Effect of High Glucose-Induced Oxidative Stress on Paraoxonase 2 Expression and Activity in Caco-2 Cells

 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture and Incubation with Glucose
2.3. Cell Extracts
2.4. Western Blot Analysis
2.5. Quantitative Real-Time PCR
2.6. Intracellular ROS Levels
2.7. Cell Lipid Peroxidation
2.8. Cell Total Antioxidant Activity
2.9. Evaluation of Fluorescent Advanced Glycation End Products (AGEs)
2.10. MTT Test
2.11. Apoptosis Analysis
2.12. Determination of Caspase-3 and Caspase-8 Activity
2.13. Enzymatic Activity Assays
2.13.1. Paroxonase 2 (PON2) Activity
2.13.2. Glutathione Reductase (GR) Activity
2.13.3. Glutathione Peroxidase (GPX) Activity
2.13.4. Catalase (CAT) Activity
2.14. Statistical Analysis
3. Results
3.1. Effect of High Glucose (HG) Treatment on ROS Levels and AGEs in Caco-2 Cells
3.2. Effect of High Glucose (HG) Treatment on PON2 Expression and Activity
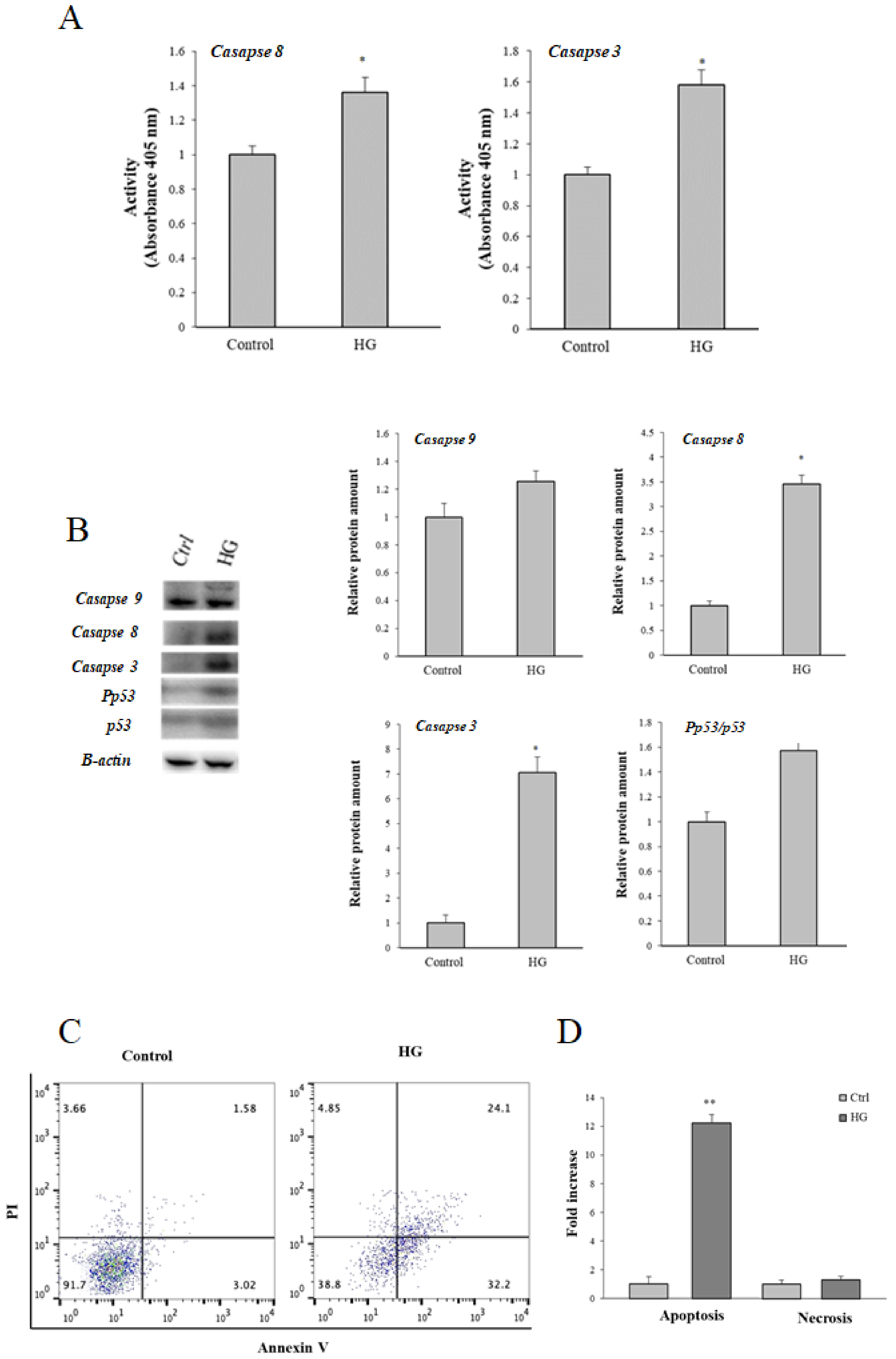
3.3. Effect of High Glucose (HG) Treatment on Caspases and Apoptosis
3.4. Effect of High Glucose (HG)-Treatment on p53
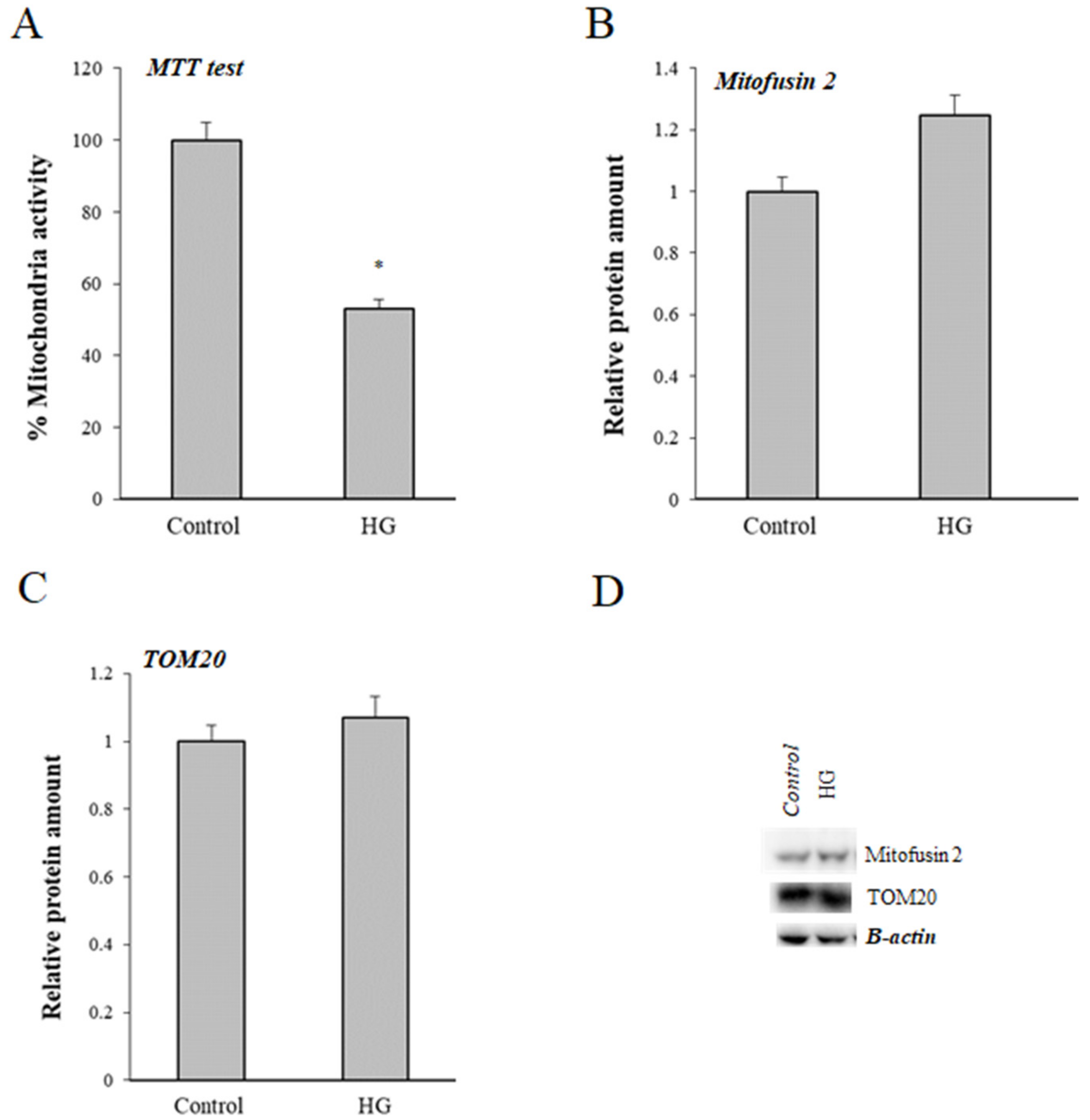
3.5. Effect of High Glucose (HG) Treatment on Mitochondria
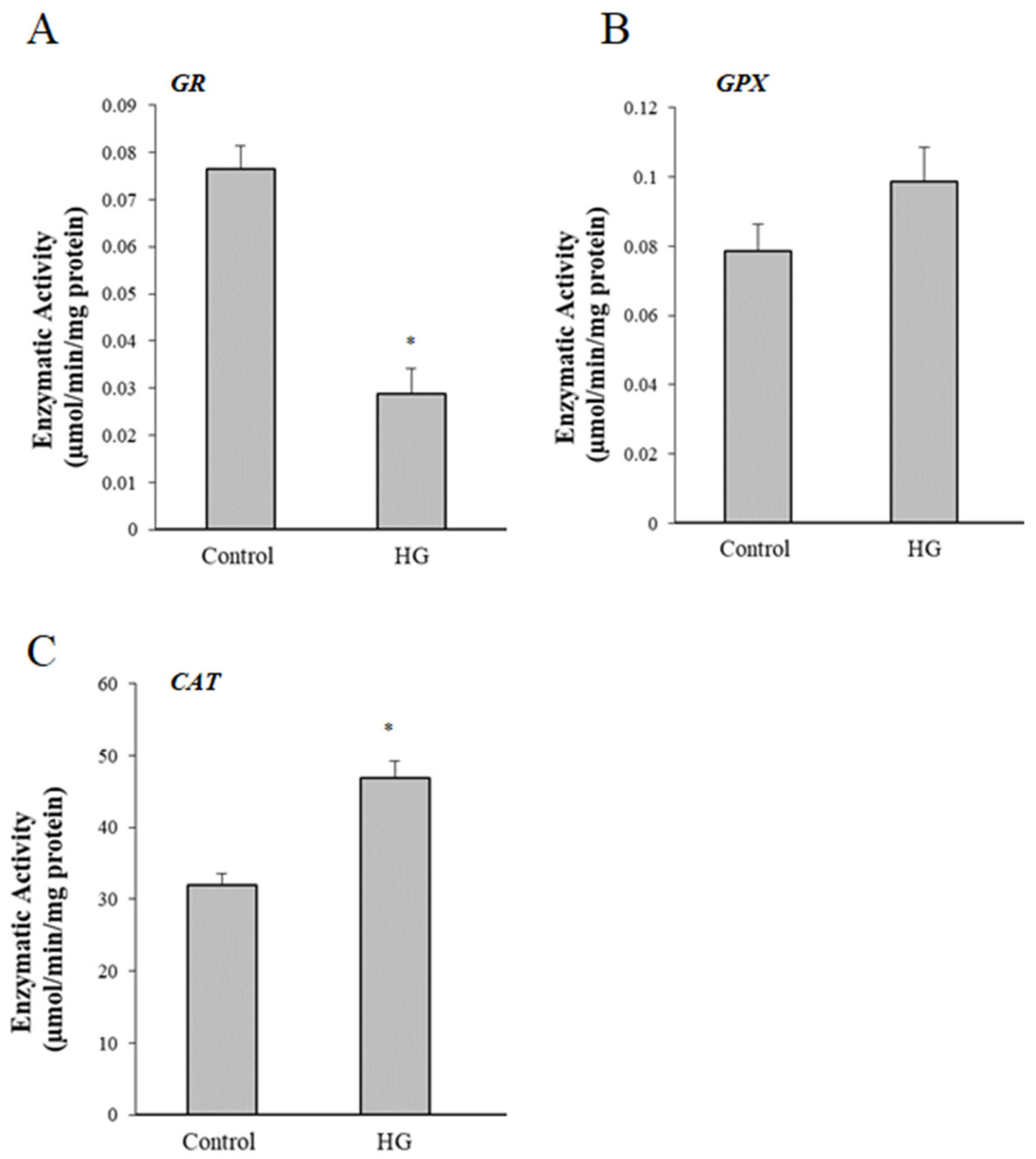
3.6. Effect of High Glucose (HG) Treatment on Activity of Glutathione Peroxidase (GPX), Glutathione Reductase (GR), and Catalase (CAT)
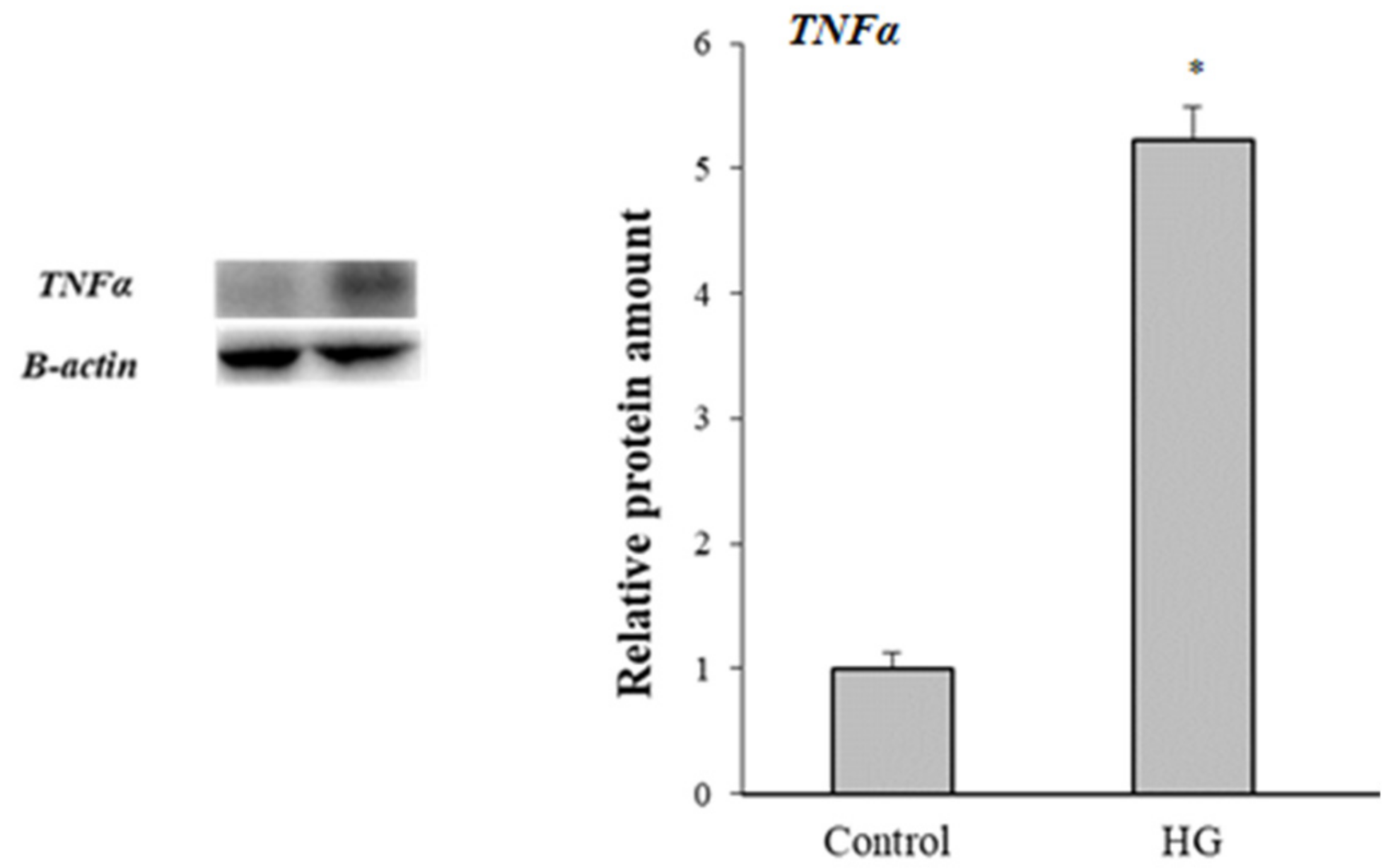
3.7. Inflammatory Response
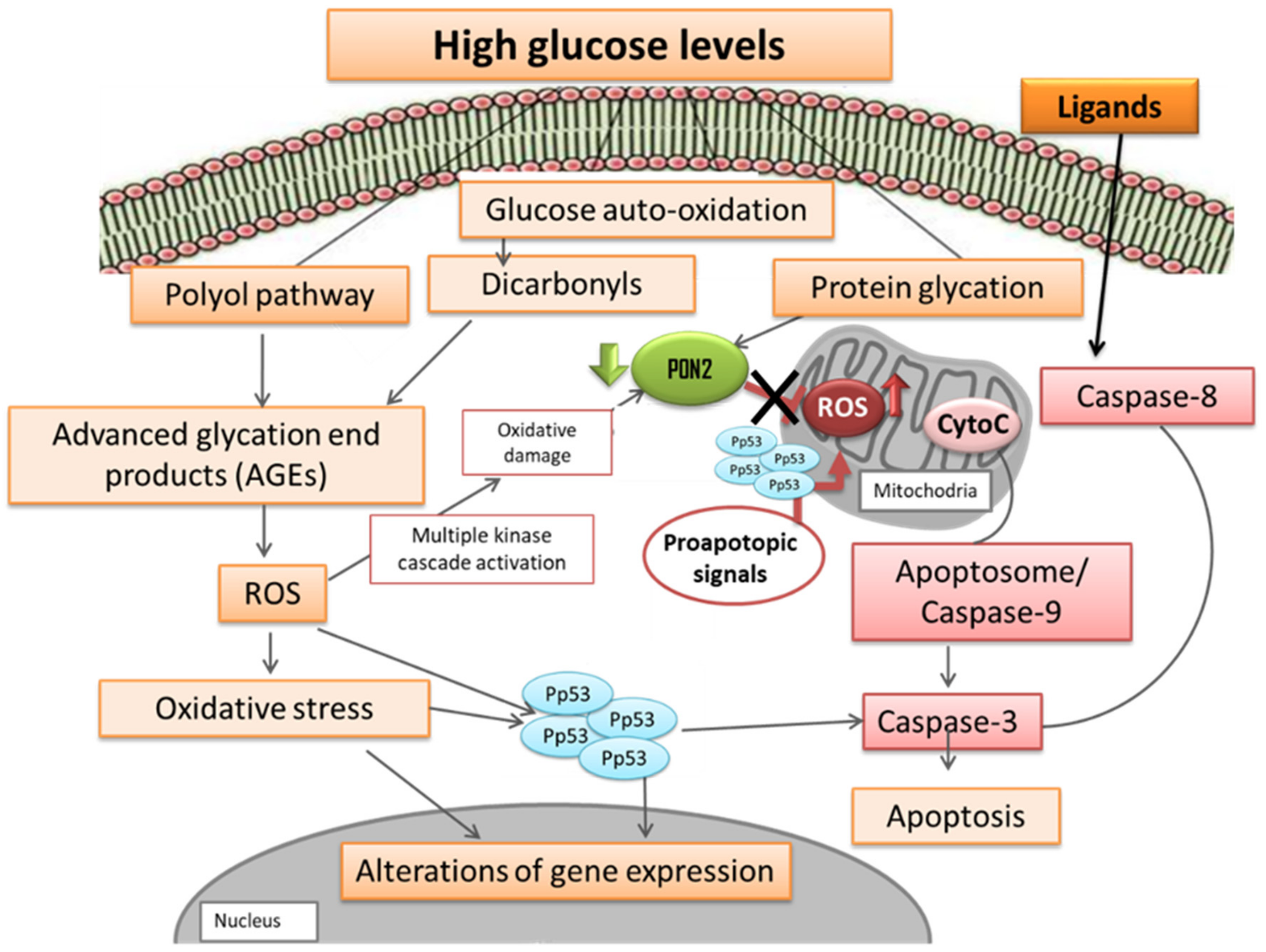
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Aw, T.Y. Molecular and cellular responses to oxidative stress and changes in oxidation-reduction imbalance in the intestine. Am. J. Clin. Nutr. 1999, 70, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.A. Oxygen radicals: Mediators of gastrointestinal pathophysiology. Gut 1989, 30, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Sottero, B.; Rossin, D.; Poli, G.; Biasi, F. Lipid Oxidation Products in the Pathogenesis of Inflammation-related Gut Diseases. Curr. Med. Chem. 2018, 25, 1311–1326. [Google Scholar] [CrossRef]
