Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs


Abstract
:1. Introduction
2. Overview of Cdc42
3. Cdc42 in Mammary Epithelial Cells Morphogenesis
3.1. Cdc42 Is Essential for MECs Morphogenesis
3.2. Deregulation of Cdc42 in Breast Cancer during MECs Morphogenesis
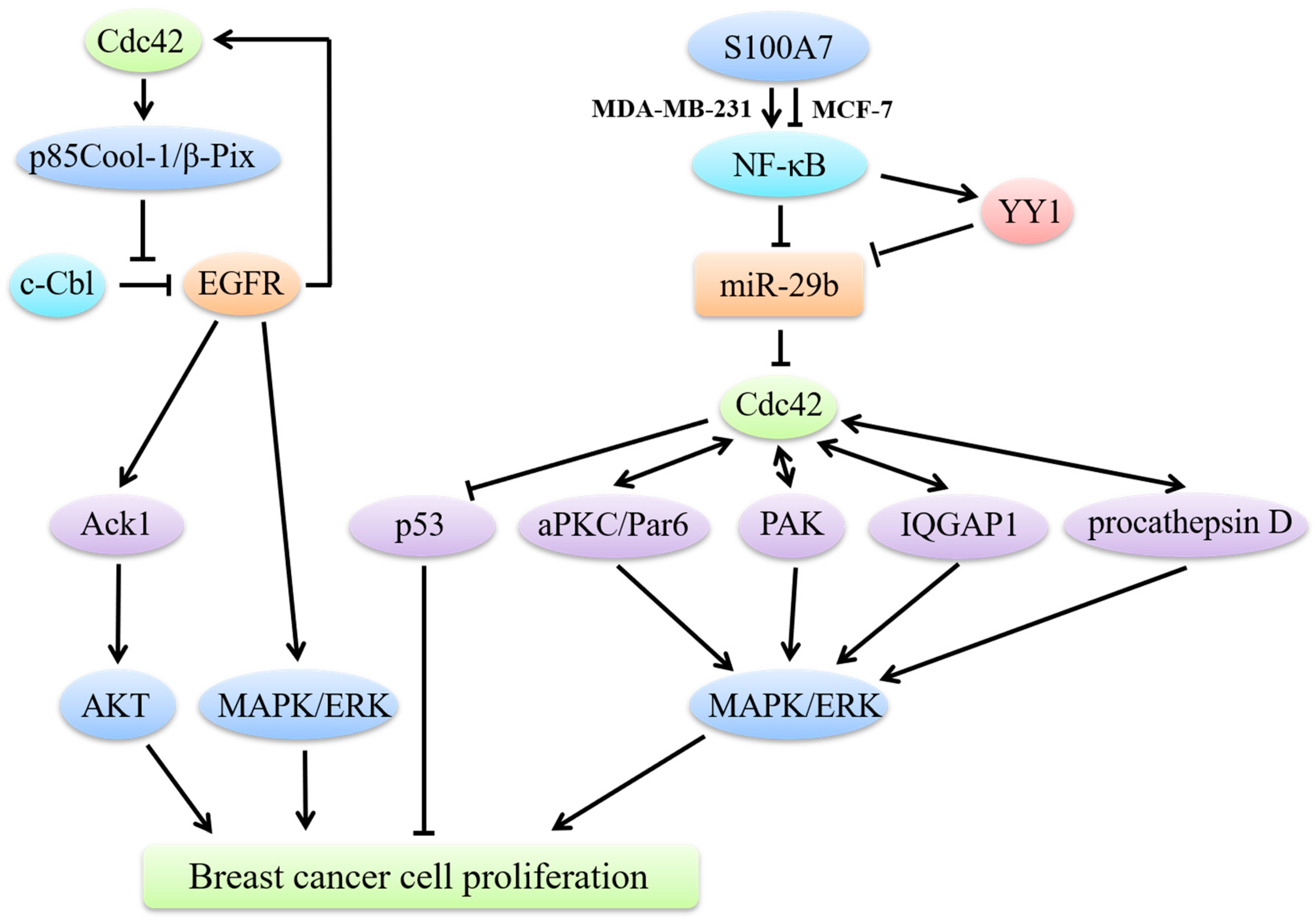
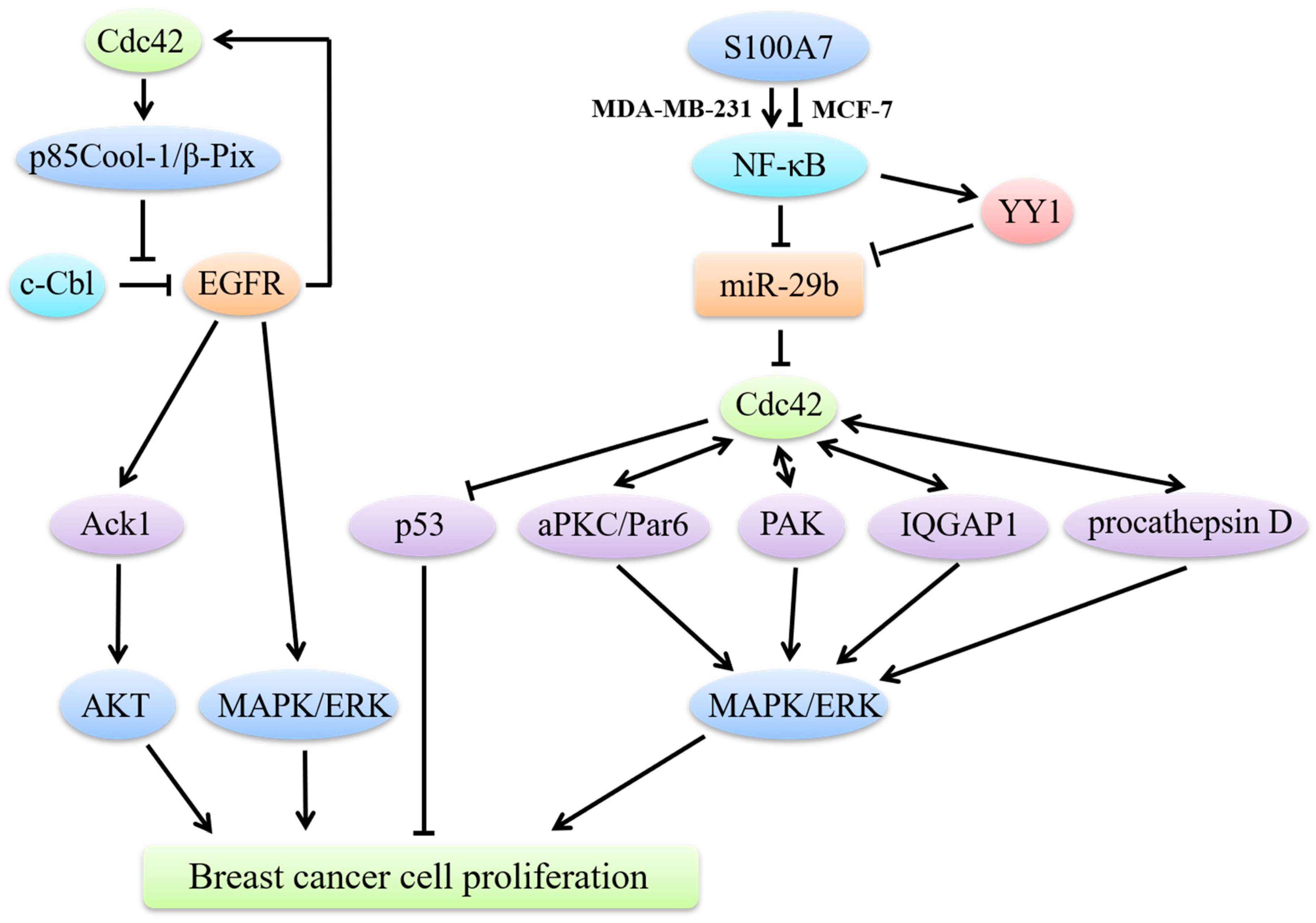
4. Cdc42 and Breast Cancer Cell Proliferation
4.1. Cdc42 Regulates Breast Cancer Cell Proliferation through MAPK Signaling
4.2. Cdc42/p53 Signaling in Breast Cancer Cell Proliferation
5. Cdc42 and Breast Cancer Cell Motility
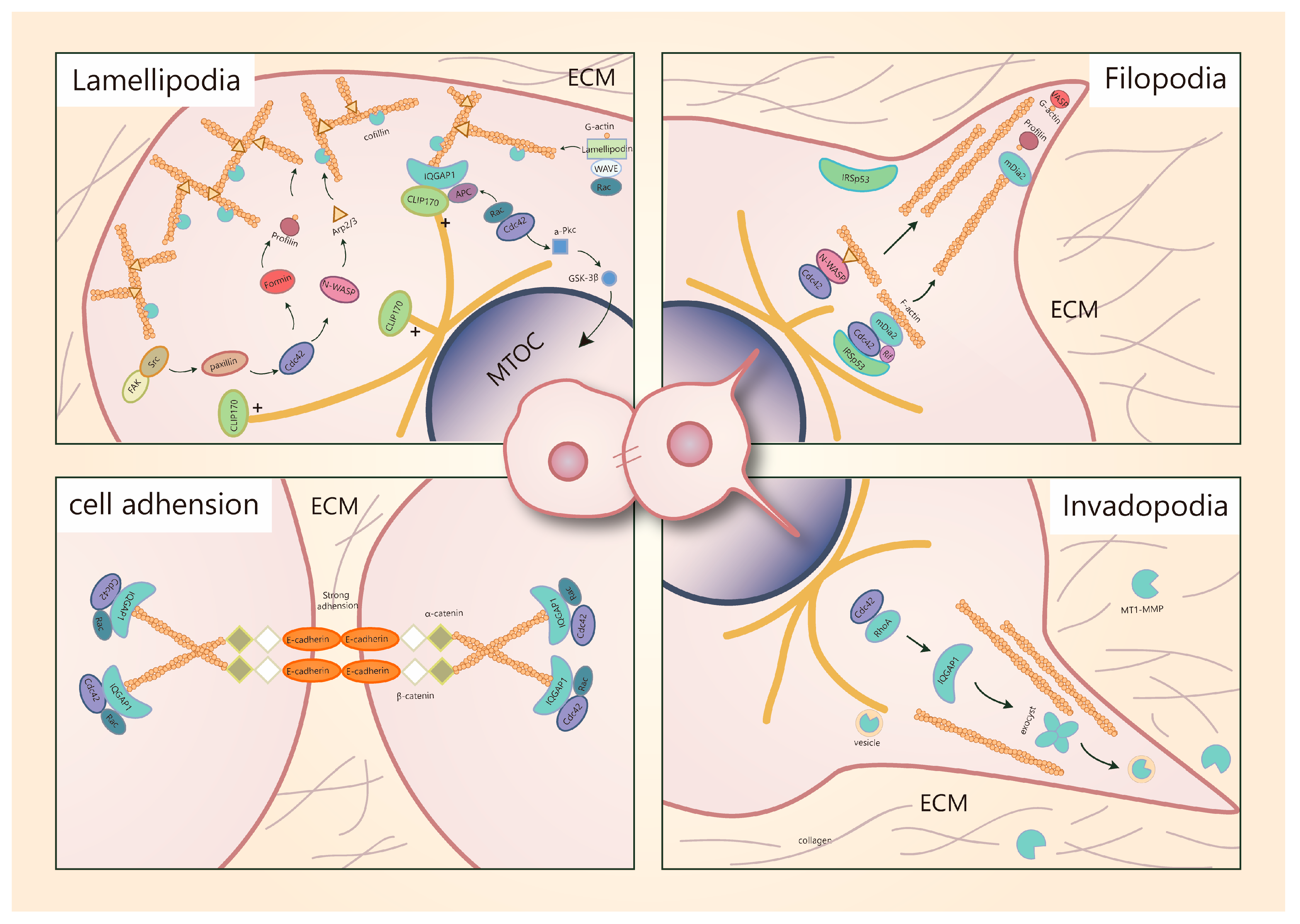
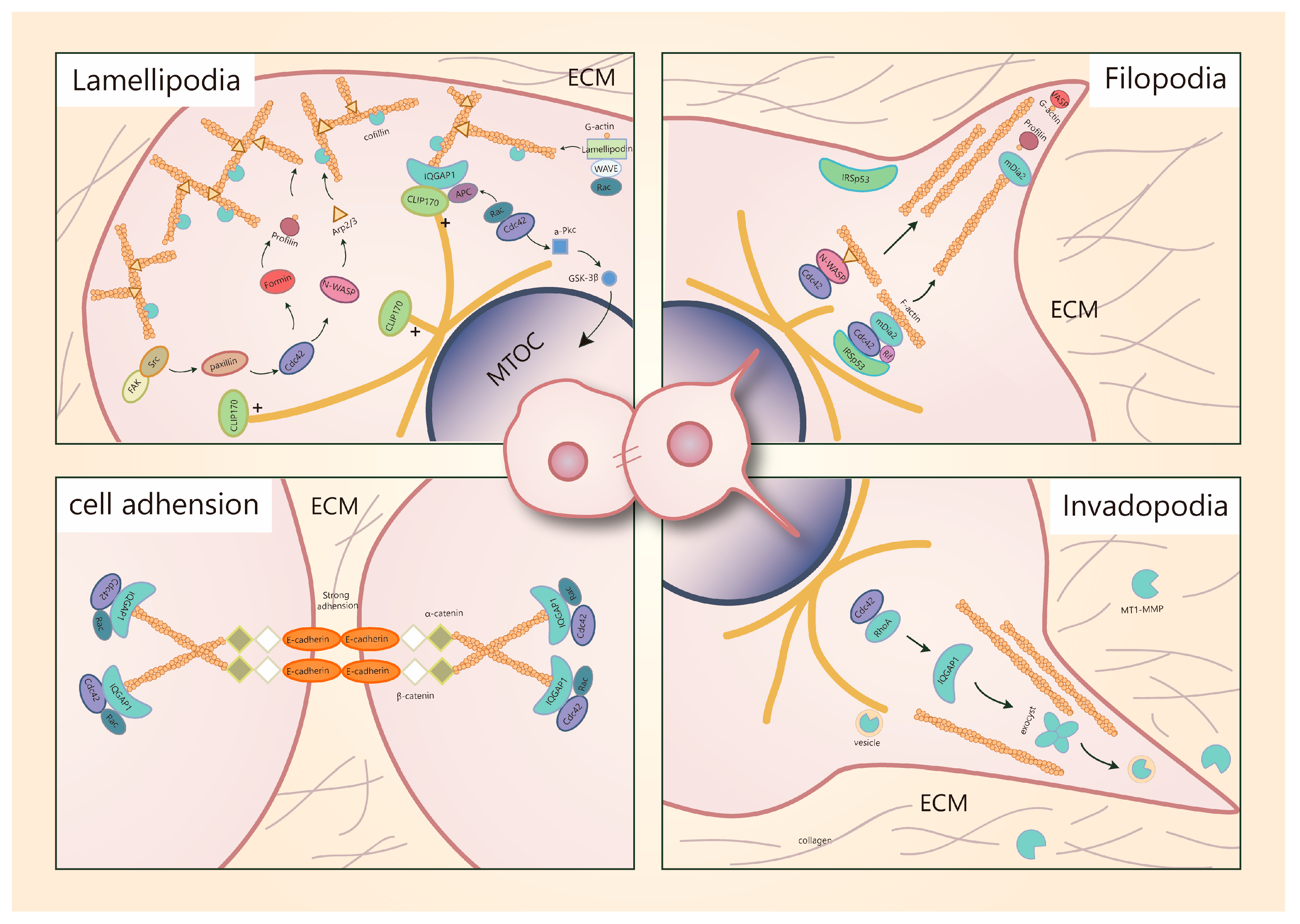
5.1. Cdc42 Is a Key Regulator of Migratory Protrusion Formation
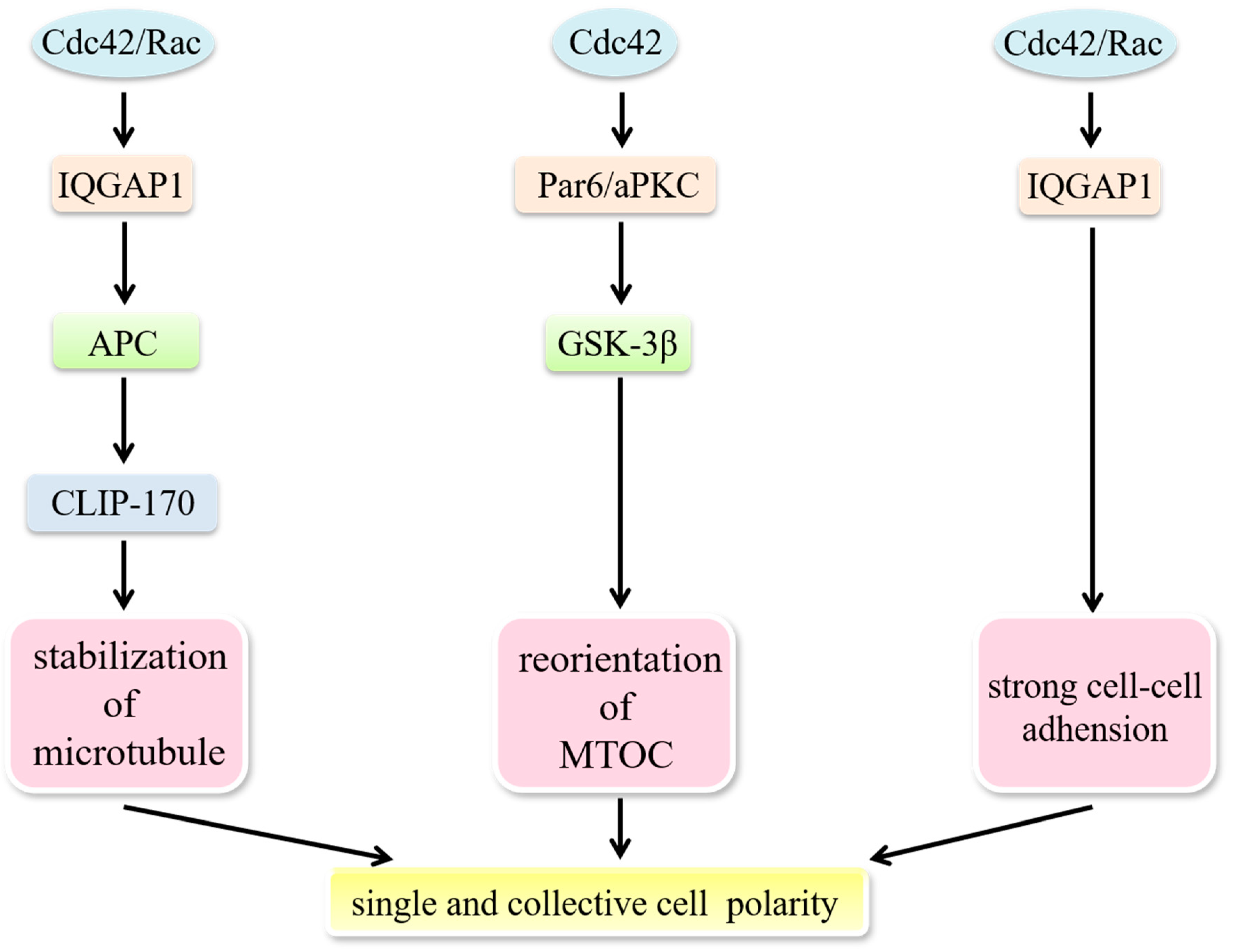
5.2. Cdc42 Modulates the Establishment of Cell Polarity
5.3. Cdc42 Involves the Progression of EMT
5.4. Cdc42 Regulates Breast Cancer Cells Motility via Various Effectors
6. Cdc42 and Breast Cancer Angiogenesis
7. Survival of Breast Cancer Cells Requires Cdc42
7.1. Cdc42 Regulates Apoptosis-Related Genes through PAK and JNK Signaling
7.2. Cdc42 Drives Actin Responses in NK Cells
7.3. Crosstalk of RhoGTPases during Breast Cancer Apoptosis
7.4. Cdc42 and Anti-Cancer Drugs Resistance
8. Current Research Advances of Cdc42-Targeted Therapies in Breast Cancer
8.1. GEF Interaction Inhibitors
8.2. Nucleotide Binding Inhibitors
8.3. RhoGDI Modulators
8.4. Metformin
8.5. Biological Extractions
9. Cdc42-Related Non-Coding RNAs in Breast Cancer
9.1. microRNA
9.2. lncRNA
10. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.; Zheng, Y. Cdc42 in oncogenic transformation, invasion and tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, B.J.; Zhou, H.; Lu, Q. Cdc42 Signaling Pathway Inhibition as a Therapeutic Target in Ras-Related Cancers. Curr. Med. Chem. 2017, 24, 3485–3507. [Google Scholar] [CrossRef] [PubMed]
- Smithers, C.C.; Overduin, M. Structural Mechanisms and Drug Discovery Prospects of Rho GTPases. Cells 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Carlson, J.H.; Jepperson, T.; Willis, S.; Leyland-Jones, B.; Dey, N. RAC1 GTP-ase signals Wnt-beta-catenin pathway mediated integrin-directed metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget 2017, 8, 3072–3103. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xiong, J.; Liu, G.; Wu, J.; Wen, L.; Zhang, Q.; Zhang, C. High expression of Rac1 is correlated with partial reversed cell polarity and poor prognosis in invasive ductal carcinoma of the breast. Tumour Biol. 2017, 39, 1010428317710908. [Google Scholar] [CrossRef]
- Chrysanthou, E.; Gorringe, K.L.; Joseph, C.; Craze, M.; Nolan, C.C.; Diez-Rodriguez, M.; Green, A.R.; Rakha, E.A.; Ellis, I.O.; Mukherjee, A. Phenotypic characterisation of breast cancer: The role of CDC42. Breast Cancer Res. Treat. 2017, 164, 317–325. [Google Scholar] [CrossRef]
- Gumuskaya, B.; Alper, M.; Hucumenoglu, S.; Altundag, K.; Uner, A.; Guler, G. EGFR expression and gene copy number in triple-negative breast carcinoma. Cancer Genet. Cytogenet. 2010, 203, 222–229. [Google Scholar] [CrossRef]
- Grob, T.J.; Heilenkotter, U.; Geist, S.; Paluchowski, P.; Wilke, C.; Jaenicke, F.; Quaas, A.; Wilczak, W.; Choschzick, M.; Sauter, G.; et al. Rare oncogenic mutations of predictive markers for targeted therapy in triple-negative breast cancer. Breast Cancer Res. Treat. 2012, 134, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.M.; Zhang, F.; Meng, G.; Wang, X.X.; Xu, H.; Yu, X.W.; Chen, L.Y.; Tse, G.M. Epidermal growth factor receptor gene amplification and protein overexpression in basal-like carcinoma of the breast. Histopathology 2011, 59, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Botta, F.; Zanellato, E.; Molinari, F.; Crippa, S.; Mazzucchelli, L.; Frattini, M. Molecular characterization of EGFR and EGFR-downstream pathways in triple negative breast carcinomas with basal like features. Histol. Histopathol. 2012, 27, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Koutras, A.; Lazaridis, G.; Koliou, G.A.; Kouvatseas, G.; Christodoulou, C.; Pectasides, D.; Kotoula, V.; Batistatou, A.; Bobos, M.; Tsolaki, E.; et al. Evaluation of the prognostic value of all four HER family receptors in patients with metastatic breast cancer treated with trastuzumab: A Hellenic Cooperative Oncology Group (HeCOG) study. PLoS ONE 2018, 13, e0207707. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jung, W.H.; Koo, J.S. Clinicopathologic features of molecular subtypes of triple negative breast cancer based on immunohistochemical markers. Histol. Histopathol. 2012, 27, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, J.; Yuan, Z.; Wang, S.; Peng, R.; Shi, Y.; Teng, X.; Qin, T. EGFR expression correlates with decreased disease-free survival in triple-negative breast cancer: A retrospective analysis based on a tissue microarray. Med. Oncol. 2012, 29, 401–405. [Google Scholar] [CrossRef]
