RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Antibodies and Reagents
2.3. Cell Transfection and Small Interfering RNA
2.4. Immunoblotting
2.5. Pull Down Assays
2.6. The Tube Formation Assay
2.7. Statistical Analysis
3. Results
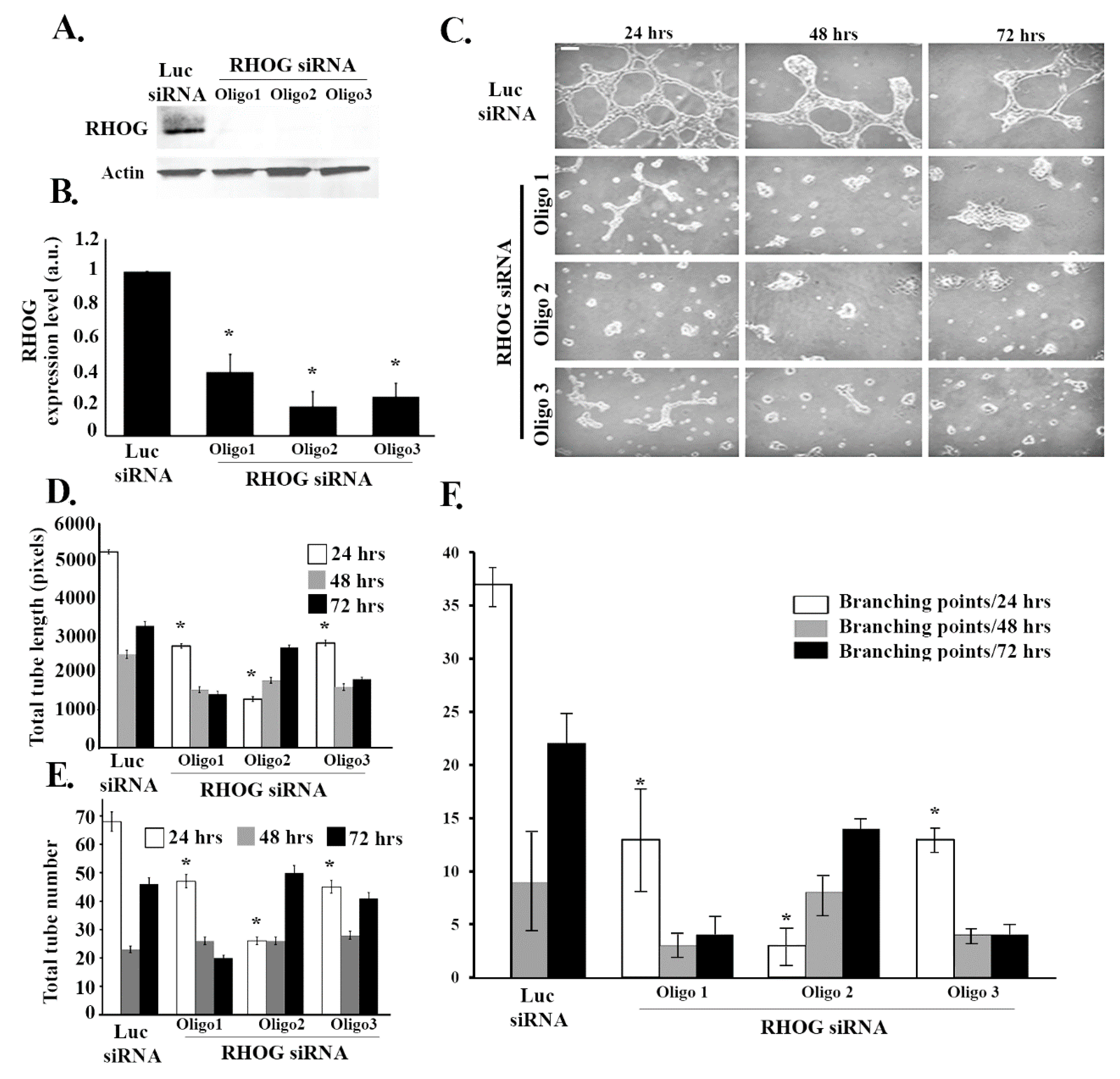
3.1. RHOG Positively Regulates Tube Formation in ECV Cells
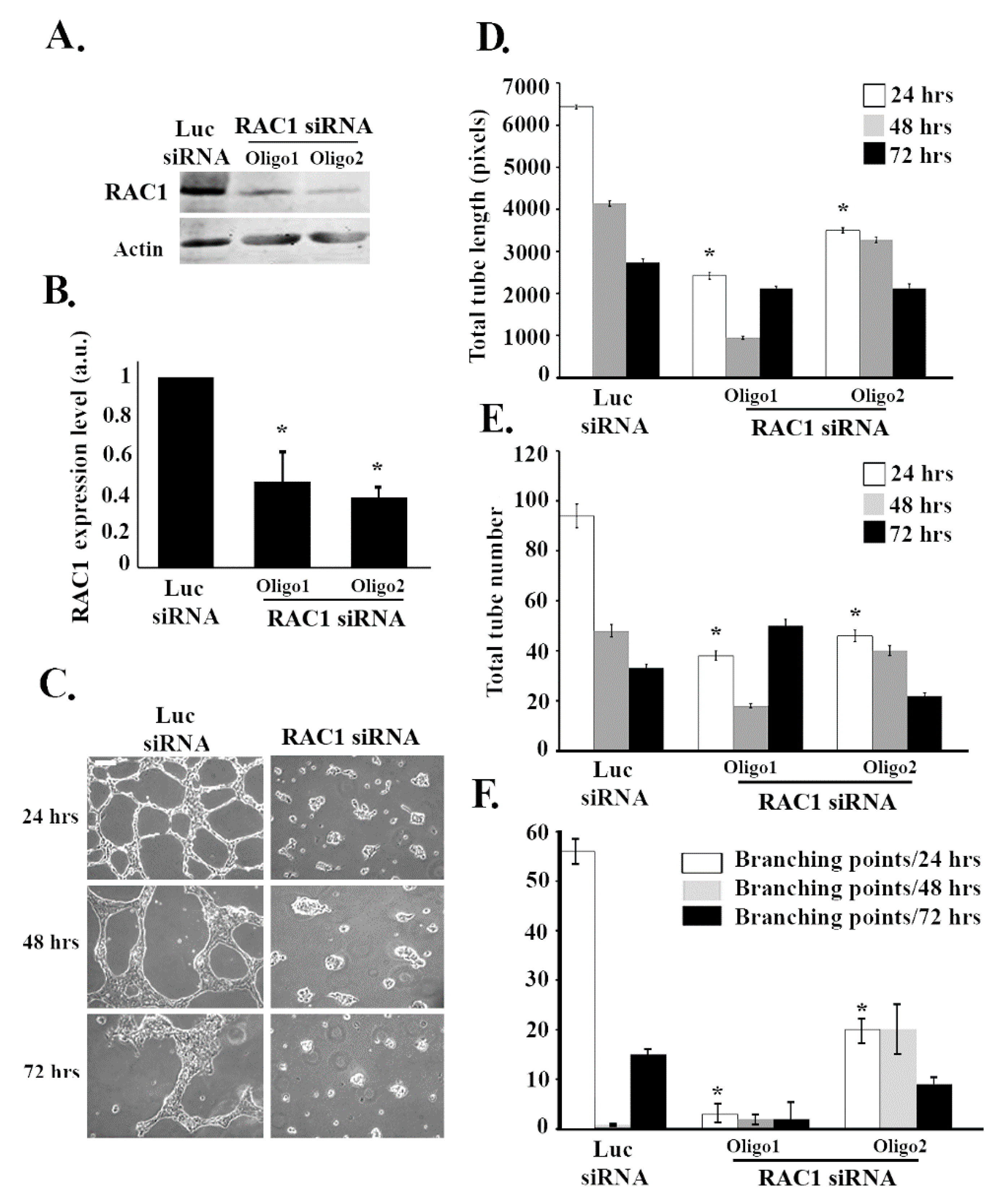
3.2. RAC1 Positively Regulates Tube Formation in ECV Cells
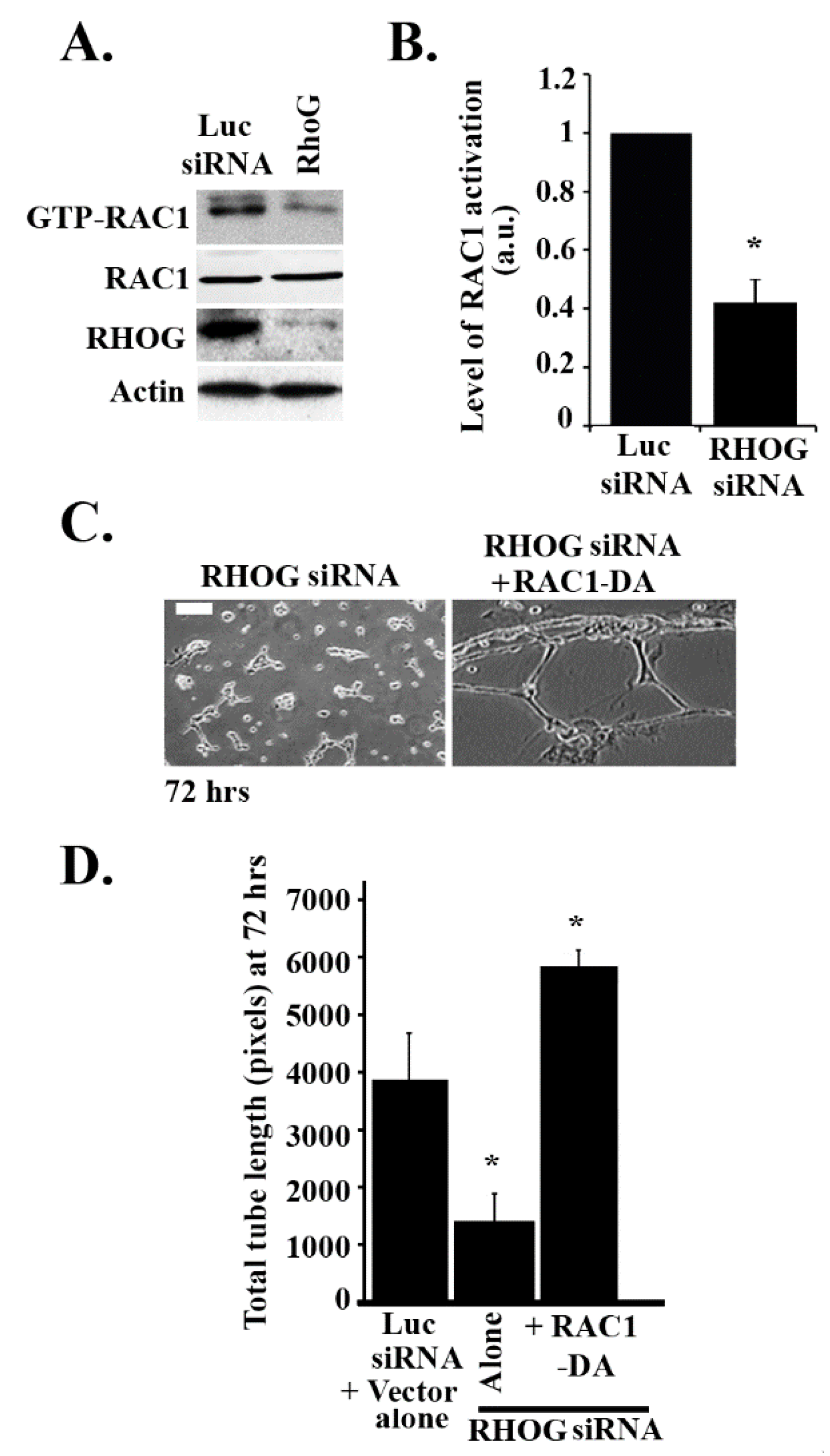
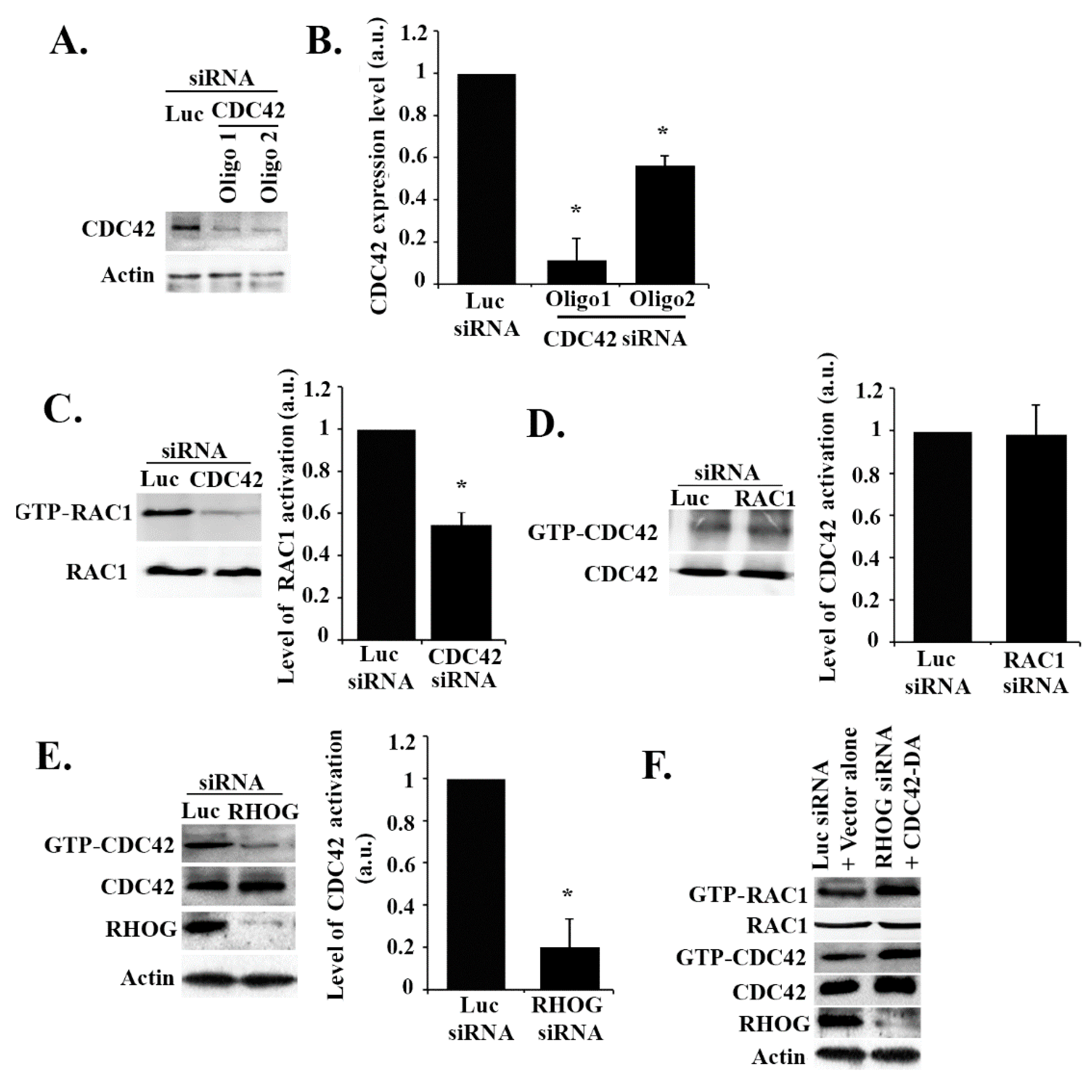
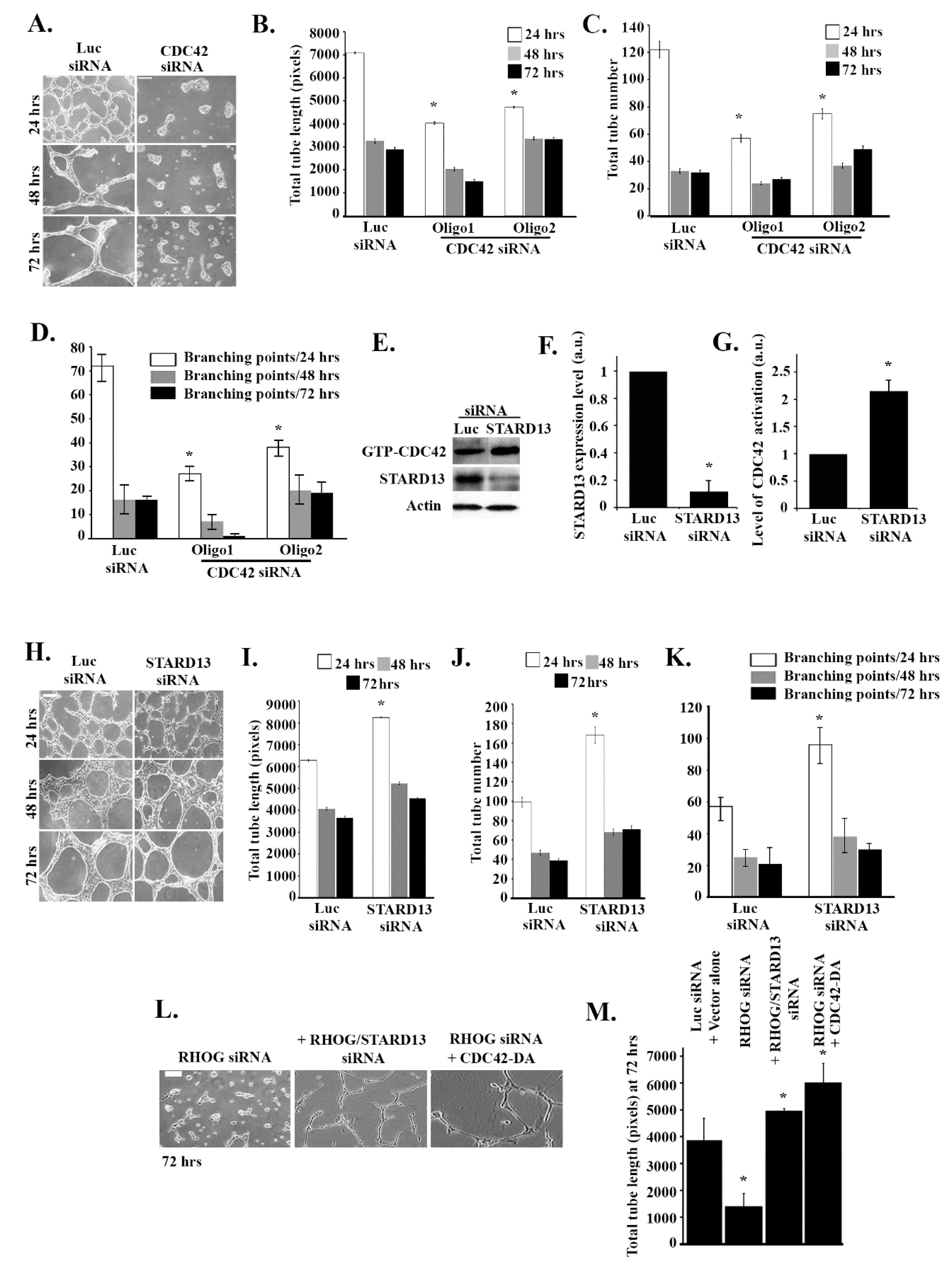
3.3. RHOG Activates RAC1 through the Activation of CDC42 in ECV Cells
3.4. RHOG Stimulates Tube Formation through CDC42 and RAC1
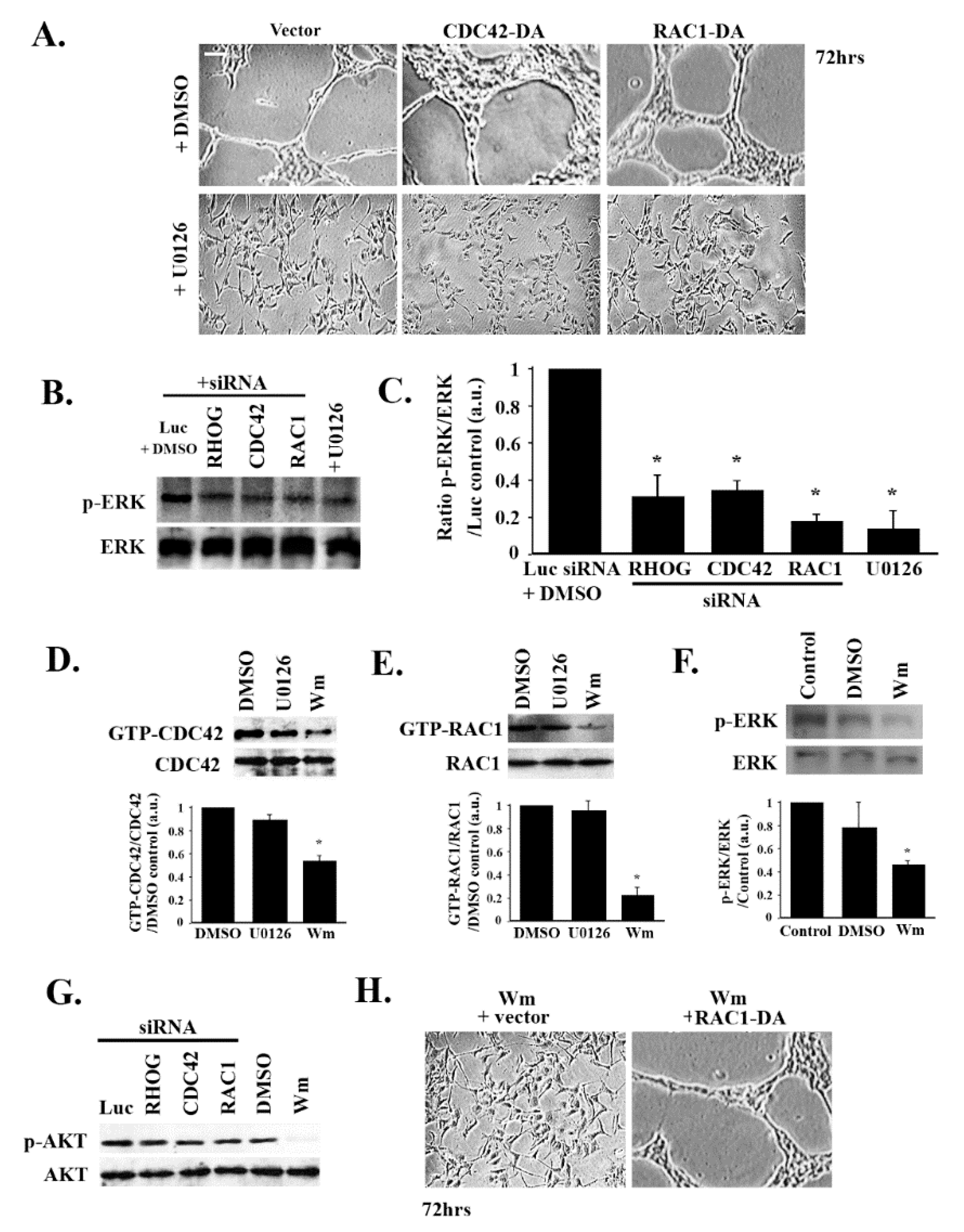
3.5. RHOG/CDC42/RAC1 Leads to Tube Formation through the ERK Pathway
3.6. RHOG/CDC42/RAC1 Leads to Tube Formation Downstream from the PI3K Pathway
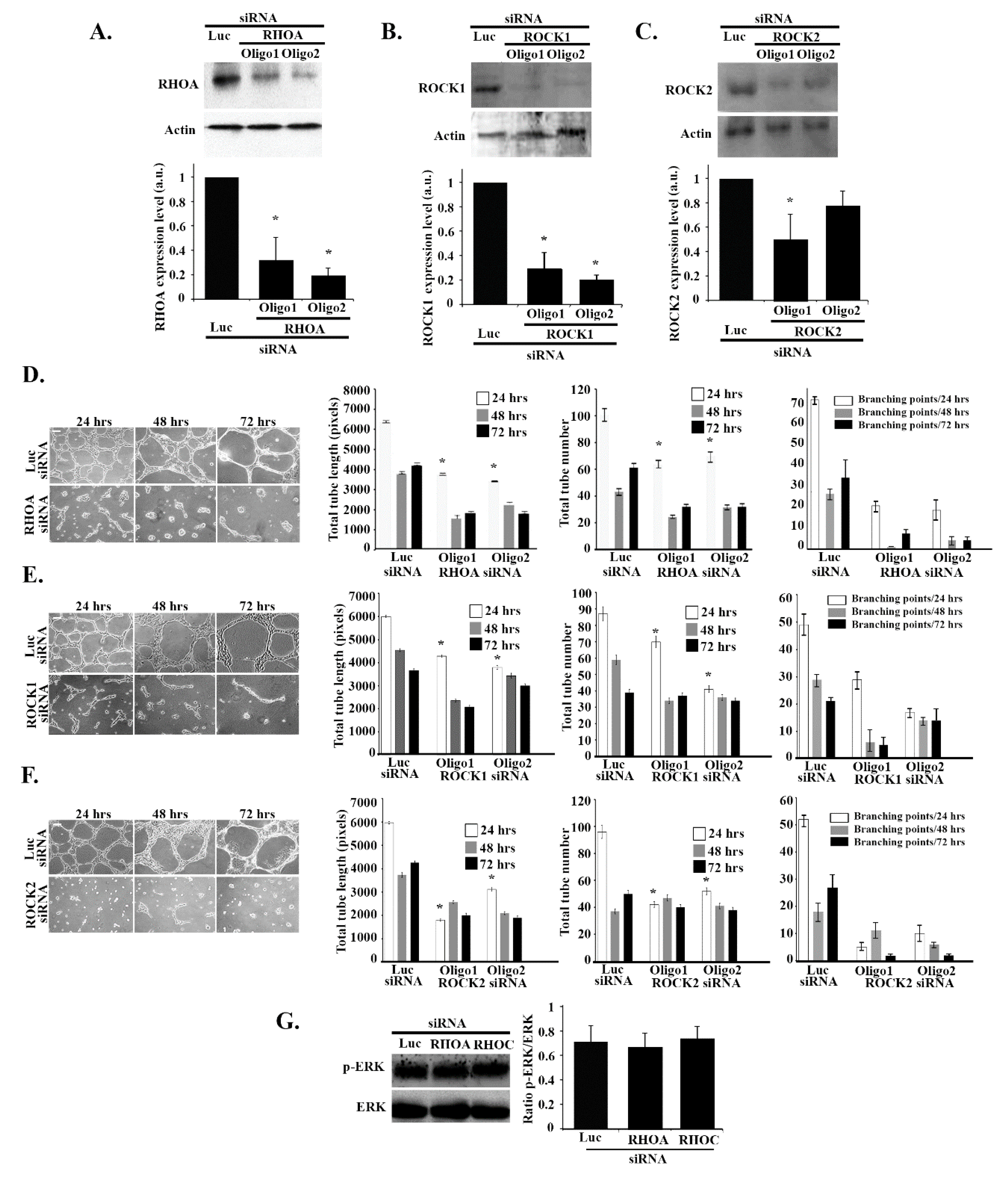
3.7. RHOA/ROCK1/ROCK2 Regulate Tube Formation in an ERK-Independent Manner
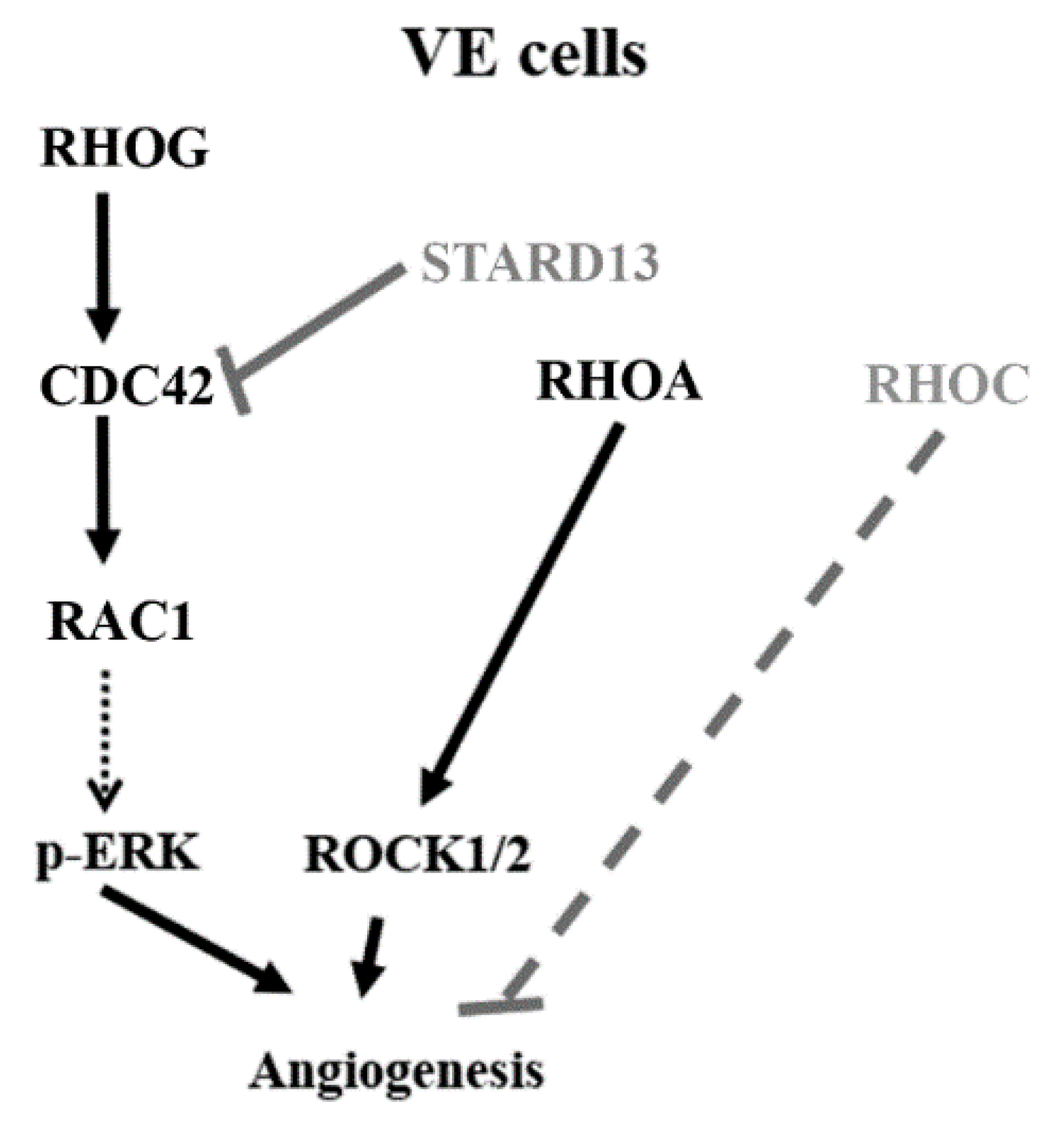
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xu, G.; Zhou, J.; Xing, H.; Wang, S.; Wu, M.; Lu, Y.P.; Ma, D. The effect of RhoA on human umbilical vein endothelial cell migration and angiogenesis in vitro. Oncol. Rep. 2006, 15, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 1998, 273, 13313–13316. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Nieves, B.J.; D’Amore, P.A.; Bryan, B.A. The function of vascular endothelial growth factor. Biofactors 2009, 35, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, F.; Fang, F.; Tao, Y.; Yang, L. RhoC is essential for angiogenesis induced by hepatocellular carcinoma cells via regulation of endothelial cell organization. Cancer Sci. 2008, 99, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; D’Amore, P.A. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell Mol. Life Sci. 2007, 64, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.P.; Liao, J.K.; D’Amore, P.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348 Pt 2, 241–255. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac and cdc42 GTPases: Regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 1995, 23, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Sharma, V.P.; Roh-Johnson, M.; Chen, X.; Eddy, R.; Condeelis, J.; Hodgson, L. Spatial regulation of RhoC activity defines protrusion formation in migrating cells. J. Cell Sci. 2013, 126, 3356–3369. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.C.; El-Sibai, M.; Coniglio, S.J.; Mouneimne, G.; Eddy, R.J.; Drees, B.E.; Neilsen, P.O.; Goswami, S.; Symons, M.; Condeelis, J.S.; et al. The distinct roles of Ras and Rac in PI 3-kinase-dependent protrusion during EGF-stimulated cell migration. J. Cell. Sci. 2007, 120, 3138–3146. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Hotchin, N.A.; Hall, A. Regulation of the actin cytoskeleton, integrins and cell growth by the Rho family of small GTPases. Cancer Surv. 1996, 27, 311–322. [Google Scholar] [PubMed]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Holinstat, M.; Knezevic, N.; Broman, M.; Samarel, A.M.; Malik, A.B.; Mehta, D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: Role in regulation of endothelial permeability. J. Biol. Chem. 2006, 281, 2296–2305. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Burridge, K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 2009, 284, 25602–25611. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; van der Meel, R.; Symons, M.H.; Fens, M.H.; Pieters, E.; Wilschut, K.J.; Storm, G.; Jarzabek, M.; Gallagher, W.M.; Schiffelers, R.M.; et al. Examining the role of Rac1 in tumor angiogenesis and growth: A clinically relevant RNAi-mediated approach. Angiogenesis 