- Alzoghaibi, M.A. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J. Gastroentero. 2013, 19, 6540–6547. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxid. Med. Cell Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Et Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of reactive oxygen species in cell signalling pathways. Biochem. Soc. Trans. 2001, 29, 345–350. [Google Scholar] [CrossRef]
- Wen, Z.; Liu, W.; Li, X.; Chen, W.; Liu, Z.; Wen, J.; Liu, Z. A Protective Role of the NRF2-Keap1 Pathway in Maintaining Intestinal Barrier Function. Oxid. Med. Cell Longev. 2019, 2019, 1759149. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, L.; Li, X.; Zhang, L.; Lv, J.; Guo, X.; Chen, H.; Zhao, T. Neuroprotective effects of an Nrf2 agonist on high glucose-induced damage in HT22 cells. Biol. Res. 2019, 52, 53. [Google Scholar] [CrossRef]
- Zawada, A.E.; Moszak, M.; Skrzypczak, D.; Grzymislawski, M. Gastrointestinal complications in patients with diabetes mellitus. Adv. Clin. Exp. Med. 2018, 27, 567–572. [Google Scholar] [CrossRef]
- Krishnan, B.; Babu, S.; Walker, J.; Walker, A.B.; Pappachan, J.M. Gastrointestinal complications of diabetes mellitus. World J. Diabetes 2013, 4, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A.; Vinodkumar, P. Advanced Glycation End Products: Association with the Pathogenesis of Diseases and the Current Therapeutic Advances. Curr. Clin. Pharmacol. 2016, 11, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroentero. 2009, 15, 4137–4142. [Google Scholar] [CrossRef]
- Kaneto, H.; Fujii, J.; Myint, T.; Miyazawa, N.; Islam, K.N.; Kawasaki, Y.; Suzuki, K.; Nakamura, M.; Tatsumi, H.; Yamasaki, Y.; et al. Reducing sugars trigger oxidative modification and apoptosis in pancreatic beta-cells by provoking oxidative stress through the glycation reaction. Biochem. J. 1996, 320, 855–863. [Google Scholar] [CrossRef]