- Viale, G.; Rotmensz, N.; Maisonneuve, P.; Bottiglieri, L.; Montagna, E.; Luini, A.; Veronesi, P.; Intra, M.; Torrisi, R.; Cardillo, A.; et al. Invasive ductal carcinoma of the breast with the "triple-negative" phenotype: Prognostic implications of EGFR immunoreactivity. Breast Cancer Res. Treat. 2009, 116, 317–328. [Google Scholar] [CrossRef]
- Tan, D.S.; Marchio, C.; Jones, R.L.; Savage, K.; Smith, I.E.; Dowsett, M.; Reis-Filho, J.S. Triple negative breast cancer: Molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res. Treat. 2008, 111, 27–44. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef]
- Toyama, T.; Yamashita, H.; Kondo, N.; Okuda, K.; Takahashi, S.; Sasaki, H.; Sugiura, H.; Iwase, H.; Fujii, Y. Frequently increased epidermal growth factor receptor (EGFR) copy numbers and decreased BRCA1 mRNA expression in Japanese triple-negative breast cancers. BMC Cancer 2008, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Meseure, D.; Vacher, S.; Drak Alsibai, K.; Trassard, M.; Susini, A.; Le Ray, C.; Lerebours, F.; Le Scodan, R.; Spyratos, F.; Marc Guinebretiere, J.; et al. Profiling of EGFR mRNA and protein expression in 471 breast cancers compared with 10 normal tissues: A candidate biomarker to predict EGFR inhibitor effectiveness. Int. J. Cancer 2012, 131, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Ishikawa, Y.; Furuya, M.; Sano, T.; Ohno, Y.; Horiguchi, J.; Oyama, T. Protein expression, gene amplification and mutational analysis of EGFR in triple-negative breast cancer. Breast Cancer (Tokyo, Japan) 2014, 21, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, A.E.; Rashed, H.E.; Abdelgawad, M.; Abdelhamid, M.I. Prognostic impact of EGFR and cytokeratin 5/6 immunohistochemical expression in triple-negative breast cancer. Ann. Diagnost. Pathol. 2017, 28, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Li, L.; Wang, N.; Xiong, Y.; Li, Y.; Gu, Y. Relationship of Epidermal Growth Factor Receptor Expression with Clinical Symptoms and Metastasis of Invasive Breast Cancer. J. Int. Cytokine Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 2005, 6, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.; Thornton, L.; McNally, S.; McCarthy, S.; Lanigan, F.; Gallagher, W.M.; Stein, T.; Martin, F. PKCzeta regulates cell polarisation and proliferation restriction during mammary acinus formation. J. Cell Sci. 2010, 123, 3316–3328. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef]
- Caldon, C.E.; Sutherland, R.L.; Musgrove, E. Cell cycle proteins in epithelial cell differentiation: Implications for breast cancer. Cell Cycle 2010, 9, 1918–1928. [Google Scholar] [CrossRef]
- Bray, K.; Brakebusch, C.; Vargo-Gogola, T. The Rho GTPase Cdc42 is required for primary mammary epithelial cell morphogenesis in vitro. Small GTPases 2011, 2, 247–258. [Google Scholar] [CrossRef]
- Liberto, M.; Cobrinik, D.; Minden, A. Rho regulates p21(CIP1), cyclin D1 and checkpoint control in mammary epithelial cells. Oncogene 2002, 21, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Maroto, B.; Ye, M.B.; von Lohneysen, K.; Schnelzer, A.; Knaus, U.G. P21-activated kinase is required for mitotic progression and regulates Plk1. Oncogene 2008, 27, 4900–4908. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fraticelli, A.E.; Vergarajauregui, S.; Eastburn, D.J.; Datta, A.; Alonso, M.A.; Mostov, K.; Martin-Belmonte, F. The Cdc42 GEF Intersectin 2 controls mitotic spindle orientation to form the lumen during epithelial morphogenesis. J. Cell. Biol 2010, 189, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Kaji, N.; Durgan, J.; Hall, A. Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J. Cell Biol. 2008, 183, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Kaji, N.; Jin, D.; Hall, A. Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J. Biol. Chem. 2011, 286, 12461–12474. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ‘invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Bray, K.; Gillette, M.; Young, J.; Loughran, E.; Hwang, M.; Sears, J.C.; Vargo-Gogola, T. Cdc42 overexpression induces hyperbranching in the developing mammary gland by enhancing cell migration. Breast Cancer Res. 2013, 15, R91. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef]
- Paszek, M.J.; Weaver, V.M. The tension mounts: Mechanics meets morphogenesis and malignancy. J. Mammary Gland Biol. Neoplasia 2004, 9, 325–342. [Google Scholar] [CrossRef]
