Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells


Abstract
1. Introduction
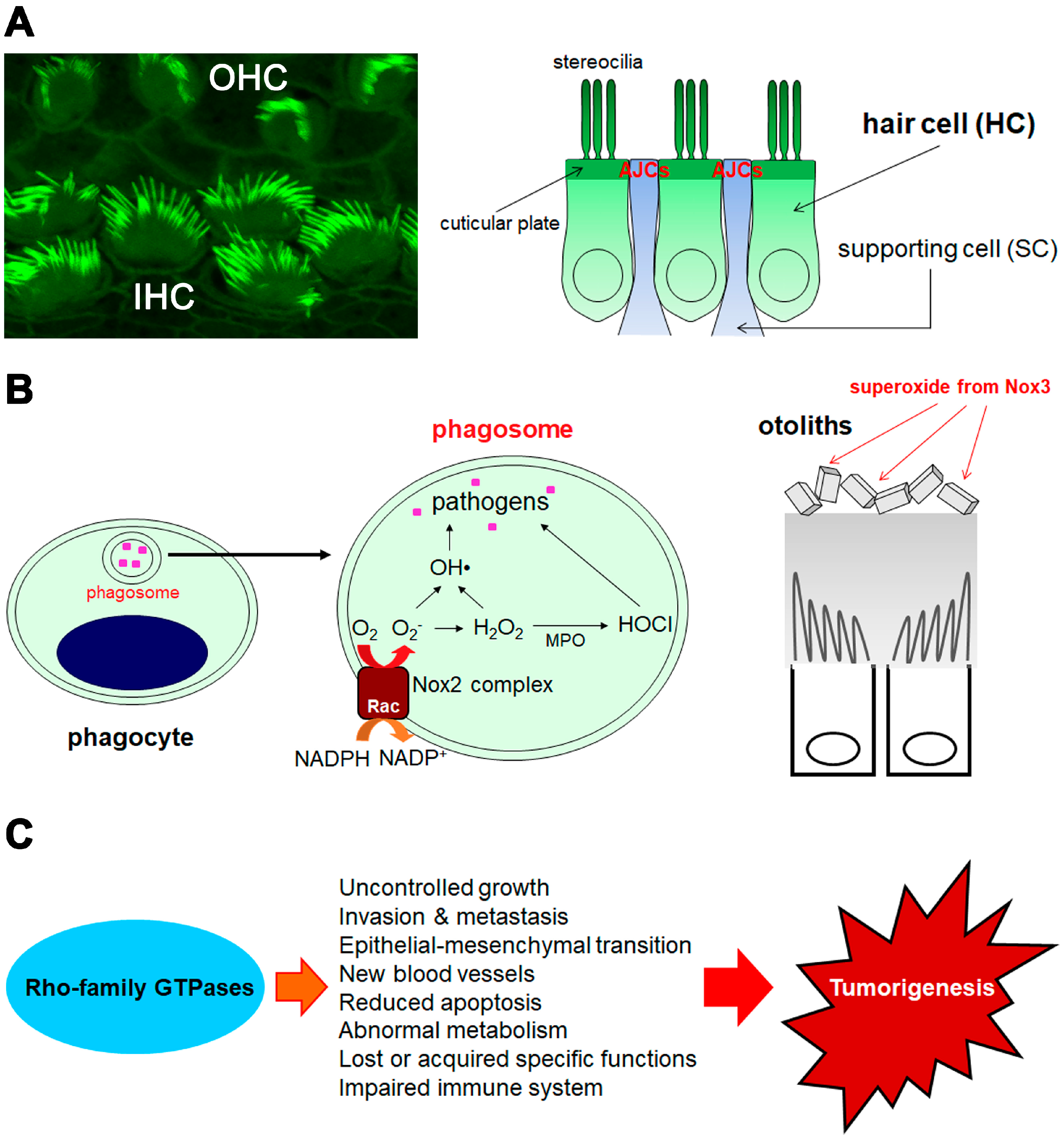
2. Hearing Function and Beyond
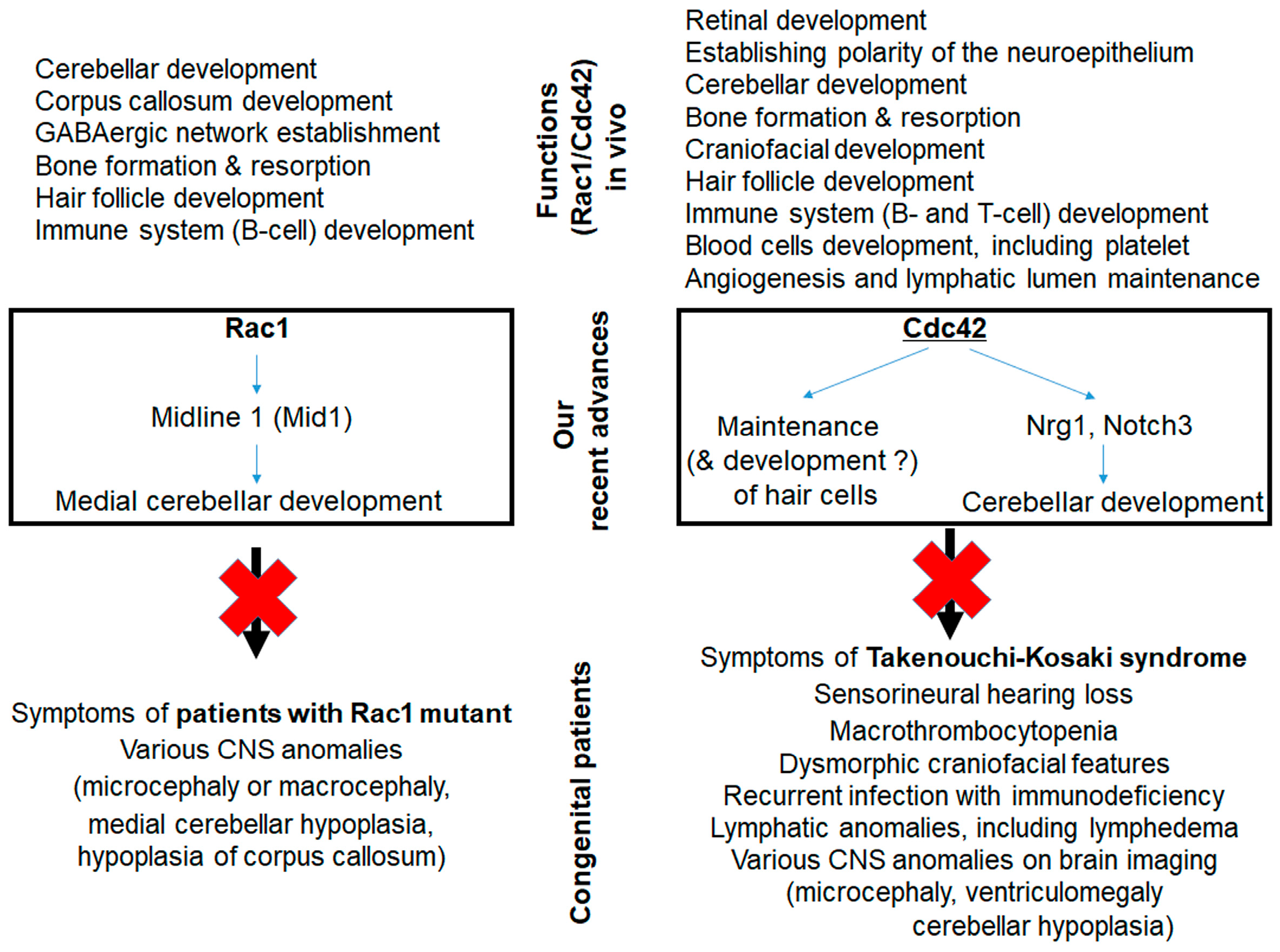
2.1. Role and Function of Rac in Hearing
2.2. Role and Function of Cdc42 in Hearing
2.3. Hearing in Patients with RAC1 or CDC42 Mutations
2.4. Deafness in Patients with Active DIA1 Mutations Downstream of RhoA
2.5. Macrothrombocytopenia in Patients and Mice Associated with Rho-Family GTPases
2.6. Deafness Associated with Rho-Family GTPases Other than Rac and Cdc42
2.7. Roles of Rho-Family GTPases in Other Sensory Organs
3. Host Defenses through Superoxide Generation and Arrangements of Actin and Membranes
3.1. Superoxide Production from Rac-Dependent Nox2-Based Oxidase
3.2. Superoxide/Reactive Oxygen Species (ROS) Production from Novel Noxs
3.3. Regulation of Superoxide Production by RhoGDIs
4. Tumorigenesis
4.1. Recent Advances in Rac Involvement in Tumorigenesis
4.2. A Novel Downstream Target of Rac Signaling, GSPT1, Is Associated with Tumorigenesis
4.3. Involvement of Other Rho-Family GTPases in Tumorigenesis
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, S.M.; Awadia, S.; Garcia-Mata, R. I’m coming to GEF you: Regulation of RhoGEFs during cell migration. Cell Adh. Migr. 2014, 8, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The “invisible hand”: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Rivero, F. Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells 2016, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Peyrollier, K.; Pedersen, E.; Basse, A.; Karlsson, R.; Wang, Z.; Lefever, T.; Ochsenbein, A.M.; Schmidt, G.; Aktories, K.; et al. RhoA is dispensable for skin development, but crucial for contraction and directed migration of keratinocytes. Mol. Biol. Cell 2011, 22, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Couronne, L.; Khiabanian, H.; Kim, M.Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef]
- Rohde, M.; Richter, J.; Schlesner, M.; Betts, M.J.; Claviez, A.; Bonn, B.R.; Zimmermann, M.; Damm-Welk, C.; Russell, R.B.; Borkhardt, A.; et al. Recurrent RHOA mutations in pediatric Burkitt lymphoma treated according to the NHL-BFM protocols. Genes Chromosomes Cancer 2014, 53, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.X.; Rane, N.; Liu, J.P.; Prendergast, G.C. RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cells. Mol. Cell Biol. 2001, 21, 6906–6912. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.A.; Gilkes, D.M. RhoB: Team Oncogene or Team Tumor Suppressor? Genes (Basel) 2018, 9, 67. [Google Scholar] [CrossRef]
- Calvayrac, O.; Nowosad, A.; Cabantous, S.; Lin, L.P.; Figarol, S.; Jeannot, P.; Serres, M.; Callot, C.; Perchey, R.T.; Creff, J.; et al. Cytoplasmic p27(Kip1) promotes tumorigenesis via suppression of RhoB activity. J. Pathol. 2018. [Google Scholar] [CrossRef]
- Hakem, A.; Sanchez-Sweatman, O.; You-Ten, A.; Duncan, G.; Wakeham, A.; Khokha, R.; Mak, T.W. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes. Dev. 2005, 19, 1974–1979. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.K.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Ambruso, D.R.; Knall, C.; Abell, A.N.; Panepinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; deBoer, M.; Harbeck, R.J.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad Sci. USA 2000, 97, 4654–4659. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Tao, W.; Yang, F.; Kim, C.; Gu, Y.; Mansfield, P.; Levine, J.E.; Petryniak, B.; Derrow, C.W.; Harris, C.; et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood 2000, 96, 1646–1654. [Google Scholar] [PubMed]
- Accetta, D.; Syverson, G.; Bonacci, B.; Reddy, S.; Bengtson, C.; Surfus, J.; Harbeck, R.; Huttenlocher, A.; Grossman, W.; Routes, J.; et al. Human phagocyte defect caused by a Rac2 mutation detected by means of neonatal screening for T-cell lymphopenia. J. Allergy Clin. Immunol. 2011, 127, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Alkhairy, O.K.; Rezaei, N.; Graham, R.R.; Abolhassani, H.; Borte, S.; Hultenby, K.; Wu, C.; Aghamohammadi, A.; Williams, D.A.; Behrens, T.W.; et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J. Allergy Clin. Immunol. 2015, 135, 1380–1384. [Google Scholar] [CrossRef] [PubMed]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, E.; Bell, S.; Hebeis, B.J.; Reynolds, H.; McAdam, S.; Emson, P.C.; McKenzie, A.; Turner, M. Immunological function in mice lacking the Rac-related GTPase RhoG. Mol. Cell Biol. 2004, 24, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Kosaki, R.; Niizuma, T.; Hata, K.; Kosaki, K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am. J. Med. Genet. A 2015, 167A, 2822–2825. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Okamoto, N.; Ida, S.; Uehara, T.; Kosaki, K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am. J. Med. Genet. A 2016, 170A, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Motokawa, M.; Watanabe, S.; Nakatomi, A.; Kondoh, T.; Matsumoto, T.; Morifuji, K.; Sawada, H.; Nishimura, T.; Nunoi, H.; Yoshiura, K.I.; et al. A hot-spot mutation in CDC42 (p.Tyr64Cys) and novel phenotypes in the third patient with Takenouchi-Kosaki syndrome. J. Hum. Genet. 2018, 63, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, S.; Krumbach, O.H.F.; Pantaleoni, F.; Coppola, S.; Amin, E.; Pannone, L.; Nouri, K.; Farina, L.; Dvorsky, R.; Lepri, F.; et al. Functional Dysregulation of CDC42 Causes Diverse Developmental Phenotypes. Am. J. Hum. Genet. 2018, 102, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Yang, H.; Fukushima, Y.; Saw, P.E.; Lee, J.; Park, J.S.; Park, I.; Jung, J.; Kataoka, H.; Lee, D.; et al. Vascular RhoJ is an effective and selective target for tumor angiogenesis and vascular disruption. Cancer Cell 2014, 25, 102–117. [Google Scholar] [CrossRef]
