Gen. Virol. 2000, 81, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Smirnova, O.; Ivanov, A.V.; Kilpelainen, A.; Kuzmenko, Y.; Petkov, S.; Latanova, A.; Krotova, O.; Engstrom, G.; Karpov, V.; et al. Oxidative stress induced by hiv-1 reverse transcriptase modulates the enzyme’s performance in gene immunization. Hum. Vaccines Immunother. 2013, 9, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis c and b virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Ivanova, O.N.; Mukhtarov, F.S.; Tunitskaya, V.L.; Jansons, J.; Isaguliants, M.G.; Kochetkov, S.N.; Ivanov, A.V. Analysis of the domains of hepatitis c virus core and NS5A proteins that activate the NRF2/ARE cascade. Acta Naturae 2016, 8, 155–159. [Google Scholar]
- Aleksunes, L.M.; Manautou, J.E. Emerging role of NRF2 in protecting against hepatic and gastrointestinal disease. Toxicol. Pathol. 2007, 35, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. Landmark Ed. 2011, 16, 2372–2388. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Ke, B.; Shen, X.D.; Buelow, R.; Melinek, J.; Amersi, F.; Gao, F.; Ritter, T.; Volk, H.D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Heme oxygenase-1 gene transfer prevents CD95/FASL-mediated apoptosis and improves liver allograft survival via carbon monoxide signaling pathway. Transplant. Proc. 2002, 34, 1465–1466. [Google Scholar] [CrossRef]
- Devadas, K.; Hewlett, I.K.; Dhawan, S. Lipopolysaccharide suppresses hiv-1 replication in human monocytes by protein kinase c-dependent heme oxygenase-1 induction. J. Leukoc. Biol. 2010, 87, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.O.; Oh, G.S.; Choi, B.M.; Chae, S.C.; Chung, H.T. Differential expressions of heme oxygenase-1 gene in cd25- and cd25+ subsets of human cd4+ t cells. Biochem. Biophys. Res. Commun. 2003, 306, 701–705. [Google Scholar] [CrossRef]
- Ke, B.; Shen, X.D.; Melinek, J.; Gao, F.; Ritter, T.; Volk, H.D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Heme oxygenase-1 gene therapy: A novel immunomodulatory approach in liver allograft recipients? Transplant. Proc. 2001, 33, 581–582. [Google Scholar] [CrossRef]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y. Nad(p)h: Quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc. Toxicol. 2012, 12, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Iskander, K.; Li, J.; Han, S.; Zheng, B.; Jaiswal, A.K. Nqo1 and nqo2 regulation of humoral immunity and autoimmunity. J. Biol. Chem. 2006, 281, 30917–30924. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. Cyp2e1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of hepatotoxicity. Toxicol. Sci. 2002, 65, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Rigamonti, C.; Vidali, M.; Albano, E. Cyp2e1 autoantibodies in liver diseases. Redox Biol. 2014, 3, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.U.; Kono, H.; Isayama, F.; Kosyk, O.; Wheeler, M.D.; Akiyama, T.E.; Bleye, L.; Krausz, K.W.; Gonzalez, F.J.; Koop, D.R.; et al. Cytochrome p450 cyp2e1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology 2005, 41, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Chouteau, P.; Lerat, H. ‘Liver let die’: Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Gilbert, K.M.; Pumford, N.R. Inhibition of cyp2e1 reverses cd4+ t-cell alterations in trichloroethylene-treated MRL+/+ mice. Toxicol. Sci. 2000, 54, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Blossom, S.J.; Pumford, N.R.; Gilbert, K.M. Activation and attenuation of apoptosis of cd4+ t cells following in vivo exposure to two common environmental toxicants, trichloroacetaldehyde hydrate and trichloroacetic acid. J. Autoimmun. 2004, 23, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wu, C.; Deng, W.; Pei, R.; Wang, Y.; Cao, L.; Qin, B.; Lu, M.; Chen, X. Inhibition of the hcv core protein on the immune response to hbv surface antigen and on hbv gene expression and replication in vivo. PLoS ONE 2012, 7, e45146. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; White, R.L.; Todero, S.; McVicker, B.L.; Thiele, G.M.; Clemens, D.L.; Tuma, D.J.; Donohue, T.M., Jr. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology 2007, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Marusawa, H.; Hijikata, M.; Chiba, T.; Shimotohno, K. Hepatitis c virus core protein inhibits fas- and tumor necrosis factor alpha-mediated apoptosis via nf-kappab activation. J. Virol. 1999, 73, 4713–4720. [Google Scholar] [PubMed]
