Phosphatidylinositol Monophosphates Regulate Optimal Vav1 Signaling Output

 , , ,
, , ,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
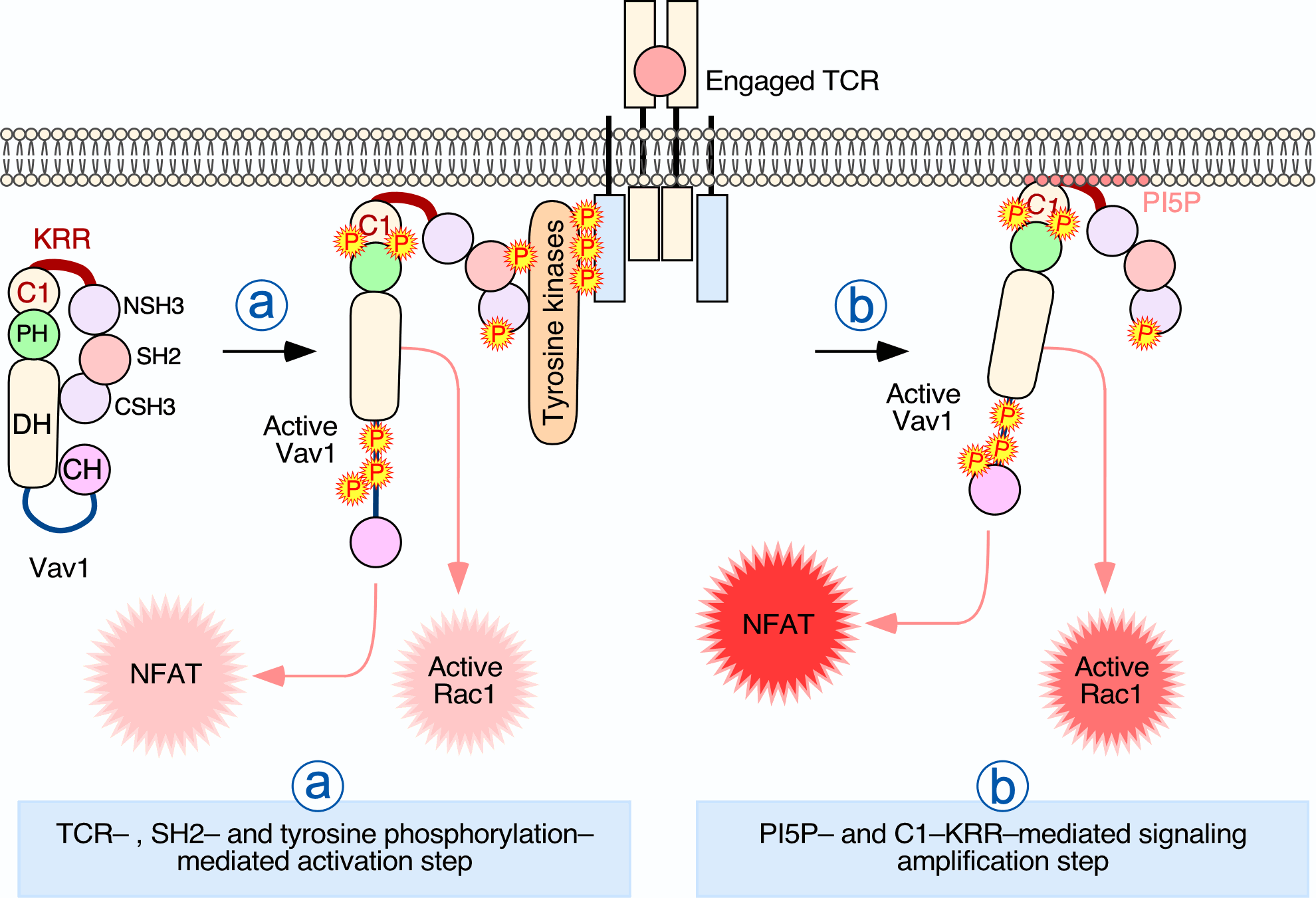
1. Introduction
2. Materials and Methods
2.1. Mammalian Expression Vectors
2.2. Bacterial Expression Vectors
2.3. Immunological Reagents
2.4. Phylogenetic Analyses
2.5. Calculation of Isoelectric Points
2.6. Cell Culture and Treatments
2.7. Luciferase Reporter, Immunoprecipitation, and Western Blot Analyses
2.8. Confocal Microscopy Analyses
2.9. Quantification of Confocal Images
2.10. Subcellular Fractionation
2.11. Purification of Maltose Binding Fusion Proteins
2.12. Phospholipid Binding Assays
2.13. Lipid Sedimentation Assays
2.14. Purification of Vav1 from Insect Cells and Lipid Binding Assays
2.15. Alignment of Amino Acid Sequences
2.16. Statistical Analyses
3. Results
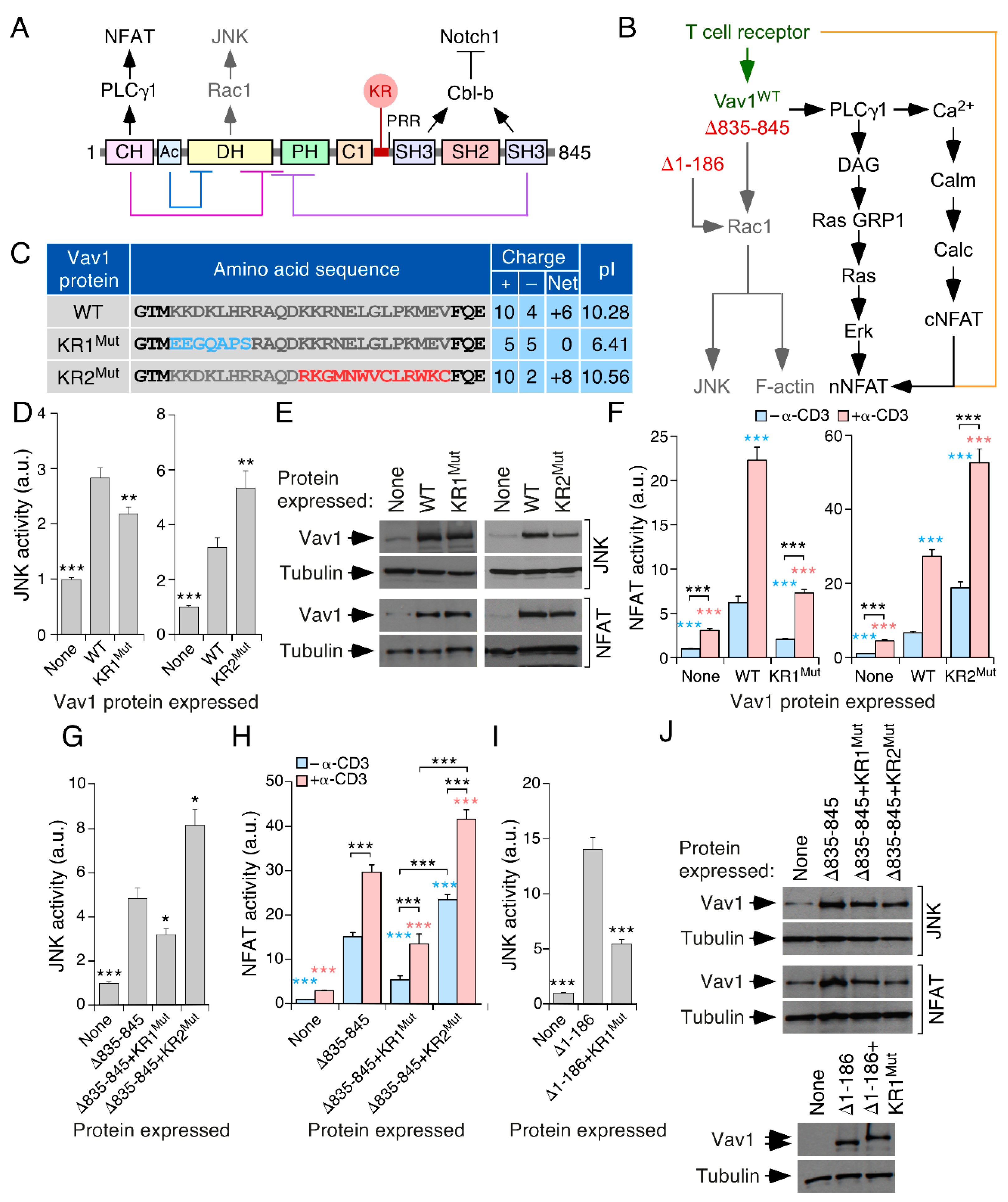
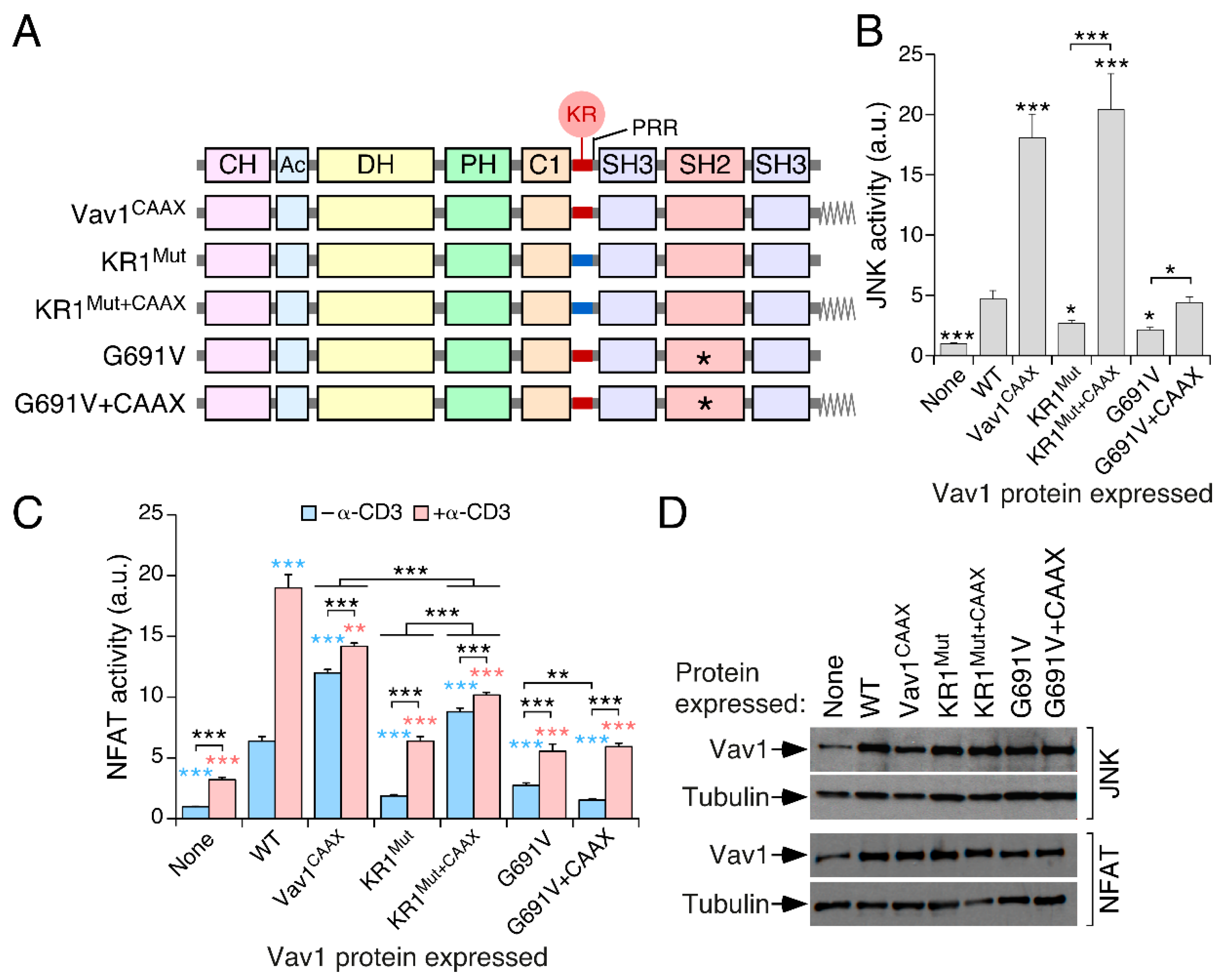
3.1. A Lysine-Rich Region Contributes to Vav1 Optimal Signaling Output in Lymphocytes
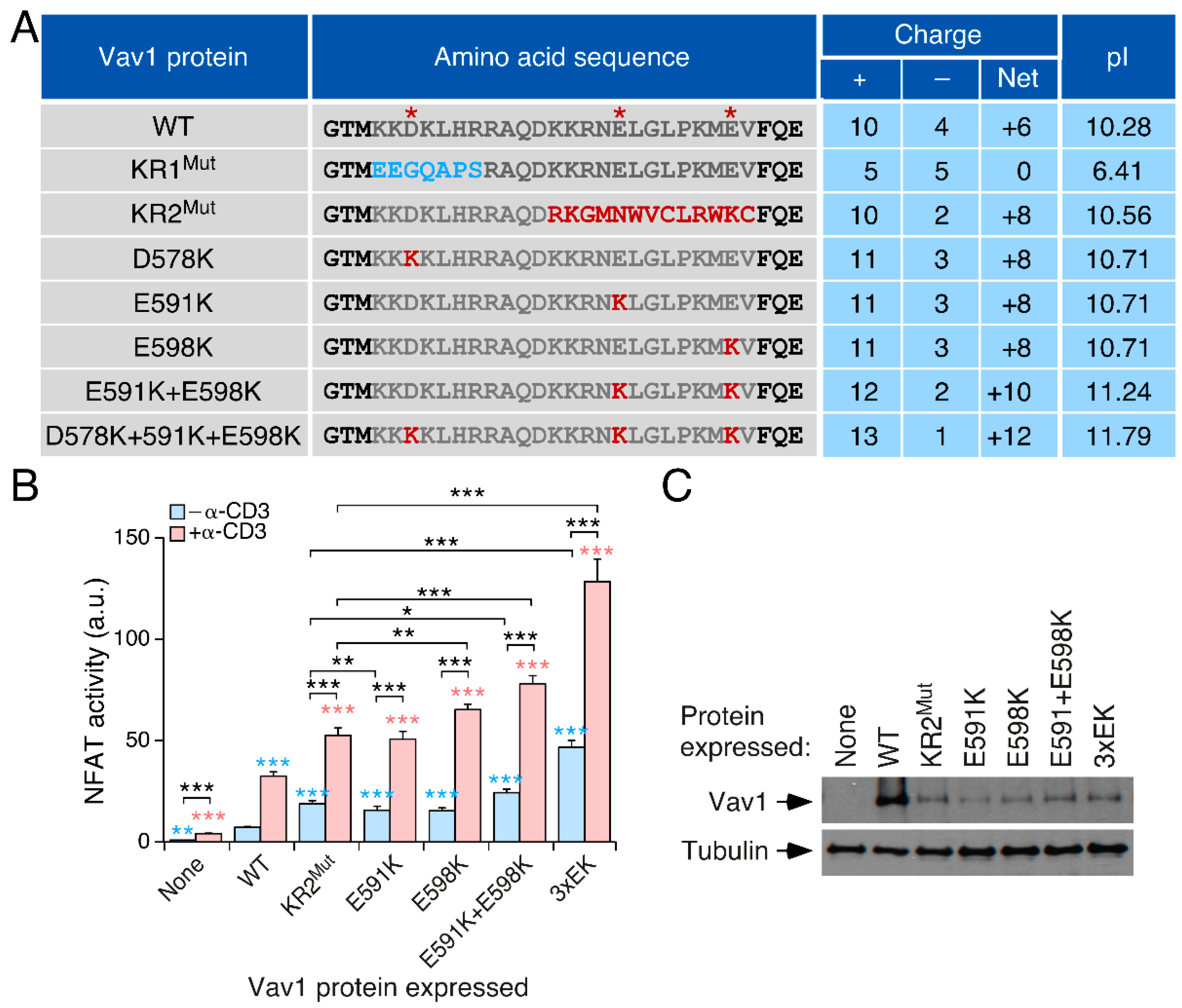
3.2. The Effect of the KR Region on Vav1 Activity Is Related to Its Electrostatic Properties
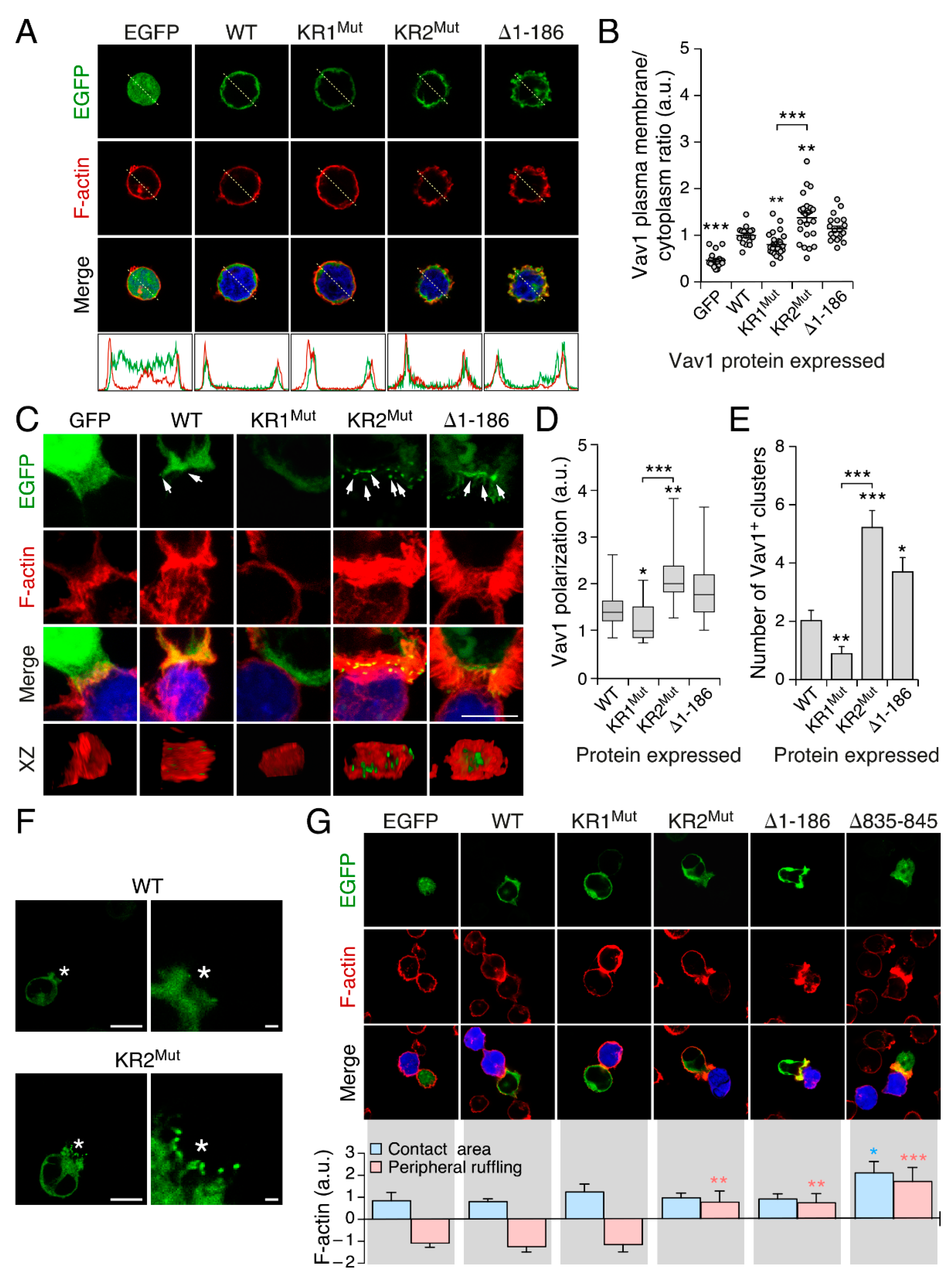
3.3. The KR Region Is Important for the Localization of Vav1 in Lymphocytes
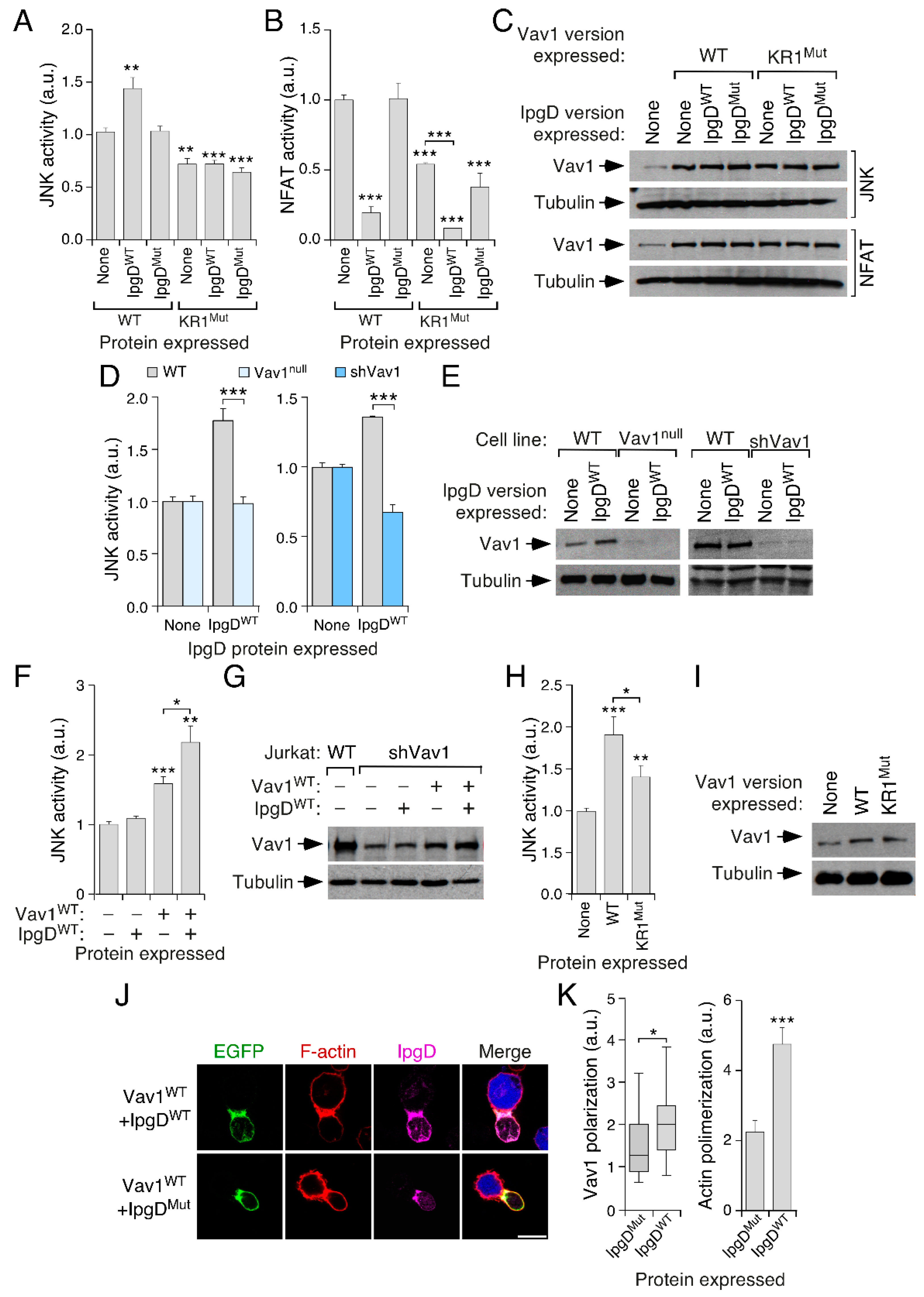
3.4. The Inclusion of the H-Ras CAAX Box Rescues the Activity of Vav1 KR1Mut
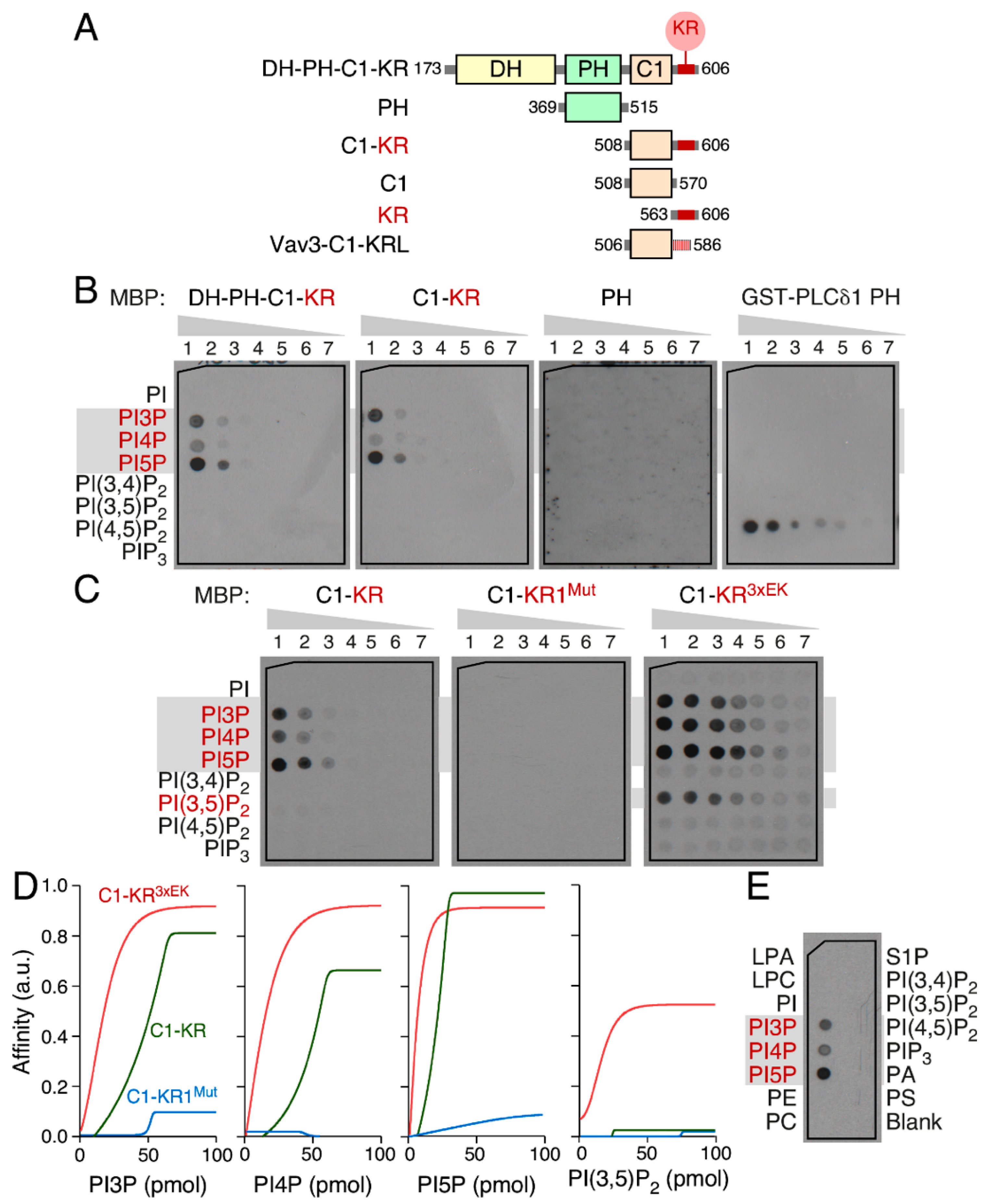
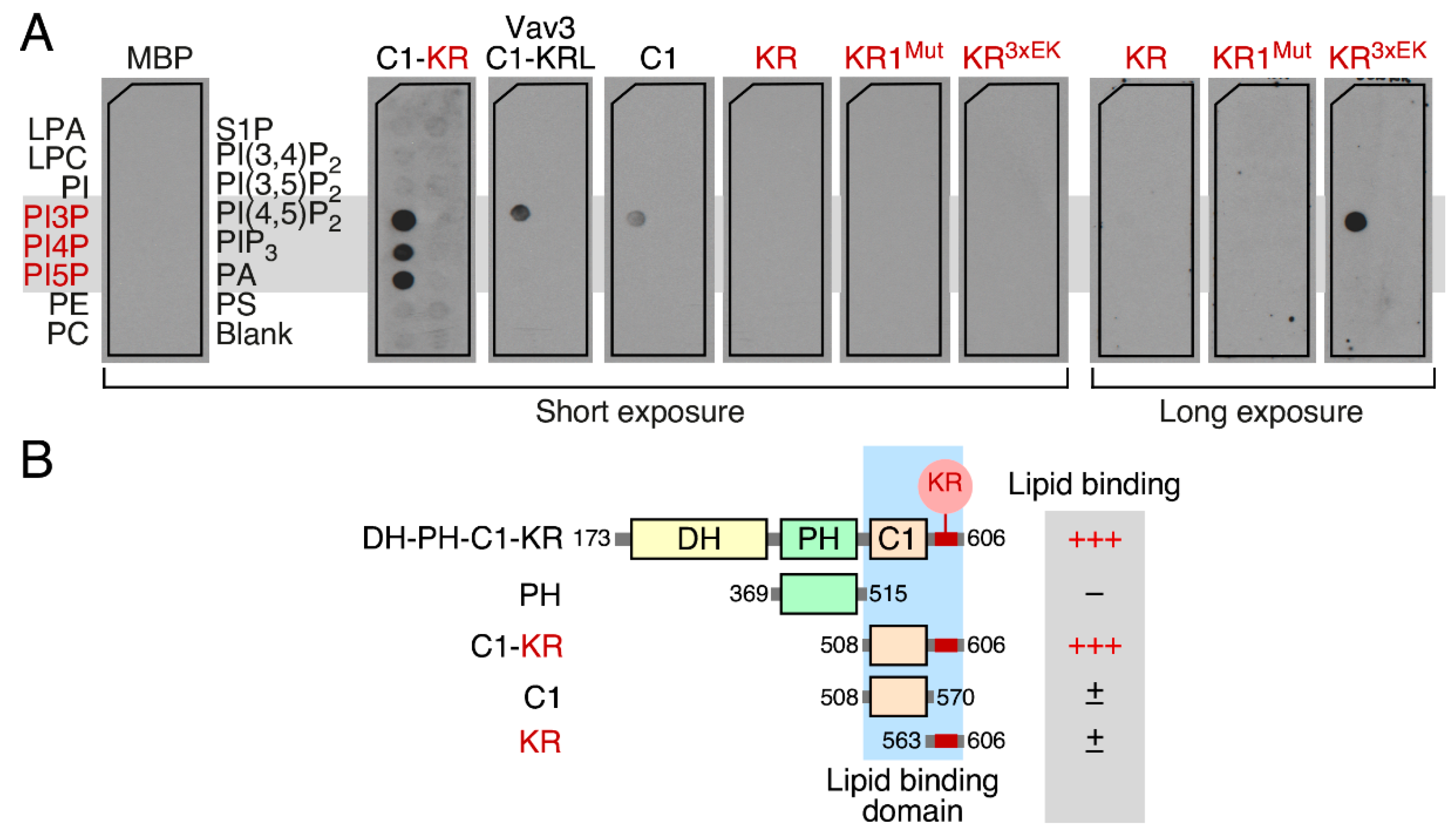
3.5. The Vav1 KR Region Contributes to the Binding of Specific Mono-PIs
3.6. Phospholipid Binding Requires Cooperation of the Vav1 C1 and KR Regions