- Burbage, M.; Keppler, S.J.; Montaner, B.; Mattila, P.K.; Batista, F.D. The Small Rho GTPase TC10 Modulates B Cell Immune Responses. J. Immunol. 2017, 199, 1682–1695. [Google Scholar] [CrossRef] [PubMed]
- Goggs, R.; Savage, J.S.; Mellor, H.; Poole, A.W. The small GTPase Rif is dispensable for platelet filopodia generation in mice. PLoS ONE 2013, 8, e54663. [Google Scholar] [CrossRef]
- Gu, Y.; Chae, H.D.; Siefring, J.E.; Jasti, A.C.; Hildeman, D.A.; Williams, D.A. RhoH GTPase recruits and activates Zap70 required for T cell receptor signaling and thymocyte development. Nat. Immunol. 2006, 7, 1182–1190. [Google Scholar] [CrossRef]
- Preudhomme, C.; Roumier, C.; Hildebrand, M.P.; Dallery-Prudhomme, E.; Lantoine, D.; Lai, J.L.; Daudignon, A.; Adenis, C.; Bauters, F.; Fenaux, P.; et al. Nonrandom 4p13 rearrangements of the RhoH/TTF gene, encoding a GTP-binding protein, in non-Hodgkin’s lymphoma and multiple myeloma. Oncogene 2000, 19, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.; Konrad, E.D.H.; Gruner, J.; Toutain, A.; Bok, L.A.; Cho, M.T.; Crawford, H.P.; Dubbs, H.; Douglas, G.; Jobling, R.; et al. Missense Variants in RHOBTB2 Cause a Developmental and Epileptic Encephalopathy in Humans, and Altered Levels Cause Neurological Defects in Drosophila. Am. J. Hum. Genet. 2018, 102, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Grimm-Gunter, E.M.; Joshi, P.; Rivero, F. Expression analysis of mouse Rhobtb3 using a LacZ reporter and preliminary characterization of a knockout strain. Histochem. Cell Biol. 2014, 142, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Bosco, E.E.; Mulloy, J.C.; Zheng, Y. Rac1 GTPase: A “Rac” of all trades. Cell Mol. Life Sci. 2009, 66, 370–374. [Google Scholar] [CrossRef]
- Funahashi, Y.; Namba, T.; Nakamuta, S.; Kaibuchi, K. Neuronal polarization in vivo: Growing in a complex environment. Curr. Opin. Neurobiol. 2014, 27, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Chircop, M. Rho GTPases as regulators of mitosis and cytokinesis in mammalian cells. Small GTPases 2014, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Mack, N.A.; Georgiou, M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Michalski, N.; Petit, C. Genetics of auditory mechano-electrical transduction. Pflugers Arch. 2015, 467, 49–72. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Roy, P.; Perrin, B.J. Stereocilia morphogenesis and maintenance through regulation of actin stability. Semin Cell Dev. Biol. 2017, 65, 88–95. [Google Scholar] [CrossRef]
- Goutman, J.D.; Elgoyhen, A.B.; Gomez-Casati, M.E. Cochlear hair cells: The sound-sensing machines. FEBS Lett. 2015, 589, 3354–3361. [Google Scholar] [CrossRef]
- Pollock, L.M.; McDermott, B.M., Jr. The cuticular plate: A riddle, wrapped in a mystery, inside a hair cell. Birth Defects Res. C Embryo Today 2015, 105, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Grimsley-Myers, C.M.; Sipe, C.W.; Geleoc, G.S.; Lu, X. The small GTPase Rac1 regulates auditory hair cell morphogenesis. J. Neurosci. 2009, 29, 15859–15869. [Google Scholar] [CrossRef] [PubMed]
- Grimsley-Myers, C.M.; Sipe, C.W.; Wu, D.K.; Lu, X. Redundant functions of Rac GTPases in inner ear morphogenesis. Dev. Biol. 2012, 362, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.M.; McConnell, S.K. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 2000, 222, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Pirvola, U.; Ylikoski, J.; Trokovic, R.; Hebert, J.M.; McConnell, S.K.; Partanen, J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron 2002, 35, 671–680. [Google Scholar] [CrossRef]
- Ohyama, T.; Groves, A.K. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 2004, 38, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.W. Regulation of cell fate in the sensory epithelia of the inner ear. Nat. Rev. Neurosci. 2006, 7, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.C.; Liu, Z.; Lagarde, M.M.; Zuo, J. Conditional gene expression in the mouse inner ear using Cre-loxP. J. Assoc. Res. Otolaryngol. 2012, 13, 295–322. [Google Scholar] [CrossRef]
- Ueyama, T.; Sakaguchi, H.; Nakamura, T.; Goto, A.; Morioka, S.; Shimizu, A.; Nakao, K.; Hishikawa, Y.; Ninoyu, Y.; Kassai, H.; et al. Maintenance of stereocilia and apical junctional complexes by Cdc42 in cochlear hair cells. J. Cell Sci. 2014, 127, 2040–2052. [Google Scholar] [CrossRef]