- Bergqvist, A.; Rice, C.M. Transcriptional activation of the interleukin-2 promoter by hepatitis c virus core protein. J. Virol. 2001, 75, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Scatizzi, J.C.; Mavers, M.; Hutcheson, J.; Young, B.; Shi, B.; Pope, R.M.; Ruderman, E.M.; Samways, D.S.; Corbett, J.A.; Egan, T.M.; et al. The cdk domain of p21 is a suppressor of il-1beta-mediated inflammation in activated macrophages. Eur. J. Immunol. 2009, 39, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Trakala, M.; Arias, C.F.; Garcia, M.I.; Moreno-Ortiz, M.C.; Tsilingiri, K.; Fernandez, P.J.; Mellado, M.; Diaz-Meco, M.T.; Moscat, J.; Serrano, M.; et al. Regulation of macrophage activation and septic shock susceptibility via p21(waf1/cip1). Eur. J. Immunol. 2009, 39, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.F.; Ballesteros-Tato, A.; Garcia, M.I.; Martin-Caballero, J.; Flores, J.M.; Martinez, A.C.; Balomenos, D. P21cip1/waf1 controls proliferation of activated/memory t cells and affects homeostasis and memory t cell responses. J. Immunol. 2007, 178, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, C.; Huang, J.; Cung, T.; Seiss, K.; Beamon, J.; Carrington, M.F.; Porter, L.C.; Burke, P.S.; Yang, Y.; et al. Cd4+ t cells from elite controllers resist hiv-1 infection by selective upregulation of p21. J. Clin. Investig. 2011, 121, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. P21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yoshida, I.; Takamatsu, M.; Ishido, S.; Fujita, T.; Oka, K.; Hotta, H. Complex formation between hepatitis c virus core protein and p21waf1/cip1/sdi1. Biochem. Biophys. Res. Commun. 2000, 273, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Oka, K.; Nagano-Fujii, M.; Yoshida, I.; Hidajat, R.; Deng, L.; Akutsu, M.; Hotta, H. Hepatitis c virus core protein selectively inhibits synthesis and accumulation of p21/waf1 and certain nuclear proteins. Microbiol. Immunol. 2003, 47, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Brault, C.; Levy, P.L.; Bartosch, B. Hepatitis c virus-induced mitochondrial dysfunctions. Viruses 2013, 5, 954–980. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis c virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ros) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis c virus core triggers apoptosis in liver cells by inducing er stress and er calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of hepatitis c virus core protein in viral-induced mitochondrial dysfunction. J. Viral Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Correlates | ||||
|---|---|---|---|---|---|
| Total IFN-γ Production to All Peptides | IFN-γ Core 1-152 | IFN-γ Core Peptide Pool 1 1 | IFN-γ Core Peptide Pool 2 2 | IFN-γ Core Peptide Pool 3 3 | |
| ROS | 0.857143 | 0.882919 | 0.637748 | 0.600000 | 0.637748 |
| Nqo1 | 0.750000 | 0.774806 | 0.637748 | 0.600000 | 0.637748 |
| HO-1 | 0.571429 | 0.540562 | 0.985611 | 0.942857 | 0.811679 |
| ROS normalized to vector | 0.535714 | 0.504525 | 0.927634 | 0.885714 | 0.753702 |
| Nqo1 normalized to vector | 0.821429 | 0.792825 | 0.985611 | 0.942857 | 0.811679 |
| HO-1 normalized to vector | 0.785714 | 0.756787 | 0.927634 | 0.885714 | 0.753702 |
| ROS normalized to effect of core152v | −0.750000 | −0.774806 | −0.637748 | −0.600000 | −0.637748 |
| Nqo1 normalized to effect of core152v | −0.857143 | −0.882919 | −0.811679 | −0.771429 | −0.637748 |
| HO-1 normalized to effect of core152v | 0.857143 | 0.882919 | 0.637748 | 0.600000 | 0.637748 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansons, J.; Sominskaya, I.; Petrakova, N.; Starodubova, E.S.; Smirnova, O.A.; Alekseeva, E.; Bruvere, R.; Eliseeva, O.; Skrastina, D.; Kashuba, E.; et al. The Immunogenicity in Mice of HCV Core Delivered as DNA Is Modulated by Its Capacity to Induce Oxidative Stress and Oxidative Stress Response. Cells 2019, 8, 208. https://doi.org/10.3390/cells8030208
Jansons J, Sominskaya I, Petrakova N, Starodubova ES, Smirnova OA, Alekseeva E, Bruvere R, Eliseeva O, Skrastina D, Kashuba E, et al. The Immunogenicity in Mice of HCV Core Delivered as DNA Is Modulated by Its Capacity to Induce Oxidative Stress and Oxidative Stress Response. Cells. 2019; 8(3):208. https://doi.org/10.3390/cells8030208
Chicago/Turabian StyleJansons, Juris, Irina Sominskaya, Natalia Petrakova, Elizaveta S. Starodubova, Olga A. Smirnova, Ekaterina Alekseeva, Ruta Bruvere, Olesja Eliseeva, Dace Skrastina, Elena Kashuba, and et al. 2019. "The Immunogenicity in Mice of HCV Core Delivered as DNA Is Modulated by Its Capacity to Induce Oxidative Stress and Oxidative Stress Response" Cells 8, no. 3: 208. https://doi.org/10.3390/cells8030208
APA StyleJansons, J., Sominskaya, I., Petrakova, N., Starodubova, E. S., Smirnova, O. A., Alekseeva, E., Bruvere, R., Eliseeva, O., Skrastina, D., Kashuba, E., Mihailova, M., Kochetkov, S. N., Ivanov, A. V., & Isaguliants, M. G. (2019). The Immunogenicity in Mice of HCV Core Delivered as DNA Is Modulated by Its Capacity to Induce Oxidative Stress and Oxidative Stress Response. Cells, 8(3), 208. https://doi.org/10.3390/cells8030208