3.7. PI5P Contributes to Vav1 Activity in a KR Region-Dependent Manner in Lymphocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Irvine, R.F. A short history of inositol lipids. J. Lipid Res. 2016, 57, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Viaud, J.; Mansour, R.; Antkowiak, A.; Mujalli, A.; Valet, C.; Chicanne, G.; Xuereb, J.M.; Terrisse, A.D.; Severin, S.; Gratacap, M.P.; et al. Phosphoinositides: Important lipids in the coordination of cell dynamics. Biochimie 2016, 125, 250–258. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Wilson, C.; D’Angelo, G. Phosphatidylinositol-4-phosphate: The Golgi and beyond. Bioessays 2013, 35, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Viaud, J.; Boal, F.; Tronchere, H.; Gaits-Iacovoni, F.; Payrastre, B. Phosphatidylinositol 5-phosphate: A nuclear stress lipid and a tuner of membranes and cytoskeleton dynamics. Bioessays 2014, 36, 260–272. [Google Scholar] [CrossRef]
- Vicinanza, M.; Korolchuk, V.I.; Ashkenazi, A.; Puri, C.; Menzies, F.M.; Clarke, J.H.; Rubinsztein, D.C. PI(5)P regulates autophagosome biogenesis. Mol. Cell 2015, 57, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.A.; Guittard, G. An Emerging Role for PI5P in T Cell Biology. Front. Immunol. 2013, 4, 80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guittard, G.; Gerard, A.; Dupuis-Coronas, S.; Tronchere, H.; Mortier, E.; Favre, C.; Olive, D.; Zimmermann, P.; Payrastre, B.; Nunes, J.A. Cutting edge: Dok-1 and Dok-2 adaptor molecules are regulated by phosphatidylinositol 5-phosphate production in T cells. J. Immunol. 2009, 182, 3974–3978. [Google Scholar] [CrossRef] [PubMed]
- Guittard, G.; Mortier, E.; Tronchere, H.; Firaguay, G.; Gerard, A.; Zimmermann, P.; Payrastre, B.; Nunes, J.A. Evidence for a positive role of PtdIns5P in T-cell signal transduction pathways. FEBS Lett. 2010, 584, 2455–2460. [Google Scholar] [CrossRef]