- King, C.R.; Kraus, M.H.; Williams, L.T.; Merlino, G.T.; Pastan, I.H.; Aaronson, S.A. Human tumor cell lines with EGF receptor gene amplification in the absence of aberrant sized mRNAs. Nucleic Acids Res. 1985, 13, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, D.S.; Shen, Y.; Wu, W.J. Growth and motility inhibition of breast cancer cells by epidermal growth factor receptor degradation is correlated with inactivation of Cdc42. Cancer Res. 2006, 66, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Cerione, R.A. Cdc42 is a substrate for caspases and influences Fas-induced apoptosis. J. Biol. Chem. 2001, 276, 19656–19663. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.J.; Tu, S.; Cerione, R.A. Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 2003, 114, 715–725. [Google Scholar] [CrossRef]
- Hou, Y.; Zhou, M.; Xie, J.; Chao, P.; Feng, Q.; Wu, J. High glucose levels promote the proliferation of breast cancer cells through GTPases. Breast Cancer (Dove Med. Press) 2017, 9, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, M.; Pietschmann, K.; Brand, P.; Gunther, A.; Mahajan, N.P.; Heinzel, T.; Kramer, O.H. SIAH ubiquitin ligases target the nonreceptor tyrosine kinase ACK1 for ubiquitinylation and proteasomal degradation. Oncogene 2013, 32, 4913–4920. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zahari, M.S.; Renuse, S.; Kelkar, D.S.; Barbhuiya, M.A.; Rojas, P.L.; Stearns, V.; Gabrielson, E.; Malla, P.; Sukumar, S.; et al. The non-receptor tyrosine kinase TNK2/ACK1 is a novel therapeutic target in triple negative breast cancer. Oncotarget 2017, 8, 2971–2983. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Mahajan, N.P. Shepherding AKT and androgen receptor by Ack1 tyrosine kinase. J. Cell. Physiol. 2010, 224, 327–333. [Google Scholar] [CrossRef]
- Nolan, M.E.; Aranda, V.; Lee, S.; Lakshmi, B.; Basu, S.; Allred, D.C.; Muthuswamy, S.K. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 2008, 68, 8201–8209. [Google Scholar] [CrossRef]
- Vadlamudi, R.K.; Adam, L.; Wang, R.A.; Mandal, M.; Nguyen, D.; Sahin, A.; Chernoff, J.; Hung, M.C.; Kumar, R. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J. Biol. Chem. 2000, 275, 36238–36244. [Google Scholar] [CrossRef]
- Bokoch, G.M. Biology of the p21-activated kinases. Annu. Rev. Biochem. 2003, 72, 743–781. [Google Scholar] [CrossRef] [PubMed]
- Ohri, S.S.; Vashishta, A.; Proctor, M.; Fusek, M.; Vetvicka, V. Depletion of procathepsin D gene expression by RNA interference: A potential therapeutic target for breast cancer. Cancer Biol. Ther. 2007, 6, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Jadeski, L.; Mataraza, J.M.; Jeong, H.W.; Li, Z.; Sacks, D.B. IQGAP1 stimulates proliferation and enhances tumorigenesis of human breast epithelial cells. J. Biol. Chem. 2008, 283, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xue, Y.; Liu, W.; Yue, C.; Bi, F.; Xu, J.; Zhang, J.; Li, Y.; Zhong, C.; Chen, Y. Role of activated Rac1/Cdc42 in mediating endothelial cell proliferation and tumor angiogenesis in breast cancer. PLoS ONE 2013, 8, e66275. [Google Scholar] [CrossRef]
- Mott, J.L.; Kurita, S.; Cazanave, S.C.; Bronk, S.F.; Werneburg, N.W.; Fernandez-Zapico, M.E. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog and NF-kappaB. J. Cell. Biochem. 2010, 110, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar] [CrossRef]
- Zhao, H.; Wilkie, T.; Deol, Y.; Sneh, A.; Ganju, A.; Basree, M.; Nasser, M.W.; Ganju, R.K. miR-29b defines the pro-/anti-proliferative effects of S100A7 in breast cancer. Mol. Cancer 2015, 14, 11. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Zegers, M.M.; Friedl, P. Rho GTPases in collective cell migration. Small GTPases 2014, 5, e28997. [Google Scholar] [CrossRef]
- Insall, R.H.; Machesky, L.M. Actin dynamics at the leading edge: From simple machinery to complex networks. Dev. Cell 2009, 17, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.B.; Dujardin, D.; Noulet, F.; Martin, S.; Vauchelles, R.; Takeda, K.; Ronde, P. Altering FAK-paxillin interactions reduces adhesion, migration and invasion processes. PLoS ONE 2014, 9, e92059. [Google Scholar] [CrossRef] [PubMed]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Milo, R.; Kam, Z.; Geiger, B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 2007, 120, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Shortrede, J.E.; Uzair, I.D.; Neira, F.J.; Flamini, M.I.; Sanchez, A.M. Paxillin, a novel controller in the signaling of estrogen to FAK/N-WASP/Arp2/3 complex in breast cancer cells. Mol. Cell. Endocrinol. 2016, 430, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Law, A.L.; Vehlow, A.; Kotini, M.; Dodgson, L.; Soong, D.; Theveneau, E.; Bodo, C.; Taylor, E.; Navarro, C.; Perera, U.; et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J. Cell Biol. 2013, 203, 673–689. [Google Scholar] [CrossRef]
- Michael, M.; Vehlow, A.; Navarro, C.; Krause, M. c-Abl, Lamellipodin and Ena/VASP proteins cooperate in dorsal ruffling of fibroblasts and axonal morphogenesis. Curr. Biol. 2010, 20, 783–791. [Google Scholar] [CrossRef]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef]