- Wolff, S.P.; Jiang, Z.Y.; Hunt, J.V. Protein Glycation and Oxidative Stress in Diabetes-Mellitus and Aging. Free Radic. Bio. Med. 1991, 10, 339–352. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef]
- Di Loreto, S.; Zimmitti, V.; Sebastiani, P.; Cervelli, C.; Falone, S.; Amicarelli, F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int. J. Biochem. Cell Biol. 2008, 40, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Rosenblat, M. Paraoxonases 1, 2, and 3, oxidative stress, and macrophage foam cell formation during atherosclerosis development. Free Radic. Biol. Med. 2004, 37, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.I.; La Du, B.N. Pharmacogenetics of paraoxonases: A brief review. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 369, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, K.; Socha, E.; Milnerowicz, H. Review: The role of paraoxonase in cardiovascular diseases. Ann. Clin. Lab. Sci. 2015, 45, 226–233. [Google Scholar]
- Shih, D.M.; Lusis, A.J. The roles of PON1 and PON2 in cardiovascular disease and innate immunity. Curr. Opin. Lipidol. 2009, 20, 288–292. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Masciangelo, S.; Bicchiega, V. HDL-paraoxonase and membrane lipid peroxidation: A comparison between healthy and obese subjects. Obesity 2010, 18, 1079–1084. [Google Scholar] [CrossRef]
- Precourt, L.P.; Marcil, V.; Ntimbane, T.; Taha, R.; Lavoie, J.C.; Delvin, E.; Seidman, E.G.; Beaulieu, J.F.; Levy, E. Antioxidative properties of paraoxonase 2 in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G623–634. [Google Scholar] [CrossRef]