- DeFord-Watts, L.M.; Dougall, D.S.; Belkaya, S.; Johnson, B.A.; Eitson, J.L.; Roybal, K.T.; Barylko, B.; Albanesi, J.P.; Wulfing, C.; van Oers, N.S. The CD3 zeta subunit contains a phosphoinositide-binding motif that is required for the stable accumulation of TCR-CD3 complex at the immunological synapse. J. Immunol. 2011, 186, 6839–6847. [Google Scholar] [CrossRef]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef]
- Tarakhovsky, A.; Turner, M.; Schaal, S.; Mee, P.J.; Duddy, L.P.; Rajewsky, K.; Tybulewicz, V.L. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature 1995, 374, 467–470. [Google Scholar] [CrossRef]
- Turner, M.; Mee, P.J.; Walters, A.E.; Quinn, M.E.; Mellor, A.L.; Zamoyska, R.; Tybulewicz, V.L. A requirement for the Rho-family GTP exchange factor Vav in positive and negative selection of thymocytes. Immunity 1997, 7, 451–460. [Google Scholar] [CrossRef]
- Costello, P.S.; Walters, A.E.; Mee, P.J.; Turner, M.; Reynolds, L.F.; Prisco, A.; Sarner, N.; Zamoyska, R.; Tybulewicz, V.L. The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK, and NF-kappaB pathways. Proc. Natl. Acad. Sci. USA 1999, 96, 3035–3040. [Google Scholar] [CrossRef]
- Gulbranson-Judge, A.; Tybulewicz, V.L.; Walters, A.E.; Toellner, K.M.; MacLennan, I.C.; Turner, M. Defective immunoglobulin class switching in Vav-deficient mice is attributable to compromised T cell help. Eur. J. Immunol. 1999, 29, 477–487. [Google Scholar] [CrossRef]
- Rodriguez-Fdez, S.; Bustelo, X.R. The Vav GEF family: An evolutionary and functional perspective. Cells 2019, 8, 465. [Google Scholar] [CrossRef]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef]
- Crespo, P.; Schuebel, K.E.; Ostrom, A.A.; Gutkind, J.S.; Bustelo, X.R. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 1997, 385, 169–172. [Google Scholar] [CrossRef]
- Wu, J.; Katzav, S.; Weiss, A. A functional T-cell receptor signaling pathway is required for p95vav activity. Mol. Cell. Biol. 1995, 15, 4337–4346. [Google Scholar] [CrossRef]