- Zhang, D.S.; Piazza, V.; Perrin, B.J.; Rzadzinska, A.K.; Poczatek, J.C.; Wang, M.; Prosser, H.M.; Ervasti, J.M.; Corey, D.P.; Lechene, C.P. Multi-isotope imaging mass spectrometry reveals slow protein turnover in hair-cell stereocilia. Nature 2012, 481, 520–524. [Google Scholar] [CrossRef]
- Narayanan, P.; Chatterton, P.; Ikeda, A.; Ikeda, S.; Corey, D.P.; Ervasti, J.M.; Perrin, B.J. Length regulation of mechanosensitive stereocilia depends on very slow actin dynamics and filament-severing proteins. Nat. Commun. 2015, 6, 6855. [Google Scholar] [CrossRef]
- Drummond, M.C.; Barzik, M.; Bird, J.E.; Zhang, D.S.; Lechene, C.P.; Corey, D.P.; Cunningham, L.L.; Friedman, T.B. Live-cell imaging of actin dynamics reveals mechanisms of stereocilia length regulation in the inner ear. Nat. Commun. 2015, 6, 6873. [Google Scholar] [CrossRef]
- Scheffer, D.I.; Shen, J.; Corey, D.P.; Chen, Z.Y. Gene Expression by Mouse Inner Ear Hair Cells during Development. J. Neurosci. 2015, 35, 6366–6380. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, T.; Kirjavainen, A.; Belevich, I.; Laos, M.; Richardson, W.D.; Jokitalo, E.; Brakebusch, C.; Pirvola, U. Cdc42-dependent structural development of auditory supporting cells is required for wound healing at adulthood. Sci. Rep. 2012, 2, 978. [Google Scholar] [CrossRef] [PubMed]
- Kirjavainen, A.; Laos, M.; Anttonen, T.; Pirvola, U. The Rho GTPase Cdc42 regulates hair cell planar polarity and cellular patterning in the developing cochlea. Biol. Open 2015, 4, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Matei, V.; Pauley, S.; Kaing, S.; Rowitch, D.; Beisel, K.W.; Morris, K.; Feng, F.; Jones, K.; Lee, J.; Fritzsch, B. Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 2005, 234, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, X.; Deng, M.; Chen, X.; Gan, L. Generation and characterization of Atoh1-Cre knock-in mouse line. Genesis 2010, 48, 407–413. [Google Scholar] [CrossRef]
- Nakamura, T.; Ueyama, T.; Ninoyu, Y.; Sakaguchi, H.; Choijookhuu, N.; Hishikawa, Y.; Kiyonari, H.; Kohta, M.; Sakahara, M.; de Curtis, I.; et al. Novel role of Rac-Mid1 signaling in medial cerebellar development. Development 2017, 144, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Tahirovic, S.; Hellal, F.; Neukirchen, D.; Hindges, R.; Garvalov, B.K.; Flynn, K.C.; Stradal, T.E.; Chrostek-Grashoff, A.; Brakebusch, C.; Bradke, F. Rac1 regulates neuronal polarization through the WAVE complex. J. Neurosci. 2010, 30, 6930–6943. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liao, G.; Waclaw, R.R.; Burns, K.A.; Linquist, D.; Campbell, K.; Zheng, Y.; Kuan, C.Y. Rac1 controls the formation of midline commissures and the competency of tangential migration in ventral telencephalic neurons. J. Neurosci. 2007, 27, 3884–3893. [Google Scholar] [CrossRef]
- Kassai, H.; Terashima, T.; Fukaya, M.; Nakao, K.; Sakahara, M.; Watanabe, M.; Aiba, A. Rac1 in cortical projection neurons is selectively required for midline crossing of commissural axonal formation. Eur. J. Neurosci. 2008, 28, 257–267. [Google Scholar] [CrossRef]
- Chen, L.; Melendez, J.; Campbell, K.; Kuan, C.Y.; Zheng, Y. Rac1 deficiency in the forebrain results in neural progenitor reduction and microcephaly. Dev. Biol. 2009, 325, 162–170. [Google Scholar] [CrossRef]
- Sakamoto, I.; Ueyama, T.; Hayashibe, M.; Nakamura, T.; Mohri, H.; Kiyonari, H.; Shigyo, M.; Tohda, C.; Saito, N. Roles of Cdc42 and Rac in Bergmann glia during cerebellar corticogenesis. Exp. Neurol. 2018, 302, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Pennucci, R.; Talpo, F.; Astro, V.; Montinaro, V.; More, L.; Cursi, M.; Castoldi, V.; Chiaretti, S.; Bianchi, V.; Marenna, S.; et al. Loss of Either Rac1 or Rac3 GTPase Differentially Affects the Behavior of Mutant Mice and the Development of Functional GABAergic Networks. Cereb. Cortex. 2016, 26, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Croke, M.; Ross, F.P.; Korhonen, M.; Williams, D.A.; Zou, W.; Teitelbaum, S.L. Rac deletion in osteoclasts causes severe osteopetrosis. J. Cell Sci. 2011, 124, 3811–3821. [Google Scholar] [CrossRef] [PubMed]
- Touaitahuata, H.; Blangy, A.; Vives, V. Modulation of osteoclast differentiation and bone resorption by Rho GTPases. Small GTPases 2014, 5, e28119. [Google Scholar] [CrossRef] [PubMed]
- Benitah, S.A.; Frye, M.; Glogauer, M.; Watt, F.M. Stem cell depletion through epidermal deletion of Rac1. Science 2005, 309, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Chrostek, A.; Wu, X.; Quondamatteo, F.; Hu, R.; Sanecka, A.; Niemann, C.; Langbein, L.; Haase, I.; Brakebusch, C. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol. Cell Biol 2006, 26, 6957–6970. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.M.; Squarize, C.H.; Patel, V.; Millar, S.E.; Zheng, Y.; Molinolo, A.; Gutkind, J.S. Requirement of Rac1 distinguishes follicular from interfollicular epithelial stem cells. Oncogene 2007, 26, 5078–5085. [Google Scholar] [CrossRef]
