CVm6A: A Visualization and Exploration Database for m6As in Cell Lines

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Data Collection and Database Content
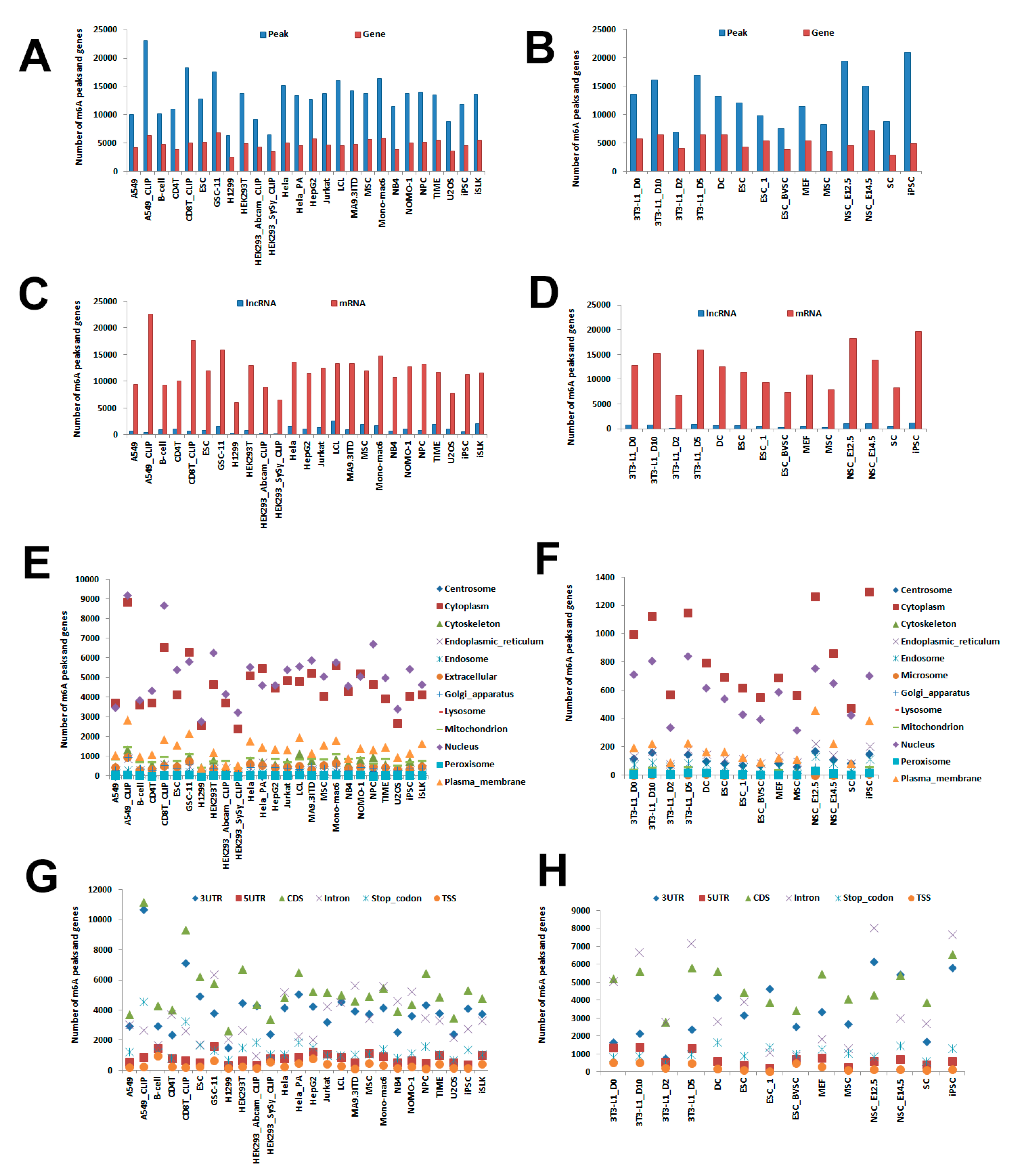
2.1. Cell Line Samples in CVm6A
2.2. Identification of Cell m6A Peaks
2.3. Prediction of m6A lncRNA and mRNA
2.4. Prediction of Subcellular Location
2.5. Prediction of Gene Regions
2.6. Classification of Cancer and Non-Cancer m6A
2.7. Validated m6A Sites
3. Database Organization and Web Interface
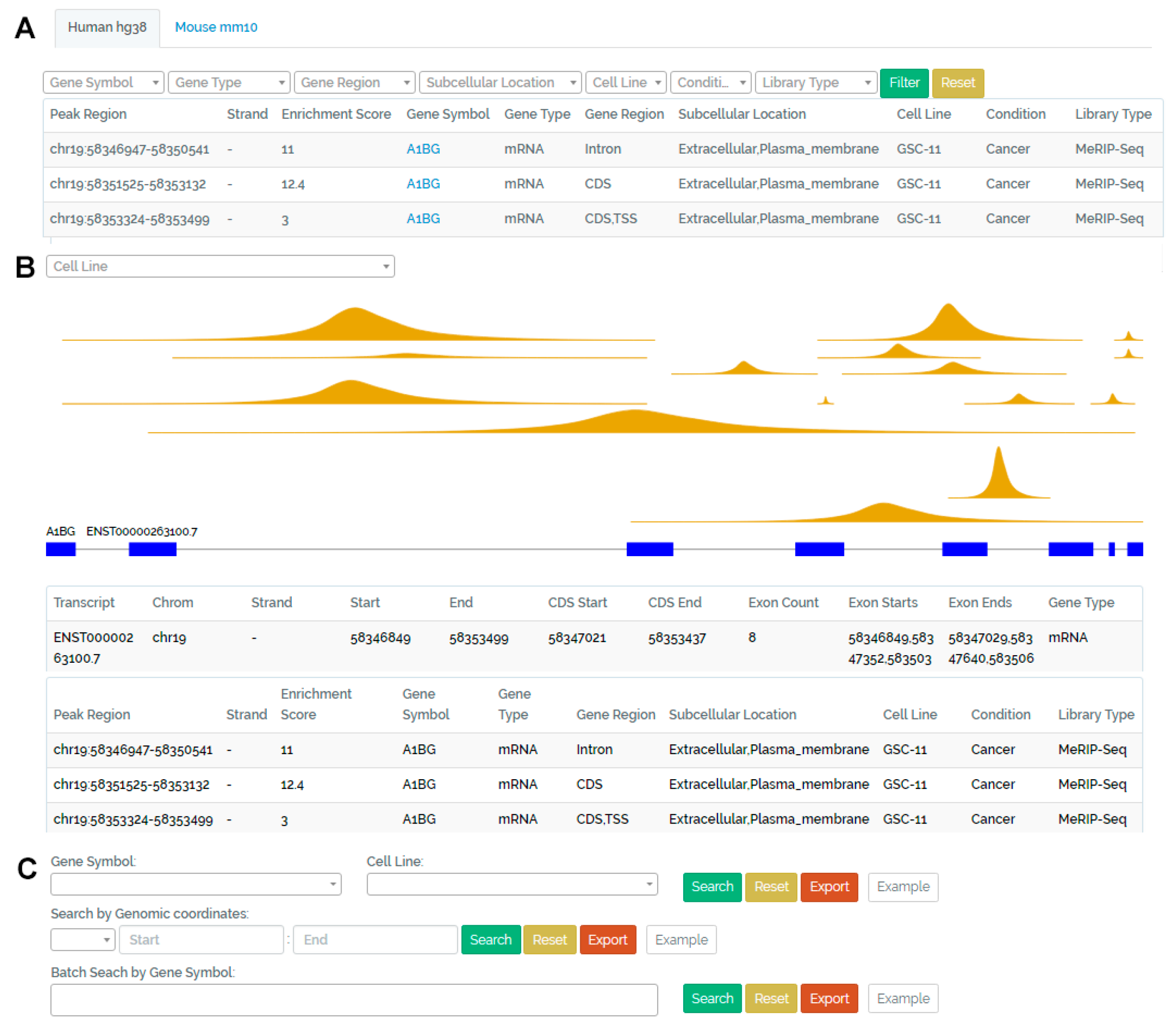
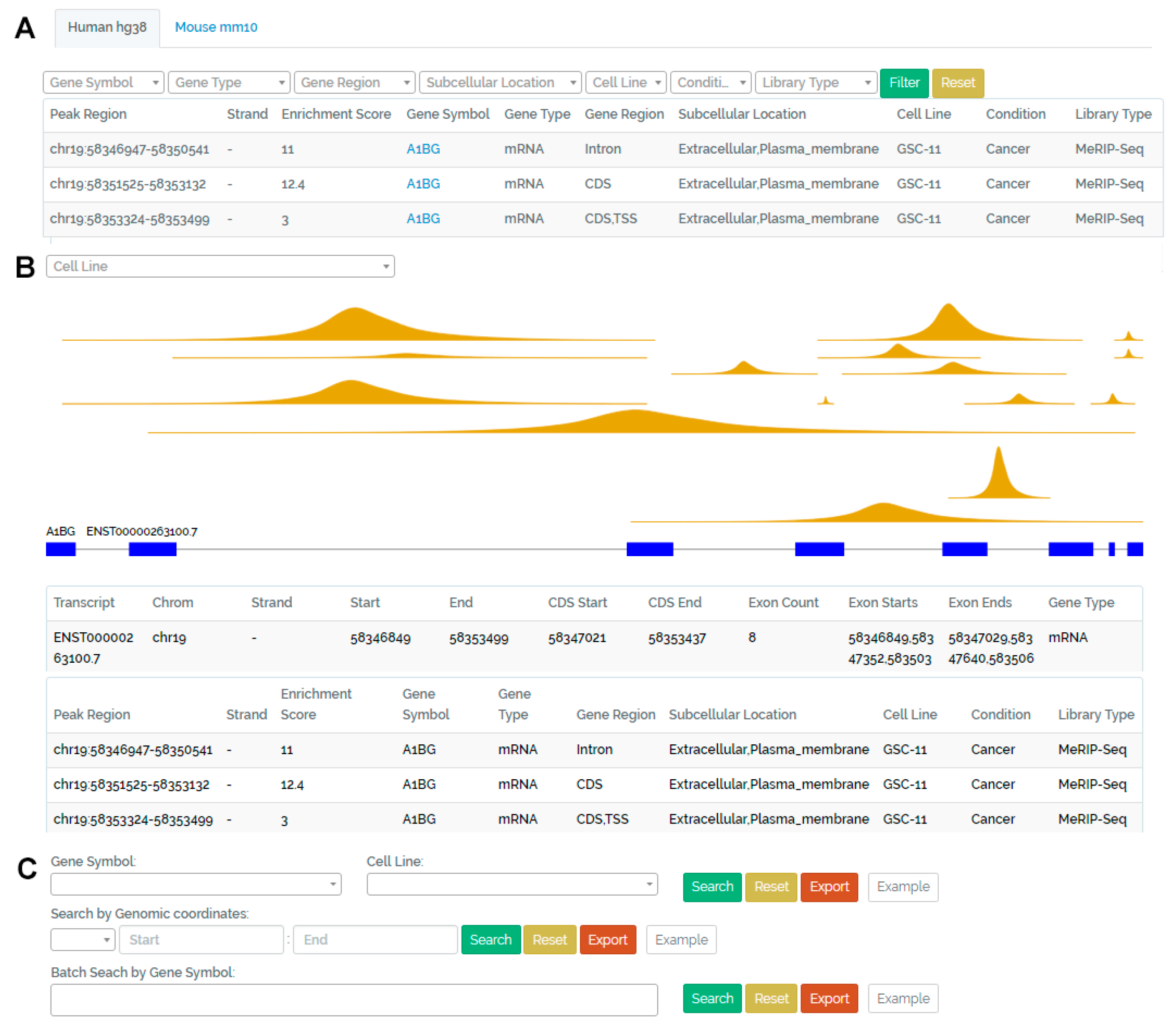
3.1. Browse Page
3.2. Visualization Page
3.3. Search Page
4. Summary and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6a RNA methylomes revealed by m6a-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mrna methylation reveals enrichment in 3’ utrs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhao, B.; Chen, K.; Thomas, A.; Tuteja, J.H.; He, X.; He, C.; White, K.P. Evolution of transcript modification by n6-methyladenosine in primates. Genome Res. 2017, 27, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Tang, K.; Zhang, D.; Xie, S.; Zhu, X.; Wang, Z.; Lang, Z. Transcriptome-wide high-throughput deep m(6)a-seq reveals unique differential m(6)a methylation patterns between three organs in arabidopsis thaliana. Genome Biol. 2015, 16, 272. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The epigenomic landscape of prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A mettl3-mettl14 complex mediates mammalian nuclear RNA n6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. M6a demethylase alkbh5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining foxm1 expression and cell proliferation program. Cancer Cell 2017, 31, 591–606. [Google Scholar] [CrossRef]
- Li, A.; Chen, Y.S.; Ping, X.L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6a reader ythdf3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. Hnrnpa2b1 is a mediator of m(6)a-dependent nuclear RNA processing events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Lence, T.; Akhtar, J.; Bayer, M.; Schmid, K.; Spindler, L.; Ho, C.H.; Kreim, N.; Andrade-Navarro, M.A.; Poeck, B.; Helm, M.; et al. M6a modulates neuronal functions and sex determination in drosophila. Nature 2016, 540, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. M6a mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. M(6)a RNA methylation promotes xist-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6a methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017. [Google Scholar]
- Zhang, S.Y.; Zhang, S.W.; Liu, L.; Meng, J.; Huang, Y. M6a-driver: Identifying context-specific mRNA m6a methylation-driven gene interaction networks. PLoS Comput. Biol. 2016, 12, e1005287. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ji, X.; Guo, X.; Ji, S. Regulatory role of n6 -methyladenosine (m6 a) methylation in RNA processing and human diseases. J. Cell. Biochem. 