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 2004, 304, 743–746. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Torka, R.; Thuma, F.; Herzog, V.; Kirfel, G. ROCK signaling mediates the adoption of different modes of migration and invasion in human mammary epithelial tumor cells. Exp. Cell. Res. 2006, 312, 3857–3871. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Eddy, R.; Condeelis, J. The cofilin pathway in breast cancer invasion and metastasis. Nat. Rev. Cancer 2007, 7, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Bear, J.E.; Gertler, F.B. Ena/VASP: Towards resolving a pointed controversy at the barbed end. J. Cell Sci. 2009, 122, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Goh, W.I.; Bu, W. I-BAR domains, IRSp53 and filopodium formation. Semin. Cell Dev. Biol. 2010, 21, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Faix, J.; Rottner, K. The making of filopodia. Curr. Opin. Cell Biol. 2006, 18, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Poincloux, R.; Lizarraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009, 122, 3015–3024. [Google Scholar] [CrossRef]
- Sakurai-Yageta, M.; Recchi, C.; Le Dez, G.; Sibarita, J.B.; Daviet, L.; Camonis, J.; D’Souza-Schorey, C.; Chavrier, P. The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J. Cell Biol. 2008, 181, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Drubin, D.G.; Barnes, G. Functional cooperation between the microtubule and actin cytoskeletons. Curr. Opin. Cell Biol. 2000, 12, 63–71. [Google Scholar] [CrossRef]
- Gomes, E.R.; Jani, S.; Gundersen, G.G. Nuclear movement regulated by Cdc42, MRCK, myosin and actin flow establishes MTOC polarization in migrating cells. Cell 2005, 121, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Noritake, J.; Watanabe, T.; Sato, K.; Wang, S.; Kaibuchi, K. IQGAP1: A key regulator of adhesion and migration. J. Cell Sci. 2005, 118, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature 2003, 421, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, G.G. Evolutionary conservation of microtubule-capture mechanisms. Nat. Rev. Mol. Cell Biol. 2002, 3, 296–304. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; McKinlay, K.J.; Jones, P.; Sablitzky, F. DEF6, a novel PH-DH-like domain protein, is an upstream activator of the Rho GTPases Rac1, Cdc42 and RhoA. Exp. Cell Res. 2004, 294, 335–344. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, M.; Chen, R.; Su, W.; Li, P.; Chen, S.; Chen, Z.; Chen, A.; Li, S.; Hu, C. IBP regulates epithelial-to-mesenchymal transition and the motility of breast cancer cells via Rac1, RhoA and Cdc42 signaling pathways. Oncogene 2014, 33, 3374–3382. [Google Scholar] [CrossRef]
- Keely, P.J.; Westwick, J.K.; Whitehead, I.P.; Der, C.J.; Parise, L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature 1997, 390, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Devanand, S.; Habib, B.; Zao-Zhong, S.; Fisher, P.B. mda-9/Syntenin: More than just a simple adapter protein when it comes to cancer metastasis. Cancer Research 2008, 68, 3087–3093. [Google Scholar]
- Menezes, M.E.; Shen, X.N.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B. MDA-9/Syntenin (SDCBP) modulates small GTPases RhoA and Cdc42 via transforming growth factor β1 to enhance epithelial-mesenchymal transition in breast cancer. Oncotarget 2016, 7, 80175. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhou, Z.; Liang, H.; Wu, J.; Shi, P.; Li, F.; Wang, Z.; Wang, C.; Chen, W.; Zhang, H.; et al. KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene 2016, 35, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Wu, Y.; Wehrli, B.; Chakrabarti, S.; Chakraborty, C. Modulation of ERK5 is a novel mechanism by which Cdc42 regulates migration of breast cancer cells. J. Cell. Biochem. 2015, 116, 124–132. [Google Scholar] [CrossRef]
- Matter, C.; Pribadi, M.; Liu, X.; Trachtenberg, J.T. Delta-catenin is required for the maintenance of neural structure and function in mature cortex in vivo. Neuron 2009, 64, 320–327. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, J.Y.; Wang, E.H. delta-catenin promotes the malignant phenotype in breast cancer. Tumour Biology 2015, 36, 569–575. [Google Scholar] [CrossRef]
- Kikuchi, K.; Li, X.; Yang, Z.; Takano, Y. Invasion of breast cancer cells into collagen matrix requires TGF-α and Cdc42 signaling. FEBS Lett. 2011, 585, 286–290. [Google Scholar] [CrossRef]
- Reynolds, A.B.; Daniel, J.; McCrea, P.D.; Wheelock, M.J.; Wu, J.; Zhang, Z. Identification of a new catenin: The tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol. Cell. Biol. 1994, 14, 8333–8342. [Google Scholar] [CrossRef] [PubMed]
- Emhonta, J.; Seachrist, D.D.; Deleon-Rodriguez, C.M.; Lozada, K.L.; John, M.; Abdul-Karim, F.W.; Keri, R.A. HER2/ErbB2-induced breast cancer cell migration and invasion require p120 catenin activation of Rac1 and Cdc42. J. Biol. Chem. 2011, 285, 29491–29501. [Google Scholar]