- Walmsley, M.J.; Ooi, S.K.; Reynolds, L.F.; Smith, S.H.; Ruf, S.; Mathiot, A.; Vanes, L.; Williams, D.A.; Cancro, M.P.; Tybulewicz, V.L. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science 2003, 302, 459–462. [Google Scholar] [CrossRef]
- Choi, S.Y.; Chacon-Heszele, M.F.; Huang, L.; McKenna, S.; Wilson, F.P.; Zuo, X.; Lipschutz, J.H. Cdc42 deficiency causes ciliary abnormalities and cystic kidneys. J. Am. Soc. Nephrol. 2013, 24, 1435–1450. [Google Scholar] [CrossRef]
- Choi, S.Y.; Baek, J.I.; Zuo, X.; Kim, S.H.; Dunaief, J.L.; Lipschutz, J.H. Cdc42 and sec10 Are Required for Normal Retinal Development in Zebrafish. Invest. Ophthalmol. Vis. Sci. 2015, 56, 3361–3370. [Google Scholar] [CrossRef]
- Cappello, S.; Attardo, A.; Wu, X.; Iwasato, T.; Itohara, S.; Wilsch-Brauninger, M.; Eilken, H.M.; Rieger, M.A.; Schroeder, T.T.; Huttner, W.B.; et al. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat. Neurosci. 2006, 9, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liao, G.; Yang, L.; Campbell, K.; Nakafuku, M.; Kuan, C.Y.; Zheng, Y. Cdc42 deficiency causes Sonic hedgehog-independent holoprosencephaly. PNAS 2006, 103, 16520–16525. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Teitelbaum, S.L.; Zou, W.; Zheng, Y.; Johnson, J.F.; Chappel, J.; Ross, F.P.; Zhao, H. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J. Clin. Invest. 2010, 120, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, R.; Yamada, A.; Suzuki, D.; Iimura, T.; Kassai, H.; Harada, T.; Tsukasaki, M.; Yamamoto, G.; Tachikawa, T.; Nakao, K.; et al. Cdc42 is required for chondrogenesis and interdigital programmed cell death during limb development. Mech. Dev. 2012, 129, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Oshima-Nakayama, M.; Yamada, A.; Kurosawa, T.; Aizawa, R.; Suzuki, D.; Saito, Y.; Kassai, H.; Sato, Y.; Yamamoto, M.; Shirota, T.; et al. Cdc42 is crucial for facial and palatal formation during craniofacial development. Bone Rep. 2016, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Quondamatteo, F.; Lefever, T.; Czuchra, A.; Meyer, H.; Chrostek, A.; Paus, R.; Langbein, L.; Brakebusch, C. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 2006, 20, 571–585. [Google Scholar] [CrossRef]
- Guo, F.; Velu, C.S.; Grimes, H.L.; Zheng, Y. Rho GTPase Cdc42 is essential for B-lymphocyte development and activation. Blood 2009, 114, 2909–2916. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Hildeman, D.; Tripathi, P.; Velu, C.S.; Grimes, H.L.; Zheng, Y. Coordination of IL-7 receptor and T-cell receptor signaling by cell-division cycle 42 in T-cell homeostasis. Proc. Natl. Acad Sci. USA 2010, 107, 18505–18510. [Google Scholar] [CrossRef] [PubMed]
- Melendez, J.; Grogg, M.; Zheng, Y. Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 2011, 286, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Pleines, I.; Eckly, A.; Elvers, M.; Hagedorn, I.; Eliautou, S.; Bender, M.; Wu, X.; Lanza, F.; Gachet, C.; Brakebusch, C.; et al. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood 2010, 115, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Pleines, I.; Dutting, S.; Cherpokova, D.; Eckly, A.; Meyer, I.; Morowski, M.; Krohne, G.; Schulze, H.; Gachet, C.; Debili, N.; et al. Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood 2013, 122, 3178–3187. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, Y.; Fukuhara, S.; Ando, K.; Matsuda, M.; Mochizuki, N. Cdc42 mediates Bmp-induced sprouting angiogenesis through Fmnl3-driven assembly of endothelial filopodia in zebrafish. Dev. Cell 2015, 32, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gu, X.; Ma, W.; Oxendine, M.; Gil, H.J.; Davis, G.E.; Cleaver, O.; Oliver, G. Rasip1 controls lymphatic vessel lumen maintenance by regulating endothelial cell junctions. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e29513. [Google Scholar] [CrossRef] [PubMed]
- Lynch, E.D.; Lee, M.K.; Morrow, J.E.; Welcsh, P.L.; Leon, P.E.; King, M.C. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 1997, 278, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Ninoyu, Y.; Nishio, S.Y.; Miyoshi, T.; Torii, H.; Nishimura, K.; Sugahara, K.; Sakata, H.; Thumkeo, D.; Sakaguchi, H.; et al. Constitutive activation of DIA1 (DIAPH1) via C-terminal truncation causes human sensorineural hearing loss. EMBO Mol. Med. 2016, 8, 1310–1324. [Google Scholar] [CrossRef] [PubMed]
- Stritt, S.; Nurden, P.; Turro, E.; Greene, D.; Jansen, S.B.; Westbury, S.K.; Petersen, R.; Astle, W.J.; Marlin, S.; Bariana, T.K.; et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood 2016, 127, 2903–2914. [Google Scholar] [CrossRef]
- Ganaha, A.; Kaname, T.; Shinjou, A.; Chinen, Y.; Yanagi, K.; Higa, T.; Kondo, S.; Suzuki, M. Progressive macrothrombocytopenia and hearing loss in a large family with DIAPH1 related disease. Am. J. Med. Genet. A 2017, 173, 2826–2830. [Google Scholar] [CrossRef]
- Neuhaus, C.; Lang-Roth, R.; Zimmermann, U.; Heller, R.; Eisenberger, T.; Weikert, M.; Markus, S.; Knipper, M.; Bolz, H.J. Extension of the clinical and molecular phenotype of DIAPH1-associated autosomal dominant hearing loss (DFNA1). Clin. Genet. 2016, 91, 892–901. [Google Scholar] [CrossRef]