2011, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- van der Meel, R.; Symons, M.H.; Kudernatsch, R.; Kok, R.J.; Schiffelers, R.M.; Storm, G.; Gallagher, W.M.; Byrne, A.T. The VEGF/Rho GTPase signalling pathway: A promising target for anti-angiogenic/anti-invasion therapy. Drug Discov. Today 2011, 16, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Fryer, B.H.; Field, J. Rho, Rac, Pak and angiogenesis: Old roles and newly identified responsibilities in endothelial cells. Cancer Lett. 2005, 229, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, E.; Billadeu, D.D.; Savoy, D.; McAdam, S.; Doody, G.; Fort, P.; Turner, M. RhoG regulates gene expression and the actin cytoskeleton in lymphocytes. Oncogene 2003, 22, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Ellerbroek, S.M.; Liu, R.Y.; Karnoub, A.E.; Burridge, K.; Der, C.J. RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 2002, 277, 47810–47817. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Rouviere, C.; Vignal, E.; Meriane, M.; Roux, P.; Montcourier, P.; Fort, P. RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol. Biol. Cell 1998, 9, 1379–1394. [Google Scholar] [CrossRef] [PubMed]
- van Buul, J.D.; Allingham, M.J.; Samson, T.; Meller, J.; Boulter, E.; Garcia-Mata, R.; Burridge, K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J. Cell Biol. 2007, 178, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Hiramoto-Yamaki, N.; Takeuchi, S.; Ueda, S.; Harada, K.; Fujimoto, S.; Negishi, M.; Katoh, H. Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J. Cell Biol. 2010, 190, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Yasui, H.; Yamaguchi, Y.; Aoki, J.; Fujita, H.; Mori, K.; Negishi, M. Small GTPase RhoG is a key regulator for neurite outgrowth in PC12 cells. Mol. Cell Biol. 2000, 20, 7378–7387. [Google Scholar] [CrossRef] [PubMed]
- deBakker, C.D.; Haney, L.B.; Kinchen, J.M.; Grimsley, C.; Lu, M.; Klingele, D.; Hsu, P.K.; Chou, B.K.; Cheng, L.C.; Blangy, A.; et al. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr. Biol. 2004, 14, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Didier, S.; Fortin, S.; Chuang, Y.; White, T.; Berens, M.E.; Rushing, E.; Eschbacher, J.; Tran, N.L.; Chan, A.; et al. The small GTPase RhoG mediates glioblastoma cell invasion. Mol. Cancer 2012, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Hiramoto, K.; Negishi, M. Activation of Rac1 by RhoG regulates cell migration. J. Cell Sci. 2006, 119, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Scarcia, M.; Bagshaw, R.D.; McMahon, K.; Grant, G.; Harvey, T.; Yeo, M.; Esteves, F.O.; Thygesen, H.H.; Jones, P.F.; et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat. Commun. 2015, 6, 7286. [Google Scholar] [CrossRef] [PubMed]
- Boettner, B.; Van Aelst, L. The role of Rho GTPases in disease development. Gene 2002, 286, 155–174. [Google Scholar] [CrossRef]
- Bosse, T.; Ehinger, J.; Czuchra, A.; Benesch, S.; Steffen, A.; Wu, X.; Schloen, K.; Niemann, H.H.; Scita, G.; Stradal, T.E.; et al. Cdc42 and phosphoinositide 3-kinase drive Rac-mediated actin polymerization downstream of c-Met in distinct and common pathways. Mol. Cell Biol. 2007, 27, 6615–6628. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Meza, H.; Castillo-Alvarez, A.; Gonzalez-Bonilla, C.; Meza, I. Cross-talk between Rac1 and Cdc42 GTPases regulates formation of filopodia required for dengue virus type-2 entry into HMEC-1 cells. J. Gen. Virol. 2009, 90, 2902–2911. [Google Scholar] [CrossRef] [PubMed]
- El-Sitt, S.; El-Sibai, M. The STAR of the DLC family. J. Recept. Signal Transduct. Res. 2013, 33, 10–13. [Google Scholar] [CrossRef] [PubMed]