- Yu, B.; Martins, I.R.; Li, P.; Amarasinghe, G.K.; Umetani, J.; Fernandez-Zapico, M.E.; Billadeau, D.D.; Machius, M.; Tomchick, D.R.; Rosen, M.K. Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell 2010, 140, 246–256. [Google Scholar] [CrossRef]
- Movilla, N.; Bustelo, X.R. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell. Biol. 1999, 19, 7870–7885. [Google Scholar] [CrossRef]
- Zugaza, J.L.; Lopez-Lago, M.A.; Caloca, M.J.; Dosil, M.; Movilla, N.; Bustelo, X.R. Structural determinants for the biological activity of Vav proteins. J. Biol. Chem. 2002, 277, 45377–45392. [Google Scholar] [CrossRef]
- Zhou, Z.; Yin, J.; Dou, Z.; Tang, J.; Zhang, C.; Cao, Y. The calponin homology domain of Vav1 associates with calmodulin and is prerequisite to T cell antigen receptor-induced calcium release in Jurkat T lymphocytes. J. Biol. Chem. 2007, 282, 23737–23744. [Google Scholar] [CrossRef]
- Robles-Valero, J.; Lorenzo-Martin, L.F.; Menacho-Marquez, M.; Fernandez-Pisonero, I.; Abad, A.; Camos, M.; Toribio, M.L.; Espinosa, L.; Bigas, A.; Bustelo, X.R. A paradoxical tumor-suppressor role for the Rac1 exchange factor Vav1 in T cell acute lymphoblastic leukemia. Cancer Cell 2017, 32, 608–623. [Google Scholar] [CrossRef]
- Barreira, M.; Fabbiano, S.; Couceiro, J.R.; Torreira, E.; Martinez-Torrecuadrada, J.L.; Montoya, G.; Llorca, O.; Bustelo, X.R. The C-terminal SH3 domain contributes to the intramolecular inhibition of Vav family proteins. Sci. Signal. 2014, 7, 35. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Ledbetter, J.A.; Barbacid, M. Product of vav proto-oncogene defines a new class of tyrosine protein kinase substrates. Nature 1992, 356, 68–71. [Google Scholar] [CrossRef]