- Precourt, L.P.; Seidman, E.; Delvin, E.; Amre, D.; Deslandres, C.; Dominguez, M.; Sinnett, D.; Levy, E. Comparative expression analysis reveals differences in the regulation of intestinal paraoxonase family members. Int. J. Biochem. Cell Biol. 2009, 41, 1628–1637. [Google Scholar] [CrossRef]
- Rothem, L.; Hartman, C.; Dahan, A.; Lachter, J.; Eliakim, R.; Shamir, R. Paraoxonases are associated with intestinal inflammatory diseases and intracellularly localized to the endoplasmic reticulum. Free Radic. Biol. Med. 2007, 43, 730–739. [Google Scholar] [CrossRef]
- Levy, E.; Trudel, K.; Bendayan, M.; Seidman, E.; Delvin, E.; Elchebly, M.; Lavoie, J.C.; Precourt, L.P.; Amre, D.; Sinnett, D. Biological role, protein expression, subcellular localization, and oxidative stress response of paraoxonase 2 in the intestine of humans and rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1252–1261. [Google Scholar] [CrossRef][Green Version]
- Shamir, R.; Hartman, C.; Karry, R.; Pavlotzky, E.; Eliakim, R.; Lachter, J.; Suissa, A.; Aviram, M. Paraoxonases (PONs) 1, 2, and 3 are expressed in human and mouse gastrointestinal tract and in Caco-2 cell line: Selective secretion of PON1 and PON2. Free Radic. Biol. Med. 2005, 39, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Witte, I.; Teiber, J.F.; Wilgenbus, P.; Pautz, A.; Li, H.; Daiber, A.; Witan, H.; Clement, A.M.; Forstermann, U.; et al. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J. Biol. Chem. 2010, 285, 24398–24403. [Google Scholar] [CrossRef] [PubMed]
- Witte, I.; Foerstermann, U.; Devarajan, A.; Reddy, S.T.; Horke, S. Protectors or Traitors: The Roles of PON2 and PON3 in Atherosclerosis and Cancer. J. Lipids 2012, 2012, 342806. [Google Scholar] [CrossRef]
- Horke, S.; Witte, I.; Wilgenbus, P.; Altenhofer, S.; Kruger, M.; Li, H.; Forstermann, U. Protective effect of paraoxonase-2 against endoplasmic reticulum stress-induced apoptosis is lost upon disturbance of calcium homoeostasis. Biochem. J. 2008, 416, 395–405. [Google Scholar] [CrossRef]