- Folkman, J. Anti-angiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Merajver, S.D.; Usmani, S.Z. Multifaceted role of Rho proteins in angiogenesis. J. Mammary Gland Biol. Neoplasia 2005, 10, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, B.M.; Tan, P.K.; Niederleithner, H.; Ferrara, N.; Petzelbauer, P.; Sibilia, M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010, 140, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Bi, F.; Zhang, X.; Zhang, S.; Pan, Y.; Liu, N.; Shi, Y.; Yao, X.; Zheng, Y.; Fan, D. Role of Rac1 and Cdc42 in hypoxia induced p53 and von Hippel-Lindau suppression and HIF1alpha activation. Int. J. Cancer 2006, 118, 2965–2972. [Google Scholar] [CrossRef] [PubMed]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J.A. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar]
- Pal, S.; Datta, K.; Mukhopadhyay, D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res. 2001, 61, 6952–6957. [Google Scholar]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef]
- Mizukami, Y.; Jo, W.S.; Duerr, E.M.; Gala, M.; Li, J.; Zhang, X.; Zimmer, M.A.; Iliopoulos, O.; Zukerberg, L.R.; Kohgo, Y.; et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat. Med. 2005, 11, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.A.; Jillian, S.A.; Michaelson, D.; Philips, M.R.; D’Eustachio, P.; Rush, M.G. Signaling mediated by the closely related mammalian Rho family GTPases TC10 and Cdc42 suggests distinct functional pathways. Cell. Growth Differ. 2001, 12, 157–167. [Google Scholar] [PubMed]
- Munoz, C.; Pascual-Salcedo, D.; Castellanos, M.C.; Alfranca, A.; Aragones, J.; Vara, A.; Redondo, J.M.; de Landazuri, M.O. Pyrrolidine dithiocarbamate inhibits the production of interleukin-6, interleukin-8 and granulocyte-macrophage colony-stimulating factor by human endothelial cells in response to inflammatory mediators: Modulation of NF-kappa B and AP-1 transcription factors activity. Blood 1996, 88, 3482–3490. [Google Scholar] [PubMed]
- Hui, Q.; Jin, Z.; Li, X.; Liu, C.; Wang, X. FGF Family: From Drug Development to Clinical Application. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Payson, R.A.; Chotani, M.A.; Chiu, I.M. Regulation of a promoter of the fibroblast growth factor 1 gene in prostate and breast cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 66, 93–103. [Google Scholar] [CrossRef]
- Kuo, C.H.; Sung, M.C.; Chen, P.K.; Chang, B.I.; Lee, F.T.; Cho, C.F.; Hsieh, T.T.; Huang, Y.C.; Li, Y.H.; Shi, G.Y.; et al. FGFR1 mediates recombinant thrombomodulin domain-induced angiogenesis. Cardiovasc. Res. 2015, 105, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Chotani, M.A.; Touhalisky, K.; Chiu, I.M. The small GTPases Ras, Rac and Cdc42 transcriptionally regulate expression of human fibroblast growth factor 1. J. Biol. Chem. 2000, 275, 30432–30438. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Schurmann, A.; Mooney, A.F.; Sanders, L.C.; Sells, M.A.; Wang, H.G.; Reed, J.C.; Bokoch, G.M. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol. Cell. Biol. 2000, 20, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. Apoptosis. CED-4 is a stranger no more. Nature 1997, 388, 714–715. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.A.; Swantek, J.L.; Stippec, S.; Yin, M.J.; Gaynor, R.; Cobb, M.H. Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J. Biol. Chem. 2000, 275, 19693–19699. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Williams-Skipp, C.; Tao, Y.; Schleicher, M.S.; Cano, L.L.; Duke, R.C.; Scheinman, R.I. NF-kappaB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell Death Differ. 1999, 6, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.H.; Wang, A.H.; Wang, C.L.; Mao, D.Z.; Lu, M.F.; Cui, Y.Q.; Jiao, R.Z. Surfactin induces apoptosis in human breast cancer MCF-7 cells through a ROS/JNK-mediated mitochondrial/caspase pathway. Chem. Biol. Interact. 2010, 183, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Coso, O.A.; Chiariello, M.; Yu, J.C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef]
- Sankpal, N.V.; Mayfield, J.D.; Willman, M.W.; Fleming, T.P.; Gillanders, W.E. Activator protein 1 (AP-1) contributes to EpCAM-dependent breast cancer invasion. Breast Cancer Res. 2011, 13, R124. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef]
- Passegue, E.; Jochum, W.; Schorpp-Kistner, M.; Mohle-Steinlein, U.; Wagner, E.F. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell 2001, 104, 21–32. [Google Scholar] [CrossRef]
- Kharbanda, S.; Saxena, S.; Yoshida, K.; Pandey, P.; Kaneki, M.; Wang, Q.; Cheng, K.; Chen, Y.N.; Campbell, A.; Sudha, T.; et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J. Biol. Chem. 2000, 275, 322–327. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Mohle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Bhoumik, A.; Krasilnikov, M.; Raz, R.; Owen-Schaub, L.B.; Levy, D.; Horvath, C.M.; Ronai, Z. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol. Cell 2001, 7, 517–528. [Google Scholar] [CrossRef]