- Westbury, S.K.; Downes, K.; Burney, C.; Lozano, M.L.; Obaji, S.G.; Toh, C.H.; Sevivas, T.; Morgan, N.V.; Erber, W.N.; Kempster, C.; et al. Phenotype description and response to thrombopoietin receptor agonist in DIAPH1-related disorder. Blood Adv. 2018, 2, 2341–2346. [Google Scholar] [CrossRef]
- Pleines, I.; Hagedorn, I.; Gupta, S.; May, F.; Chakarova, L.; van Hengel, J.; Offermanns, S.; Krohne, G.; Kleinschnitz, C.; Brakebusch, C.; et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 2012, 119, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.; Eckly, A.; Hartwig, J.H.; Elvers, M.; Pleines, I.; Gupta, S.; Krohne, G.; Jeanclos, E.; Gohla, A.; Gurniak, C.; et al. ADF/n-cofilin-dependent actin turnover determines platelet formation and sizing. Blood 2010, 116, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Sladojevic, N.; Oh, G.T.; Kim, H.H.; Beaulieu, L.M.; Falet, H.; Kaminski, K.; Freedman, J.E.; Liao, J.K. Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2-deficient platelets. Cardiovasc. Res. 2017, 113, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Pleines, I.; Cherpokova, D.; Bender, M. Rho GTPases and their downstream effectors in megakaryocyte biology. Platelets 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Thumkeo, D. Rho signaling research: History, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, A.K.; Luxford, W.M.; Mhatre, A.N.; Attaie, A.; Wilcox, E.R.; Castelein, C.M. A new locus for nonsyndromic hereditary hearing impairment, DFNA17, maps to chromosome 22 and represents a gene for cochleosaccular degeneration. Am. J. Hum. Genet. 1999, 64, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, A.K.; Goldstein, J.A.; Kelley, M.J.; Luxford, W.; Castelein, C.M.; Mhatre, A.N. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am. J. Hum. Genet. 2000, 67, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Heath, K.E.; Campos-Barros, A.; Toren, A.; Rozenfeld-Granot, G.; Carlsson, L.E.; Savige, J.; Denison, J.C.; Gregory, M.C.; White, J.G.; Barker, D.F.; et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am. J. Hum. Genet. 2001, 69, 1033–1045. [Google Scholar] [CrossRef]
- Balduini, C.L.; Pecci, A.; Savoia, A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br. J. Haematol. 2011, 154, 161–174. [Google Scholar] [CrossRef]
- Leon, C.; Eckly, A.; Hechler, B.; Aleil, B.; Freund, M.; Ravanat, C.; Jourdain, M.; Nonne, C.; Weber, J.; Tiedt, R.; et al. Megakaryocyte-restricted MYH9 inactivation dramatically affects hemostasis while preserving platelet aggregation and secretion. Blood 2007, 110, 3183–3191. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, T.; Belevich, I.; Laos, M.; Herranen, A.; Jokitalo, E.; Brakebusch, C.; Pirvola, U. Cytoskeletal Stability in the Auditory Organ In Vivo: RhoA Is Dispensable for Wound Healing but Essential for Hair Cell Development. eNeuro 2017, 4, ENEURO-0149. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wu, Z.; Muller, U. Murine Fam65b forms ring-like structures at the base of stereocilia critical for mechanosensory hair cell function. Elife 2016, 5, e14222. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Decker, A.R.; Booth, K.T.; Simpson, A.C.; Shearer, A.E.; Huygen, P.L.; Bu, F.; Hildebrand, M.S.; Ranum, P.T.; Shibata, S.B.; et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet. 2015, 11, e1005137. [Google Scholar] [CrossRef]
- Kutsche, K.; Yntema, H.; Brandt, A.; Jantke, I.; Nothwang, H.G.; Orth, U.; Boavida, M.G.; David, D.; Chelly, J.; Fryns, J.P.; et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 2000, 26, 247–250. [Google Scholar] [CrossRef]
- Zhu, C.; Cheng, C.; Wang, Y.; Muhammad, W.; Liu, S.; Zhu, W.; Shao, B.; Zhang, Z.; Yan, X.; He, Q.; et al. Loss of ARHGEF6 Causes Hair Cell Stereocilia Deficits and Hearing Loss in Mice. Front. Mol. Neurosci 2018, 11, 362. [Google Scholar] [CrossRef]
- Khanna, H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 2015, 4, 674–686. [Google Scholar] [CrossRef]
- Bokoch, G.M.; Diebold, B.A. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 2002, 100, 2692–2696. [Google Scholar] [CrossRef]
- Ueyama, T.; Lennartz, M.R.; Noda, Y.; Kobayashi, T.; Shirai, Y.; Rikitake, K.; Yamasaki, T.; Hayashi, S.; Sakai, N.; Seguchi, H.; et al. Superoxide production at phagosomal cup/phagosome through βI protein kinase C during FcγR-mediated phagocytosis in microglia. J. Immunol. 2004, 173, 4582–4589. [Google Scholar] [CrossRef]
- Ueyama, T.; Eto, M.; Kami, K.; Tatsuno, T.; Kobayashi, T.; Shirai, Y.; Lennartz, M.R.; Takeya, R.; Sumimoto, H.; Saito, N. Isoform-specific membrane targeting mechanism of Rac during FcγR-mediated phagocytosis: Positive charge-dependent and independent targeting mechanism of Rac to the phagosome. J. Immunol. 2005, 175, 2381–2390. [Google Scholar] [CrossRef]