- Horke, S.; Witte, I.; Wilgenbus, P.; Kruger, M.; Strand, D.; Forstermann, U. Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation 2007, 115, 2055–2064. [Google Scholar] [CrossRef]
- Yehuda, I.; Madar, Z.; Szuchman-Sapir, A.; Tamir, S. Glabridin, a phytoestrogen from licorice root, up-regulates manganese superoxide dismutase, catalase and paraoxonase 2 under glucose stress. Phytother. Res. Ptr 2011, 25, 659–667. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins. Antioxidants 2019, 8, 27. [Google Scholar] [CrossRef]
- Linden, A.; Gulden, M.; Martin, H.J.; Maser, E.; Seibert, H. Peroxide-induced cell death and lipid peroxidation in C6 glioma cells. Toxicol. Vitr. 2008, 22, 1371–1376. [Google Scholar] [CrossRef]
- Balcerczyk, A.; Bartosz, G. Thiols are main determinants of total antioxidant capacity of cellular homogenates. Free Radic. Res. 2003, 37, 537–541. [Google Scholar] [CrossRef] [PubMed]
- LaSelva, M.; Beltramo, E.; Pagnozzi, F.; Bena, E.; Molinatti, P.A.; Molinatti, G.M.; Porta, M. Thiamine corrects delayed replication and decreases production of lactate and advanced glycation end-products in bovine retinal and human umbilical vein endothelial cells cultured under high glucose conditions. Diabetologia 1996, 39, 1263–1268. [Google Scholar]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Armeni, T.; Ercolani, L.; Urbanelli, L.; Magini, A.; Magherini, F.; Pugnaloni, A.; Piva, F.; Modesti, A.; Emiliani, C.; Principato, G. Cellular redox imbalance and changes of protein S-glutathionylation patterns are associated with senescence induced by oncogenic H-ras. PLoS ONE 2012, 7, e52151. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Chromogenic and fluorogenic assays for the lactonase activity of serum paraoxonases. Chembiochem A Eur. J. Chem. Biol. 2006, 7, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar]