- Al Absi, A.; Wurzer, H.; Guerin, C.; Hoffmann, C.; Moreau, F.; Mao, X.; Brown-Clay, J.; Petrolli, R.; Casellas, C.P.; Dieterle, M.; et al. Actin Cytoskeleton Remodeling Drives Breast Cancer Cell Escape from Natural Killer-Mediated Cytotoxicity. Cancer Res. 2018, 78, 5631–5643. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Paterson, H.F.; Marshall, C.J. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 1998, 394, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Fabbri, A.; Donelli, G.; Cossarizza, A.; Boquet, P.; Malorni, W. Toxin-induced activation of Rho GTP-binding protein increases Bcl-2 expression and influences mitochondrial homeostasis. Exp. Cell Res. 1998, 242, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, V.; Delumeau, I.; Tocque, B. Ras-GTPase activating protein inhibition specifically induces apoptosis of tumour cells. Oncogene 1999, 18, 4884–4889. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T. Roles of multidrug resistance genes in breast cancer chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 23–30. [Google Scholar]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, L.; Xincai, Q.U.; Wenjun, H.U.; Qinggang, H.U.; Zheng, Q. Expression of Cdc42 in adriamycin-sensitivity and adriamycin-resistance MCF-7 human breast cancer cell line. Chin. J. Gen. Surg. 2010, 19, 493–496. [Google Scholar]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331s–336s. [Google Scholar] [CrossRef] [PubMed]
- Azios, N.G.; Krishnamoorthy, L.; Harris, M.; Cubano, L.A.; Cammer, M.; Dharmawardhane, S.F. Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia 2007, 9, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.C.; Zhang, Y.; Qu, X.C. Effects of Cdc42 overexpression on the estrogen-enhanced multidrug resistance in breast cancer cells. Zhonghua Zhong Liu Za Zhi 2011, 33, 489–493. [Google Scholar] [PubMed]
- Marlin, J.W.; Eaton, A.; Montano, G.T.; Chang, Y.W.; Jakobi, R. Elevated p21-activated kinase 2 activity results in anchorage-independent growth and resistance to anticancer drug-induced cell death. Neoplasia (New York, N.Y.) 2009, 11, 286–297. [Google Scholar] [CrossRef]
- Walter, B.N.; Huang, Z.; Jakobi, R.; Tuazon, P.T.; Alnemri, E.S.; Litwack, G.; Traugh, J.A. Cleavage and activation of p21-activated protein kinase gamma-PAK by CPP32 (caspase 3). Effects of autophosphorylation on activity. J. Biol. Chem. 1998, 273, 28733–28739. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xing, J.; Streuli, M.; Leto, T.L.; Zheng, Y. Trp(56) of rac1 specifies interaction with a subset of guanine nucleotide exchange factors. J. Biol. Chem. 2001, 276, 47530–47541. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, E.; De La Mota-Peynado, A.; Dharmawardhane, S.; Vlaar, C.P. Novel inhibitors of Rac1 in metastatic breast cancer. P.R. Health Sci. J. 2010, 29, 348–356. [Google Scholar]
- Dharmawardhane, S.; Hernandez, E.; Vlaar, C. Development of EHop-016: A small molecule inhibitor of Rac. Enzymes 2013, 33(Pt. A), 117–146. [Google Scholar] [CrossRef]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Trans. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Corujo-Carro, F.; Duconge, J.; Hernandez-O’Farrill, E.; Vlaar, C.; Rodriguez-Orengo, J.F.; Cubano, L.; Dharmawardhane, S. Pharmacokinetics of Rac inhibitor EHop-016 in mice by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 981–982, 19–26. [Google Scholar] [CrossRef]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Robles, M.J.; Medina-Velazquez, J.; Asencio-Torres, G.M.; Gonzalez-Crespo, S.; Rymond, B.C.; Rodriguez-Medina, J.; Dharmawardhane, S. Targeting Cdc42 with the anticancer compound MBQ-167 inhibits cell polarity and growth in the budding yeast S. cerevisiae. Small GTPases 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Tan, L.; Lim, L.; Manser, E. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 2001, 276, 17347–17353. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.C.; Sanders, L.C.; Bokoch, G.M.; Gill, G.N. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1999, 1, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Lopez-Haber, C.; Yang, C.; Wang, H.; Lemmon, M.A.; Busillo, J.M.; Luo, J.; Benovic, J.L.; Klein-Szanto, A.; Yagi, H.; et al. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 2010, 40, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Chrzanowska-Wodnicka, M.; Zhong, C. Focal adhesion assembly. Trends Cell Biol. 1997, 7, 342–347. [Google Scholar] [CrossRef]
- Manser, E.; Huang, H.Y.; Loo, T.H.; Chen, X.Q.; Dong, J.M.; Leung, T.; Lim, L. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 1997, 17, 1129–1143. [Google Scholar] [CrossRef]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef]