- Ueyama, T.; Tatsuno, T.; Kawasaki, T.; Tsujibe, S.; Shirai, Y.; Sumimoto, H.; Leto, T.L.; Saito, N. A regulated adaptor function of p40phox: Distinct p67phox membrane targeting by p40phox and by p47phox. Mol. Biol. Cell 2007, 18, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Kusakabe, T.; Karasawa, S.; Kawasaki, T.; Shimizu, A.; Son, J.; Leto, T.L.; Miyawaki, A.; Saito, N. Sequential binding of cytosolic Phox complex to phagosomes through regulated adaptor proteins: Evaluation using the novel monomeric Kusabira-Green System and live imaging of phagocytosis. J. Immunol. 2008, 181, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and Regulation of Reactive Oxygen Species Generation by Nox Family NADPH Oxidases. Antioxid. Redox. Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Nakakita, J.; Nakamura, T.; Kobayashi, T.; Son, J.; Sakuma, M.; Sakaguchi, H.; Leto, T.L.; Saito, N. Cooperation of p40phox with p47phox for Nox2-based NADPH oxidase activation during FcγR-mediated phagocytosis: Mechanism for acquisition of p40phox PI(3)P binding. J. Biol. Chem. 2011, 286, 40693–40705. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Son, J.; Kobayashi, T.; Hamada, T.; Nakamura, T.; Sakaguchi, H.; Shirafuji, T.; Saito, N. Negative Charges in the Flexible N-Terminal Domain of Rho GDP-Dissociation Inhibitors (RhoGDIs) Regulate the Targeting of the RhoGDI-Rac1 Complex to Membranes. J. Immunol. 2013, 191, 2560–2569. [Google Scholar] [CrossRef]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Bustamante, J.; Kannengiesser, C.; de Boer, M.; van Leeuwen, K.; Koker, M.Y.; Wolach, B.; Roesler, J.; et al. Hematologically important mutations: The autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol. Dis. 2010, 44, 291–299. [Google Scholar] [CrossRef]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Roesler, J.; Lopez, J.A.; Ariga, T.; Avcin, T.; de Boer, M.; Bustamante, J.; Condino-Neto, A.; et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol. Dis. 2010, 45, 246–265. [Google Scholar] [CrossRef]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Dinauer, M.C. Regulation of neutrophil function by Rac GTPases. Curr. Opin. Hematol. 2003, 10, 8–15. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G.; Heyworth, P.G.; Evans, T.; Curnutte, J.T.; Bokoch, G.M. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 1991, 254, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Kim, C.; Zhen, L.; Lowe, J.B.; Kapur, R.; Petryniak, B.; Spaetti, A.; Pollock, J.D.; Borneo, J.B.; Bradford, G.B.; et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity 1999, 10, 183–196. [Google Scholar] [CrossRef]
- Glogauer, M.; Marchal, C.C.; Zhu, F.; Worku, A.; Clausen, B.E.; Foerster, I.; Marks, P.; Downey, G.P.; Dinauer, M.; Kwiatkowski, D.J. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J. Immunol. 2003, 170, 5652–5657. [Google Scholar] [CrossRef]
- Gu, Y.; Filippi, M.D.; Cancelas, J.A.; Siefring, J.E.; Williams, E.P.; Jasti, A.C.; Harris, C.E.; Lee, A.W.; Prabhakar, R.; Atkinson, S.J.; et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 2003, 302, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Tarlinton, D.M.; Cluse, L.A.; Tuxen, A.J.; Light, A.; Yang, F.C.; Williams, D.A.; Roberts, A.W. The Rac2 guanosine triphosphatase regulates B lymphocyte antigen receptor responses and chemotaxis and is required for establishment of B-1a and marginal zone B lymphocytes. J. Immunol. 2002, 168, 3376–3386. [Google Scholar] [CrossRef]
- Hoppe, A.D.; Swanson, J.A. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol. Biol. Cell 2004, 15, 3509–3519. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, J.; Bu, X.; Cushion, M.; Kinane, T.B.; Avraham, H.; Koziel, H. Cdc42 and RhoB activation are required for mannose receptor-mediated phagocytosis by human alveolar macrophages. Mol. Biol. Cell 2005, 16, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Egami, Y.; Kawai, K.; Araki, N. RhoC regulates the actin remodeling required for phagosome formation during FcgammaR-mediated phagocytosis. J. Cell Sci. 2017, 130, 4168–4179. [Google Scholar] [CrossRef]
- Condliffe, A.M.; Webb, L.M.; Ferguson, G.J.; Davidson, K.; Turner, M.; Vigorito, E.; Manifava, M.; Chilvers, E.R.; Stephens, L.R.; Hawkins, P.T. RhoG regulates the neutrophil NADPH oxidase. J. Immunol. 2006, 176, 5314–5320. [Google Scholar] [CrossRef] [PubMed]
- Damoulakis, G.; Gambardella, L.; Rossman, K.L.; Lawson, C.D.; Anderson, K.E.; Fukui, Y.; Welch, H.C.; Der, C.J.; Stephens, L.R.; Hawkins, P.T. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J. Cell Sci. 2014, 127, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Heyworth, P.; Bohl, B.; Bokoch, G.; Curnutte, J. Rac translocates independently of the neutrophil NADPH oxidase components p47phox and p67phox. J. Biol. Chem. 1994, 269, 30749–30752. [Google Scholar] [PubMed]