- Han, J.; Luby-Phelps, K.; Das, B.; Shu, X.; Xia, Y.; Mosteller, R.D.; Krishna, U.M.; Falck, J.R.; White, M.A.; Broek, D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 1998, 279, 558–560. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Cardozo, T.; Andreev, J.; Li, Z.; Ferguson, K.M.; Abagyan, R.; Lemmon, M.A.; Aronheim, A.; Skolnik, E.Y. Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J. 1998, 17, 5374–5387. [Google Scholar] [CrossRef]
- Caloca, M.J.; Zugaza, J.L.; Bustelo, X.R. Mechanistic analysis of the amplification and diversification events induced by Vav proteins in B-lymphocytes. J. Biol. Chem. 2008, 283, 36454–36464. [Google Scholar] [CrossRef]
- Reynolds, L.F.; de Bettignies, C.; Norton, T.; Beeser, A.; Chernoff, J.; Tybulewicz, V.L. Vav1 transduces T cell receptor signals to the activation of the Ras/ERK pathway via LAT, Sos, and RasGRP1. J. Biol. Chem. 2004, 279, 18239–18246. [Google Scholar] [CrossRef]
- Reynolds, L.F.; Smyth, L.A.; Norton, T.; Freshney, N.; Downward, J.; Kioussis, D.; Tybulewicz, V.L. Vav1 transduces T cell receptor signals to the activation of phospholipase C-gamma1 via phosphoinositide 3-kinase-dependent and -independent pathways. J. Exp. Med. 2002, 195, 1103–1114. [Google Scholar] [CrossRef]
- Ruiz, S.; Santos, E.; Bustelo, X.R. RasGRF2, a guanosine nucleotide exchange factor for Ras GTPases, participates in T-cell signaling responses. Mol. Cell. Biol. 2007, 27, 8127–8142. [Google Scholar] [CrossRef]
- Schuebel, K.E.; Movilla, N.; Rosa, J.L.; Bustelo, X.R. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 1998, 17, 6608–6621. [Google Scholar] [CrossRef]
- Gozani, O.; Karuman, P.; Jones, D.R.; Ivanov, D.; Cha, J.; Lugovskoy, A.A.; Baird, C.L.; Zhu, H.; Field, S.J.; Lessnick, S.L.; et al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell 2003, 114, 99–111. [Google Scholar] [CrossRef]
- Hernandez, L.D.; Hueffer, K.; Wenk, M.R.; Galan, J.E. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 2004, 304, 1805–1807. [Google Scholar] [CrossRef]