- Morresi, C.; Cianfruglia, L.; Armeni, T.; Mancini, F.; Tenore, G.C.; D’Urso, E.; Micheletti, A.; Ferretti, G.T.B. Polyphenolic compounds and nutraceutical properties of old and new apple cultivars. J. Food Biochem. 2018, 42, e12641. [Google Scholar] [CrossRef]
- Alkhouri, R.H.; Baker, S.S.; Hashmi, H.; Liu, W.; Baker, R.D.; Zhu, L. Paraoxonase Gene Expression in Pediatric Inflammatory Bowel Disease. J. Clin. Cell. Immunol. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Yehuda, I.; Madar, Z.; Leikin-Frenkel, A.; Szuchman-Sapir, A.; Magzal, F.; Markman, G.; Tamir, S. Glabridin, an isoflavan from licorice root, upregulates paraoxonase 2 expression under hyperglycemia and protects it from oxidation. Mol. Nutr. Food Res. 2016, 60, 287–299. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell B 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef]
- Endo, T.; Kohda, D. Functions of outer membrane receptors in mitochondrial protein import. Biochim. Et Biophys. Acta 2002, 1592, 3–14. [Google Scholar] [CrossRef]
- Langer, S.; Kreutz, R.; Eisenreich, A. Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. J. Nephrol. 2016, 29, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-ssI and prooxidant enzyme NADPH oxidase. Redox Biol. 2014, 2, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Camarillo, C.; Guzman-Grenfell, A.M.; Garcia-Macedo, R.; Rosales-Torres, A.M.; Avalos-Rodriguez, A.; Duran-Reyes, G.; Medina-Navarro, R.; Cruz, M.; Diaz-Flores, M.; Kumate, J. Hyperglycemia induces apoptosis and p53 mobilization to mitochondria in RINm5F cells. Mol. Cell. Biochem. 2006, 281, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lee, S.; Bobadilla, S.; Duan, S.Z.; Liu, X. High glucose-induced p53 phosphorylation contributes to impairment of endothelial antioxidant system. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2017, 1863, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Reviews. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Flores-Lopez, L.A.; Diaz-Flores, M.; Garcia-Macedo, R.; Avalos-Rodriguez, A.; Vergara-Onofre, M.; Cruz, M.; Contreras-Ramos, A.; Konigsberg, M.; Ortega-Camarillo, C. High glucose induces mitochondrial p53 phosphorylation by p38 MAPK in pancreatic RINm5F cells. Mol. Biol. Rep. 2013, 40, 4947–4958. [Google Scholar] [CrossRef]
- Ng, C.J.; Bourquard, N.; Grijalva, V.; Hama, S.; Shih, D.M.; Navab, M.; Fogelman, A.M.; Lusis, A.J.; Young, S.; Reddy, S.T. Paraoxonase-2 deficiency aggravates atherosclerosis in mice despite lower apolipoprotein-B-containing lipoproteins: Anti-atherogenic role for paraoxonase-2. J. Biol. Chem. 2006, 281, 29491–29500. [Google Scholar] [CrossRef]
- Mastorikou, M.; Mackness, B.; Liu, Y.; Mackness, M. Glycation of paraoxonase-1 inhibits its activity and impairs the ability of high-density lipoprotein to metabolize membrane lipid hydroperoxides. Diabet. Med. A J. Br. Diabet. Assoc. 2008, 25, 1049–1055. [Google Scholar] [CrossRef]
- Bacchetti, T.; Masciangelo, S.; Armeni, T.; Bicchiega, V.; Ferretti, G. Glycation of human high density lipoprotein by methylglyoxal: Effect on HDL-paraoxonase activity. Metab. Clin. Exp. 2014, 63, 307–311. [Google Scholar] [CrossRef]
- Yu, W.; Liu, X.; Feng, L.; Yang, H.; Yu, W.; Feng, T.; Wang, S.; Wang, J.; Liu, N. Glycation of paraoxonase 1 by high glucose instigates endoplasmic reticulum stress to induce endothelial dysfunction in vivo. Sci. Rep. 2017, 7, 45827. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.B.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Volkova, N.; Aviram, M. Macrophage endoplasmic reticulum (ER) proteins and reducing elements stabilize paraoxonase 2 (PON2). Atherosclerosis 2010, 213, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.I.; Teiber, J.F.; Speelman, A.; Osawa, Y.; Sunahara, R.; La Du, B.N. Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res. 2005, 46, 1239–1247. [Google Scholar] [CrossRef]