- El-Sitt, S.; Khalil, B.D.; Hanna, S.; El-Sabban, M.; Fakhreddine, N.; El-Sibai, M. DLC2/StarD13 plays a role of a tumor suppressor in astrocytoma. Oncol. Rep. 2012, 28, 511–518. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanna, S.; Khalil, B.; Nasrallah, A.; Saykali, B.A.; Sobh, R.; Nasser, S.; El-Sibai, M. StarD13 is a tumor suppressor in breast cancer that regulates cell motility and invasion. Int. J. Oncol. 2014, 44, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.D.; Hanna, S.; Saykali, B.A.; El-Sitt, S.; Nasrallah, A.; Marston, D.; El-Sabban, M.; Hahn, K.M.; Symons, M.; El-Sibai, M. The regulation of RhoA at focal adhesions by StarD13 is important for astrocytoma cell motility. Exp. Cell Res. 2014, 321, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Otto, W.; Johannes, S.; Baumgart, J.; Nitsch, R.; Schumacher, S. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J. 2012, 31, 2908–2921. [Google Scholar] [CrossRef] [PubMed]
- Schuebel, K.E.; Movilla, N.; Rosa, J.L.; Bustelo, X.R. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 1998, 17, 6608–6621. [Google Scholar] [CrossRef] [PubMed]
- Movilla, N.; Bustelo, X.R. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell Biol. 1999, 19, 7870–7885. [Google Scholar] [CrossRef] [PubMed]