2017, 118, 2534–2543. [Google Scholar] [CrossRef]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A majority of m6a residues are in the last exons, allowing the potential for 3’ utr regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.X.; Van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. M(6)a-laic-seq reveals the census and complexity of the m(6)a epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Tirumuru, N.; Zhao, B.S.; Lu, W.; Lu, Z.; He, C.; Wu, L. N(6)-methyladenosine of hiv-1 RNA regulates viral infection and hiv-1 gag protein expression. eLife 2016, 5, e15528. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.J.; Sun, W.J.; Lin, P.H.; Zhou, K.R.; Liu, S.; Zheng, L.L.; Qu, L.H.; Yang, J.H. Rmbase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018, 46, D327–D334. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Wei, Z.; Zhang, S.; Hua, G.; Zhang, S.W.; Zhang, L.; Gao, S.J.; Meng, J.; Chen, X.; et al. Met-db v2.0: Elucidating context-specific functions of n6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018, 46, D281–D287. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6a and m6am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-resolution n(6) -methyladenosine (m(6) a) map using photo-crosslinking-assisted m(6) a sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Cui, X.; Meng, J.; Zhang, S.; Chen, Y.; Huang, Y. A novel algorithm for calling mrna m6a peaks by modeling biological variances in merip-seq data. Bioinformatics 2016, 32, i378–i385. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. Gencode: The reference human genome annotation for the encode project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. Bedtools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, Y.; Shen, H.B. Hum-mploc 3.0: Prediction enhancement of human protein subcellular localization through modeling the hidden correlations of gene ontology and functional domain features. Bioinformatics 2017, 33, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Shen, H.B. Cell-ploc: A package of web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Feng, J.; Xia, L.; Dong, X.; Zhang, X.; Zhang, S.; Miao, Y.; Xu, Q.; Xiao, S.; Zuo, Z.; et al. CVm6A: A Visualization and Exploration Database for m6As in Cell Lines. Cells 2019, 8, 168. https://doi.org/10.3390/cells8020168
Han Y, Feng J, Xia L, Dong X, Zhang X, Zhang S, Miao Y, Xu Q, Xiao S, Zuo Z, et al. CVm6A: A Visualization and Exploration Database for m6As in Cell Lines. Cells. 2019; 8(2):168. https://doi.org/10.3390/cells8020168
Chicago/Turabian StyleHan, Yujing, Jing Feng, Linjian Xia, Xin Dong, Xinyang Zhang, Shihan Zhang, Yuqi Miao, Qidi Xu, Shan Xiao, Zhixiang Zuo, and et al. 2019. "CVm6A: A Visualization and Exploration Database for m6As in Cell Lines" Cells 8, no. 2: 168. https://doi.org/10.3390/cells8020168
APA StyleHan, Y., Feng, J., Xia, L., Dong, X., Zhang, X., Zhang, S., Miao, Y., Xu, Q., Xiao, S., Zuo, Z., Xia, L., & He, C. (2019). CVm6A: A Visualization and Exploration Database for m6As in Cell Lines. Cells, 8(2), 168. https://doi.org/10.3390/cells8020168