- Kim, C.; Dinauer, M.C. Impaired NADPH oxidase activity in Rac2-deficient murine neutrophils does not result from defective translocation of p47phox and p67phox and can be rescued by exogenous arachidonic acid. J. Leukoc. Biol. 2006, 79, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Chessa, T.A.; Davidson, K.; Henderson, R.B.; Walker, S.; Tolmachova, T.; Grys, K.; Rausch, O.; Seabra, M.C.; Tybulewicz, V.L.; et al. PtdIns3P and Rac direct the assembly of the NADPH oxidase on a novel, pre-phagosomal compartment during FcR-mediated phagocytosis in primary mouse neutrophils. Blood 2010, 116, 4978–4989. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El-Hassani, R.; Morand, S.; Boucher, J.L.; Frapart, Y.M.; Apostolou, D.; Agnandji, D.; Gnidehou, S.; Ohayon, R.; Noel-Hudson, M.S.; Francon, J.; et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 2005, 280, 30046–30054. [Google Scholar] [CrossRef]
- Morand, S.; Ueyama, T.; Tsujibe, S.; Saito, N.; Korzeniowska, A.; Leto, T.L. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. Faseb J. 2009, 23, 1205–1218. [Google Scholar] [CrossRef]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Hoste, C.; Dumont, J.E.; Miot, F.; De Deken, X. The type of DUOX-dependent ROS production is dictated by defined sequences in DUOXA. Exp. Cell Res. 2012, 318, 2353–2364. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef]
- Ueyama, T.; Sakuma, M.; Ninoyu, Y.; Hamada, T.; Dupuy, C.; Geiszt, M.; Leto, T.L.; Saito, N. The extracellular A-loop of dual oxidases affects the specificity of reactive oxygen species release. J. Biol. Chem. 2015, 290, 6495–6506. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Geiszt, M.; Leto, T.L. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol. Cell Biol. 2006, 26, 2160–2174. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sumimoto, H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie 2007, 89, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef]
- Miyano, K.; Ueno, N.; Takeya, R.; Sumimoto, H. Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J. Biol. Chem. 2006, 281, 21857–21868. [Google Scholar] [CrossRef]
- Wu, W.; Hsu, Y.M.; Bi, L.; Songyang, Z.; Lin, X. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat. Immunol. 2009, 10, 1208–1214. [Google Scholar] [CrossRef]
- Pick, E. Role of the Rho GTPase Rac in the activation of the phagocyte NADPH oxidase: Outsourcing a key task. Small GTPases 2014, 5, e27952. [Google Scholar] [CrossRef]
- Ugolev, Y.; Berdichevsky, Y.; Weinbaum, C.; Pick, E. Dissociation of Rac1(GDP).RhoGDI complexes by the cooperative action of anionic liposomes containing phosphatidylinositol 3,4,5-trisphosphate, Rac guanine nucleotide exchange factor, and GTP. J. Biol. Chem. 2008, 283, 22257–22271. [Google Scholar] [CrossRef]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Dovas, A.; Choi, Y.; Yoneda, A.; Multhaupt, H.A.; Kwon, S.H.; Kang, D.; Oh, E.S.; Couchman, J.R. Serine 34 phosphorylation of rho guanine dissociation inhibitor (RhoGDIa) links signaling from conventional protein kinase C to RhoGTPase in cell adhesion. J. Biol. Chem. 2010, 285, 23296–23308. [Google Scholar] [CrossRef]
- DerMardirossian, C.; Schnelzer, A.; Bokoch, G.M. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol. Cell 2004, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Rocklin, G.; Seo, J.Y.; Bokoch, G.M. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol. Biol. Cell. 2006, 17, 4760–4768. [Google Scholar] [CrossRef] [PubMed]
- Mack, N.A.; Whalley, H.J.; Castillo-Lluva, S.; Malliri, A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011, 10, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Yukinaga, H.; Kamioka, Y.; Arakawa, Y.; Miyamoto, S.; Okada, T.; Sahai, E.; Matsuda, M. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J. Cell Sci. 2012, 125, 858–868. [Google Scholar] [CrossRef]
- Yukinaga, H.; Shionyu, C.; Hirata, E.; Ui-Tei, K.; Nagashima, T.; Kondo, S.; Okada-Hatakeyama, M.; Naoki, H.; Matsuda, M. Fluctuation of Rac1 activity is associated with the phenotypic and transcriptional heterogeneity of glioma cells. J. Cell Sci. 2014, 127, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Lehnert, H. The role of small GTPases of the Rho/Rac family in TGF-beta-induced EMT and cell motility in cancer. Dev. Dyn. 2018, 247, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Licciulli, S.; Avila, J.L.; Cho, M.; Troutman, S.; Jiang, P.; Kossenkov, A.V.; Showe, L.C.; Liu, Q.; Vachani, A.; et al. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene 2013, 32, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, L.; Walker, F.; Mamadou, G.; Lehy, T.; Jordan, P.; Chastre, E. The Rac1 splice form Rac1b favors mouse colonic mucosa regeneration and contributes to intestinal cancer progression. Oncogene 2018, 37, 6054–6058. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Ueyama, T.; Shigyo, M.; Kohta, M.; Kondoh, T.; Kuboyama, T.; Uebi, T.; Hamada, T.; Gutmann, D.H.; Aiba, A.; et al. A Novel Rac1-GSPT1 Signaling Pathway Controls Astrogliosis Following Central Nervous System Injury. J. Biol. Chem. 2016, 292, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Funakoshi, Y.; Hoshino, S.; Katada, T. The GTP-binding release factor eRF3 as a key mediator coupling translation termination to mRNA decay. J. Biol. Chem. 2004, 279, 45693–45700. [Google Scholar] [CrossRef]