- Peretti, A.S.; Dominguez, D.; Grimes, M.M.; Hathaway, H.J.; Prossnitz, E.R.; Rivera, M.R.; Wandinger-Ness, A.; Kusewitt, D.F.; Hudson, L.G. The R-Enantiomer of Ketorolac Delays Mammary Tumor Development in Mouse Mammary Tumor Virus-Polyoma Middle T Antigen (MMTV-PyMT) Mice. Am. J. Pathol. 2018, 188, 515–524. [Google Scholar] [CrossRef]
- Bidaud-Meynard, A.; Arma, D.; Taouji, S.; Laguerre, M.; Dessolin, J.; Rosenbaum, J.; Chevet, E.; Moreau, V. A novel small-molecule screening strategy identifies mitoxantrone as a RhoGTPase inhibitor. Biochem. J. 2013, 450, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Peterson, J.R.; Salvarezza, S.B.; Rodriguez-Boulan, E.; Chen, J.L.; Stamnes, M.; Macia, E.; Feng, Y.; Shair, M.D.; Kirchhausen, T. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat. Chem. Biol. 2006, 2, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, B.C.; Brown, P.H. Advances in Preventive Therapy for Estrogen-Receptor-Negative Breast Cancer. Curr. Breast Cancer Rep. 2014, 6, 96–109. [Google Scholar] [CrossRef]
- Hadad, S.M.; Coates, P.; Jordan, L.B.; Dowling, R.J.; Chang, M.C.; Done, S.J.; Purdie, C.A.; Goodwin, P.J.; Stambolic, V.; Moulder-Thompson, S.; et al. Evidence for biological effects of metformin in operable breast cancer: Biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res. Treat. 2015, 150, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Athreya, A.P.; Gaglio, A.J.; Cairns, J.; Kalari, K.R.; Weinshilboum, R.M.; Wang, L.; Kalbarczyk, Z.T.; Iyer, R.K. Machine Learning Helps Identify New Drug Mechanisms in Triple-Negative Breast Cancer. IEEE Trans. Nanobiosci. 2018, 17, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Athreya, A.P.; Kalari, K.R.; Cairns, J.; Gaglio, A.J.; Wills, Q.F.; Niu, N.; Weinshilboum, R.; Iyer, R.K.; Wang, L. Model-based unsupervised learning informs metformin-induced cell-migration inhibition through an AMPK-independent mechanism in breast cancer. Oncotarget 2017, 8, 27199–27215. [Google Scholar] [CrossRef]
- Wu, G.; Qian, Z.; Guo, J.; Hu, D.; Bao, J.; Xie, J.; Xu, W.; Lu, J.; Chen, X.; Wang, Y. Ganoderma lucidum extract induces G1 cell cycle arrest and apoptosis in human breast cancer cells. Am. J. Chin. Med. 2012, 40, 631–642. [Google Scholar] [CrossRef]
- Wu, G.S.; Lu, J.J.; Guo, J.J.; Li, Y.B.; Tan, W.; Dang, Y.Y.; Zhong, Z.F.; Xu, Z.T.; Chen, X.P.; Wang, Y.T. Ganoderic acid DM, a natural triterpenoid, induces DNA damage, G1 cell cycle arrest and apoptosis in human breast cancer cells. Fitoterapia 2012, 83, 408–414. [Google Scholar] [CrossRef]
- Wu, G.S.; Song, Y.L.; Yin, Z.Q.; Guo, J.J.; Wang, S.P.; Zhao, W.W.; Chen, X.P.; Zhang, Q.W.; Lu, J.J.; Wang, Y.T. Ganoderiol A-enriched extract suppresses migration and adhesion of MDA-MB-231 cells by inhibiting FAK-SRC-paxillin cascade pathway. PLoS ONE 2013, 8, e76620. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M.; Blankenship, K.A.; Risinger, K.E.; Bhatnagar, S.; Noisin, E.L.; Sumanasekera, W.K.; Zhao, L.; Brey, D.M.; Keynton, R.S. Resveratrol and estradiol rapidly activate MAPK signaling through estrogen receptors alpha and beta in endothelial cells. J. Biol. Chem. 2005, 280, 7460–7468. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Jeyapalan, Z.; Deng, Z.; Shatseva, T.; Fang, L.; He, C.; Yang, B.B. Expression of CD44 3’-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids Res. 2011, 39, 3026–3041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Guo, W.; Qian, J.; Wang, B. Negative regulation of CDC42 expression and cell cycle progression by miR-29a in breast cancer. Open Med. (Wars) 2016, 11, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Cao, Y.D.; Ye, W.X.; Sun, Y.Y. Effect of microRNA-206 on cytoskeleton remodelling by downregulating Cdc42 in MDA-MB-231 cells. Tumori 2010, 96, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, L.; Stebbing, J.; Braga, V.M.; Frampton, A.E.; Jacob, J.; Buluwela, L.; Jiao, L.R.; Periyasamy, M.; Madsen, C.D.; Caley, M.P.; et al. miR-23b regulates cytoskeletal remodeling, motility and metastasis by directly targeting multiple transcripts. Nucleic Acids Res. 2013, 41, 5400–5412. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Zavarella, S.; Symons, M.H. Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol. Cell. Biol. 2008, 28, 4162–4172. [Google Scholar] [CrossRef]
- Pellegrino, L.; Krell, J.; Roca-Alonso, L.; Stebbing, J.; Castellano, L. MicroRNA-23b regulates cellular architecture and impairs motogenic and invasive phenotypes during cancer progression. Bioarchitecture 2013, 3, 119–124. [Google Scholar] [CrossRef]
- Zhu, S.; Sachdeva, M.; Wu, F.; Lu, Z.; Mo, Y.Y. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene 2010, 29, 1763–1772. [Google Scholar] [CrossRef]