- El-Sibai, M.; Pertz, O.; Pang, H.; Yip, S.C.; Lorenz, M.; Symons, M.; Condeelis, J.S.; Hahn, K.M.; Backer, J.M. RhoA/ROCK-mediated switching between Cdc42- and Rac1-dependent protrusion in MTLn3 carcinoma cells. Exp. Cell Res. 2008, 314, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.R. Coupling receptor tyrosine kinases to Rho GTPases--GEFs what’s the link. Cell. Signal. 2006, 18, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.M.; Gurung, R.; Le, K.Q.; Mitchell, C.A. Effective angiogenesis requires regulation of phosphoinositide signaling. Adv. Biol. Regul. 2018. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Atat, O.; Fakih, A.; El-Sibai, M. RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells. Cells 2019, 8, 171. https://doi.org/10.3390/cells8020171
El Atat O, Fakih A, El-Sibai M. RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells. Cells. 2019; 8(2):171. https://doi.org/10.3390/cells8020171
Chicago/Turabian StyleEl Atat, Oula, Amira Fakih, and Mirvat El-Sibai. 2019. "RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells" Cells 8, no. 2: 171. https://doi.org/10.3390/cells8020171
APA StyleEl Atat, O., Fakih, A., & El-Sibai, M. (2019). RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells. Cells, 8(2), 171. https://doi.org/10.3390/cells8020171