- Hellen, C.U.T. Translation Termination and Ribosome Recycling in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Zhang, Y.; Rivera Rosado, L.A.; Chen, J.; Khan, T.; Moon, S.Y.; Zhang, B. Blockade of Rac1 activity induces G1 cell cycle arrest or apoptosis in breast cancer cells through downregulation of cyclin D1, survivin, and X-linked inhibitor of apoptosis protein. Mol. Cancer Ther. 2010, 9, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, H.; Shi, L.; Zhang, W.; Yuan, J.; Chen, X.; Liu, J.; Zhang, Y.; Wang, Z. Inhibition of Rac1 activity induces G1/S phase arrest through the GSK3/cyclin D1 pathway in human cancer cells. Oncol. Rep. 2014, 32, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Holroyd, A.; Lloyd, A.; Broderick, P.; Nsengimana, J.; Eeles, R.; Easton, D.F.; Dudakia, D.; Bishop, D.T.; Reid, A.; et al. Identification of four new susceptibility loci for testicular germ cell tumour. Nat. Commun. 2015, 6, 8690. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Li, C.; Chai, B. miRNA-144 suppresses proliferation and migration of colorectal cancer cells through GSPT1. Biomed. Pharmacother 2015, 74, 138–144. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.C.; Miller, K.; Fang, W.; Wang, N.Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Bora-Singhal, N.; Perumal, D.; Chellappan, S. Nicotine-mediated invasion and migration of non-small cell lung carcinoma cells by modulating STMN3 and GSPT1 genes in an ID1-dependent manner. Mol. Cancer 2014, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Jerbi, S.; Jolles, B.; Bouceba, T.; Jean-Jean, O. Studies on human eRF3-PABP interaction reveal the influence of eRF3a N-terminal glycin repeat on eRF3-PABP binding affinity and the lower affinity of eRF3a 12-GGC allele involved in cancer susceptibility. RNA Biol. 2016, 13, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Brito, M.; Malta-Vacas, J.; Carmona, B.; Aires, C.; Costa, P.; Martins, A.P.; Ramos, S.; Conde, A.R.; Monteiro, C. Polyglycine expansions in eRF3/GSPT1 are associated with gastric cancer susceptibility. Carcinogenesis 2005, 26, 2046–2049. [Google Scholar] [CrossRef]
- Malta-Vacas, J.; Chauvin, C.; Goncalves, L.; Nazare, A.; Carvalho, C.; Monteiro, C.; Bagrel, D.; Jean-Jean, O.; Brito, M. eRF3a/GSPT1 12-GGC allele increases the susceptibility for breast cancer development. Oncol. Rep. 2009, 21, 1551–1558. [Google Scholar] [PubMed]
- Miri, M.; Hemati, S.; Safari, F.; Tavassoli, M. GGCn polymorphism of eRF3a/GSPT1 gene and breast cancer susceptibility. Med. Oncol. 2012, 29, 1581–1585. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Enami, T.; Ogawa, S.; Sakata-Yanagimoto, M. G17V RHOA: Genetic evidence of GTP-unbound RHOA playing a role in tumorigenesis in T cells. Small GTPases 2015, 6, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Boddicker, R.L.; Razidlo, G.L.; Dasari, S.; Zeng, Y.; Hu, G.; Knudson, R.A.; Greipp, P.T.; Davila, J.I.; Johnson, S.H.; Porcher, J.C.; et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood 2016, 128, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; da Silva-Almeida, A.C.; Zairis, S.; Robles-Valero, J.; Couronne, L.; Khiabanian, H.; Quinn, S.A.; Kim, M.Y.; Laginestra, M.A.; Kim, C.; et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc. Natl. Acad Sci. USA 2017, 114, 764–769. [Google Scholar] [CrossRef]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; de Sa, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016, 53, 190–199. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name (Synonym) | KO Mice Availability | Congenital Diseases | Tumorigenesis |
|---|---|---|---|
| Rho Subfamily | |||
| RhoA | + [7] | lymphomas [8,9,10,11], gastric cancer [12,13], head & neck squamous cell carcinoma [14,15] | |
| RhoB | +C [16] | induction [17] and suppression [17,18] | |
| RhoC | +C [19] | reported [14] | |
| Rac Subfamily | |||
| Rac1 | + | + (CNS anomalies) [20] | melanoma [21,22], head & neck squamous cell carcinoma [14,15] |
| Rac2 | +C | + (HD deficiency) [23,24,25,26] | reported [14,27] |
| Rac3 | +C | reported [14] | |
| RhoG | +C [28] | ||
| Cdc42 Subfamily | |||
| Cdc42 | + | + (TKS) [29,30,31,32] | reported [14] |
| RhoJ (TCL) | + [33] | ||
| RhoQ (TC10) | + [34] | ||
| RhoD/RhoF Subfamily | |||
| RhoD | |||
| RhoF (Rif) | + [35] | ||
| Rnd Subfamily | |||
| Rnd1 | |||
| Rnd2 (RhoN) | |||
| Rnd3 (RhoE) | |||
| RhoH Subfamily | |||
| RhoH | +C [36] | lymphoma [37,38] | |
| RhoU/RhoV Subfamily | |||
| RhoU | |||
| RhoV | |||
| RhoBTB Subfamily | |||
| RhoBTB1 | |||
| RhoBTB2 | + (CNS anomalies) [39] | suppression [5] | |
| RhoBTB3 | +C [40] | ||
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueyama, T. Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells. Cells 2019, 8, 92. https://doi.org/10.3390/cells8020092
Ueyama T. Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells. Cells. 2019; 8(2):92. https://doi.org/10.3390/cells8020092
Chicago/Turabian StyleUeyama, Takehiko. 2019. "Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells" Cells 8, no. 2: 92. https://doi.org/10.3390/cells8020092
APA StyleUeyama, T. (2019). Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells. Cells, 8(2), 92. https://doi.org/10.3390/cells8020092