- Huang, S.; Cai, M.; Zheng, Y.; Zhou, L.; Wang, Q.; Chen, L. miR-888 in MCF-7 side population sphere cells directly targets E-cadherin. J. Genet. Genom. 2014, 41, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Nandy, S.B.; Orozco, A.; Lopez-Valdez, R.; Roberts, R.; Subramani, R.; Arumugam, A.; Dwivedi, A.K.; Stewart, V.; Prabhakar, G.; Jones, S.; et al. Glucose insult elicits hyperactivation of cancer stem cells through miR-424-cdc42-prdm14 signalling axis. Br. J. Cancer 2017, 117, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, M.; Ueda, J.; Hayashi, K.; Ohta, H.; Yabuta, Y.; Kurimoto, K.; Nakato, R.; Yamada, Y.; Shirahige, K.; Saitou, M. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell 2013, 12, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Liang, X.; Li, L.; Wang, B.; Ding, F.; Li, Y.; Wang, X.; Zhan, Q.; Liu, Z. MicroRNA-548j functions as a metastasis promoter in human breast cancer by targeting Tensin1. Mol. Oncol. 2016, 10, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, G.; Asmussen, H.K.; Asnaghi, L.; Vass, W.C.; Braverman, R.; Yamada, K.M.; Popescu, N.C.; Papageorge, A.G.; Lowy, D.R. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc. Natl. Acad. Sci. USA 2007, 104, 9012–9017. [Google Scholar] [CrossRef]
- Michalik, K.M.; You, X.; Manavski, Y.; Doddaballapur, A.; Zornig, M.; Braun, T.; John, D.; Ponomareva, Y.; Chen, W.; Uchida, S.; et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 2014, 114, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.S.; Wang, X.A.; Wu, W.G.; Hu, Y.P.; Li, M.L.; Ding, Q.; Weng, H.; Shu, Y.J.; Liu, T.Y.; Jiang, L.; et al. MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol. Ther. 2014, 15, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, H.; Wan, X.; Yang, X.; Zhu, C.; Wang, A.; He, L.; Miao, R.; Chen, S.; Zhao, H. Long noncoding RNA plays a key role in metastasis and prognosis of hepatocellular carcinoma. Biomed. Res. Int. 2014, 2014, 780521. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Chou, J.; Wang, B.; Zheng, T.; Li, X.; Zheng, L.; Hu, J.; Zhang, Y.; Xing, Y.; Xi, T. MALAT1 induced migration and invasion of human breast cancer cells by competitively binding miR-1 with cdc42. Biochem. Biophys. Res. Commun. 2016, 472, 262–269. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Types | Rate of Overexpression | ||
|---|---|---|---|
| Gene Amplification/mRNA | Protein | ||
| Rho GTPase family | Rac1 | >50% [7] | 61.4% [8] |
| Cdc42 | ------ | 42.5–56.9% [9] | |
| The activators of Cdc42 | EGFR | 2–37.3% [10,11,12,13,14,15] | 12.6~84.8% [10,11,14,15,16,17,18,19,20,21,22,23,24,25] |
| Inhibitors | Therapies | Cell Lines/Tissues | Inhibitory Effects | References |
|---|---|---|---|---|
| GEF interaction inhibitors | EHop-016 | MDA-MB-435 | growth, angiogenesis, metastasis | [129] |
| MBQ-167 | MDA-MB-231, MCF-7 and MDA-MB-435 | cell polarity, cell cycle progression, apoptosis and metastasis | [131] | |
| nude mice | tumor size | [131] | ||
| Nucleotide binding inhibitors | R-ketorolac | MMTV-PyMT mice | tumor progression | [140] |
| MTX | PAE | cell migration. | [141] | |
| RhoGDI modulators | secramine | Xenopus laevis cytoplasmic egg | actin polymerization | [142] |
| Antidiabetic drug | Metformin | MDA-MB-231 | proliferation and cell migration | [146] |
| Biological extractions | GAEE | MDA-MB-231 | cell migration | [151] |
| Resveratrol | MDA-MB-231 | cell migration | [122] |
| Non-Coding RNAs | RNA | Cell Lines/Tissues | Effects | Suppressor or Promoter | References |
|---|---|---|---|---|---|
| microRNAs | miR-29a | MDA-MB-453 | cell cycle progression | suppressor | [156] |
| miR-206 | MDA-MB-231 | filopodia formation and matrix degradation | suppressor | [157] | |
| miR-23b | MDA-MB-231, MCF-7 | actin cytoskeleton | suppressor | [158] | |
| EMT | suppressor | [160] | |||
| focal adhesion maturation | promoter | [160] | |||
| miR-224 | MDA-MB-231 | cell invasion | suppressor | [161] | |
| miR-888 | MCF-7 | adherens junction | suppressor | [162] | |
| miR-424 | MDA-MB-231 | CSCs pluripotency | suppressor | [163] | |
| miR-548j | MCF-7 | invasion | promoter | [165] | |
| lncRNA | MALAT1 | MDA-MB-231, MCF-7 | cell migration invasion | promoter | [171] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Li, J.; Lai, X.-N.; Jiao, X.-Q.; Xiong, J.-P.; Xiong, L.-X. Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs. Cells 2019, 8, 146. https://doi.org/10.3390/cells8020146
Zhang Y, Li J, Lai X-N, Jiao X-Q, Xiong J-P, Xiong L-X. Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs. Cells. 2019; 8(2):146. https://doi.org/10.3390/cells8020146
Chicago/Turabian StyleZhang, Yu, Jun Li, Xing-Ning Lai, Xue-Qiao Jiao, Jun-Ping Xiong, and Li-Xia Xiong. 2019. "Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs" Cells 8, no. 2: 146. https://doi.org/10.3390/cells8020146
APA StyleZhang, Y., Li, J., Lai, X.-N., Jiao, X.-Q., Xiong, J.-P., & Xiong, L.-X. (2019). Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs. Cells, 8(2), 146. https://doi.org/10.3390/cells8020146