- Coppola, J.; Bryant, S.; Koda, T.; Conway, D.; Barbacid, M. Mechanism of activation of the vav protooncogene. Cell Growth Differ. 1991, 2, 95–105. [Google Scholar]
- Lopez-Lago, M.; Lee, H.; Cruz, C.; Movilla, N.; Bustelo, X.R. Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Mol. Cell. Biol. 2000, 20, 1678–1691. [Google Scholar] [CrossRef][Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Cao, Y.; Janssen, E.M.; Duncan, A.W.; Altman, A.; Billadeau, D.D.; Abraham, R.T. Pleiotropic defects in TCR signaling in a Vav-1-null Jurkat T-cell line. EMBO J. 2002, 21, 4809–4819. [Google Scholar] [CrossRef] [PubMed]
- Barreira, M.; Rodriguez-Fdez, S.; Bustelo, X.R. New insights into the Vav1 activation cycle in lymphocytes. Cell. Signal. 2018, 45, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Robles-Valero, J.; Martin-Cofreces, N.B.; Lamana, A.; Macdonald, S.; Volkov, Y.; Sanchez-Madrid, F. Integrin and CD3/TCR activation are regulated by the scaffold protein AKAP450. Blood 2010, 115, 4174–4184. [Google Scholar] [CrossRef] [PubMed]
- Movilla, N.; Dosil, M.; Zheng, Y.; Bustelo, X.R. How Vav proteins discriminate the GTPases Rac1 and RhoA from Cdc42. Oncogene 2001, 20, 8057–8065. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef]
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immune system: Genetic events and selective pressures. Nat. Rev. Genet. 2010, 11, 47–59. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Ku, G.; Weiss, A. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J. Biol. Chem. 2000, 275, 2185–2190. [Google Scholar] [CrossRef]
- Li, S.Y.; Du, M.J.; Wan, Y.J.; Lan, B.; Liu, Y.H.; Yang, Y.; Zhang, C.Z.; Cao, Y. The N-terminal 20-amino acid region of guanine nucleotide exchange factor Vav1 plays a distinguished role in T cell receptor-mediated calcium signaling. J. Biol. Chem. 2013, 288, 3777–3785. [Google Scholar] [CrossRef]
- Doody, G.M.; Billadeau, D.D.; Clayton, E.; Hutchings, A.; Berland, R.; McAdam, S.; Leibson, P.J.; Turner, M. Vav-2 controls NFAT-dependent transcription in B- but not T-lymphocytes. EMBO J. 2000, 19, 6173–6184. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Zhou, Y.; Hancock, J.F. Rac1 nanoscale organization on the plasma membrane is driven by lipid binding specificity encoded in the membrane anchor. Mol. Cell. Biol. 2018, 38, e00186-18. [Google Scholar] [CrossRef]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.J.; Gorfe, A.A.; Hancock, J.F. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 2017, 168, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, K.; Giuriato, S.; Pedron, T.; Philpott, D.J.; Gaits, F.; Sable, J.; Sheetz, M.P.; Parsot, C.; Sansonetti, P.J.; Payrastre, B. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J. 2002, 21, 5069–5078. [Google Scholar] [CrossRef] [PubMed]
- Pendaries, C.; Tronchere, H.; Arbibe, L.; Mounier, J.; Gozani, O.; Cantley, L.; Fry, M.J.; Gaits-Iacovoni, F.; Sansonetti, P.J.; Payrastre, B. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 2006, 25, 1024–1034. [Google Scholar] [CrossRef]
- Viaud, J.; Lagarrigue, F.; Ramel, D.; Allart, S.; Chicanne, G.; Ceccato, L.; Courilleau, D.; Xuereb, J.M.; Pertz, O.; Payrastre, B.; et al. Phosphatidylinositol 5-phosphate regulates invasion through binding and activation of Tiam1. Nat. Commun. 2014, 5, 4080. [Google Scholar] [CrossRef]
- Stijf-Bultsma, Y.; Sommer, L.; Tauber, M.; Baalbaki, M.; Giardoglou, P.; Jones, D.R.; Gelato, K.A.; van Pelt, J.; Shah, Z.; Rahnamoun, H.; et al. The basal transcription complex component TAF3 transduces changes in nuclear phosphoinositides into transcriptional output. Mol. Cell 2015, 58, 453–467. [Google Scholar] [CrossRef]
- Viiri, K.M.; Janis, J.; Siggers, T.; Heinonen, T.Y.; Valjakka, J.; Bulyk, M.L.; Maki, M.; Lohi, O. DNA-binding and -bending activities of SAP30L and SAP30 are mediated by a zinc-dependent module and monophosphoinositides. Mol. Cell. Biol. 2009, 29, 342–356. [Google Scholar] [CrossRef]
- Kaadige, M.R.; Ayer, D.E. The polybasic region that follows the plant homeodomain zinc finger 1 of Pf1 is necessary and sufficient for specific phosphoinositide binding. J. Biol. Chem. 2006, 281, 28831–28836. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Fdez, S.; Citterio, C.; Lorenzo-Martín, L.F.; Baltanás-Copado, J.; Llorente-González, C.; Corbalán-García, S.; Vicente-Manzanares, M.; Bustelo, X.R. Phosphatidylinositol Monophosphates Regulate Optimal Vav1 Signaling Output. Cells 2019, 8, 1649. https://doi.org/10.3390/cells8121649
Rodríguez-Fdez S, Citterio C, Lorenzo-Martín LF, Baltanás-Copado J, Llorente-González C, Corbalán-García S, Vicente-Manzanares M, Bustelo XR. Phosphatidylinositol Monophosphates Regulate Optimal Vav1 Signaling Output. Cells. 2019; 8(12):1649. https://doi.org/10.3390/cells8121649
Chicago/Turabian StyleRodríguez-Fdez, Sonia, Carmen Citterio, L. Francisco Lorenzo-Martín, Jesús Baltanás-Copado, Clara Llorente-González, Senena Corbalán-García, Miguel Vicente-Manzanares, and Xosé R. Bustelo. 2019. "Phosphatidylinositol Monophosphates Regulate Optimal Vav1 Signaling Output" Cells 8, no. 12: 1649. https://doi.org/10.3390/cells8121649
APA StyleRodríguez-Fdez, S., Citterio, C., Lorenzo-Martín, L. F., Baltanás-Copado, J., Llorente-González, C., Corbalán-García, S., Vicente-Manzanares, M., & Bustelo, X. R. (2019). Phosphatidylinositol Monophosphates Regulate Optimal Vav1 Signaling Output. Cells, 8(12), 1649. https://doi.org/10.3390/cells8121649