- Qing, Q.; Zhang, S.; Chen, Y.; Li, R.; Mao, H.; Chen, Q. High glucose-induced intestinal epithelial barrier damage is aggravated by syndecan-1 destruction and heparanase overexpression. J. Cell Mol. Med. 2015, 19, 1366–1374. [Google Scholar] [CrossRef]
- Miyamoto, K.; Hase, K.; Takagi, T.; Fujii, T.; Taketani, Y.; Minami, H.; Oka, T.; Nakabou, Y. Differential responses of intestinal glucose transporter mRNA transcripts to levels of dietary sugars. Biochem. J. 1993, 295 Pt 1, 211–215. [Google Scholar] [CrossRef]
- Ritze, Y.; Bardos, G.; D’Haese, J.G.; Ernst, B.; Thurnheer, M.; Schultes, B.; Bischoff, S.C. Effect of high sugar intake on glucose transporter and weight regulating hormones in mice and humans. PLoS ONE 2014, 9, e101702. [Google Scholar] [CrossRef]
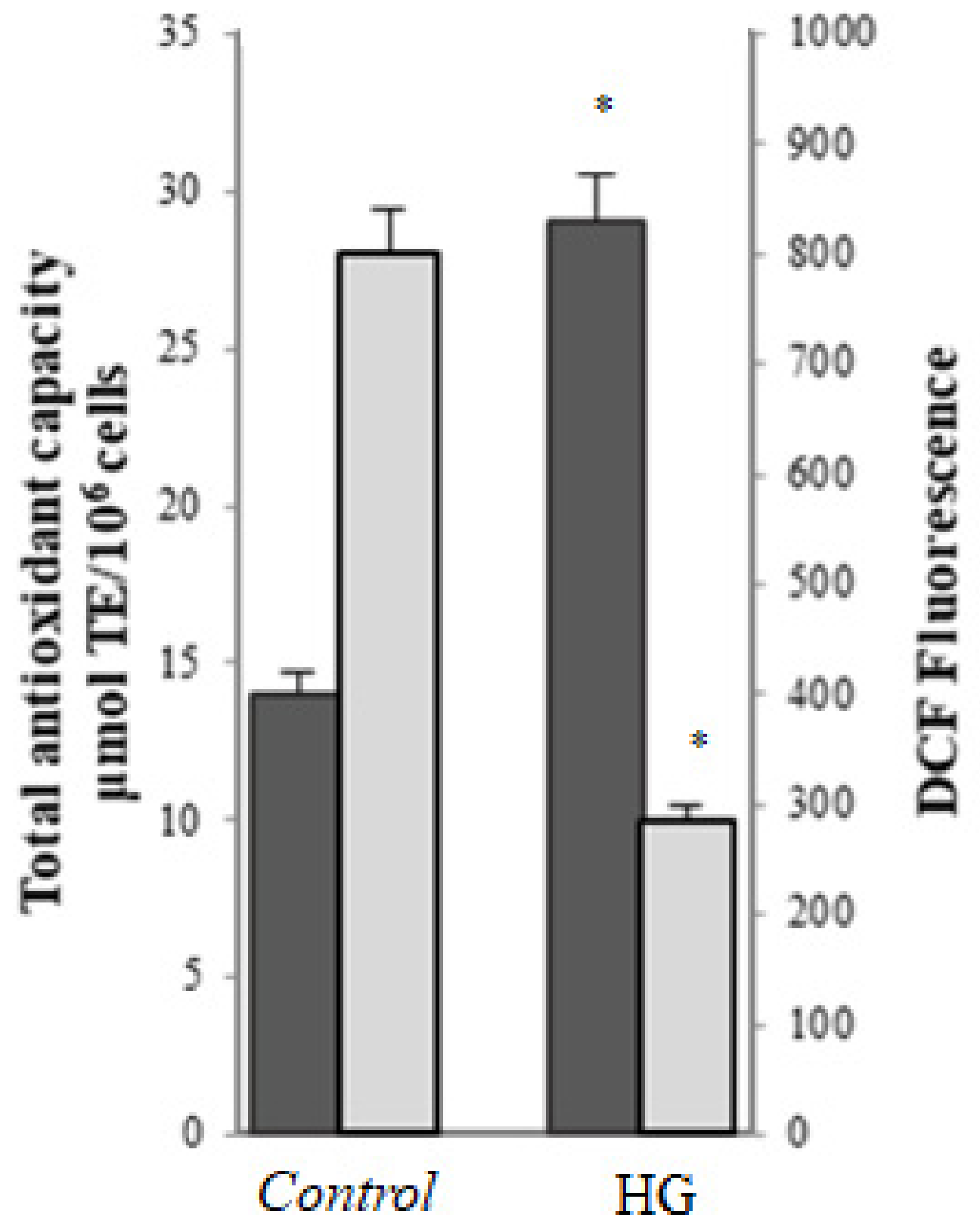
 ) and total antioxidant activity (
) and total antioxidant activity ( ) in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. Data are presented as means ± SD of three independent experiments carried out in triplicate (n = 9). * p ≤ 0.05 vs. control cells.
) and total antioxidant activity () in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. Data are presented as means ± SD of three independent experiments carried out in triplicate (n = 9). * p ≤ 0.05 vs. control cells.
) in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. Data are presented as means ± SD of three independent experiments carried out in triplicate (n = 9). * p ≤ 0.05 vs. control cells.
) and total antioxidant activity () in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. Data are presented as means ± SD of three independent experiments carried out in triplicate (n = 9). * p ≤ 0.05 vs. control cells.
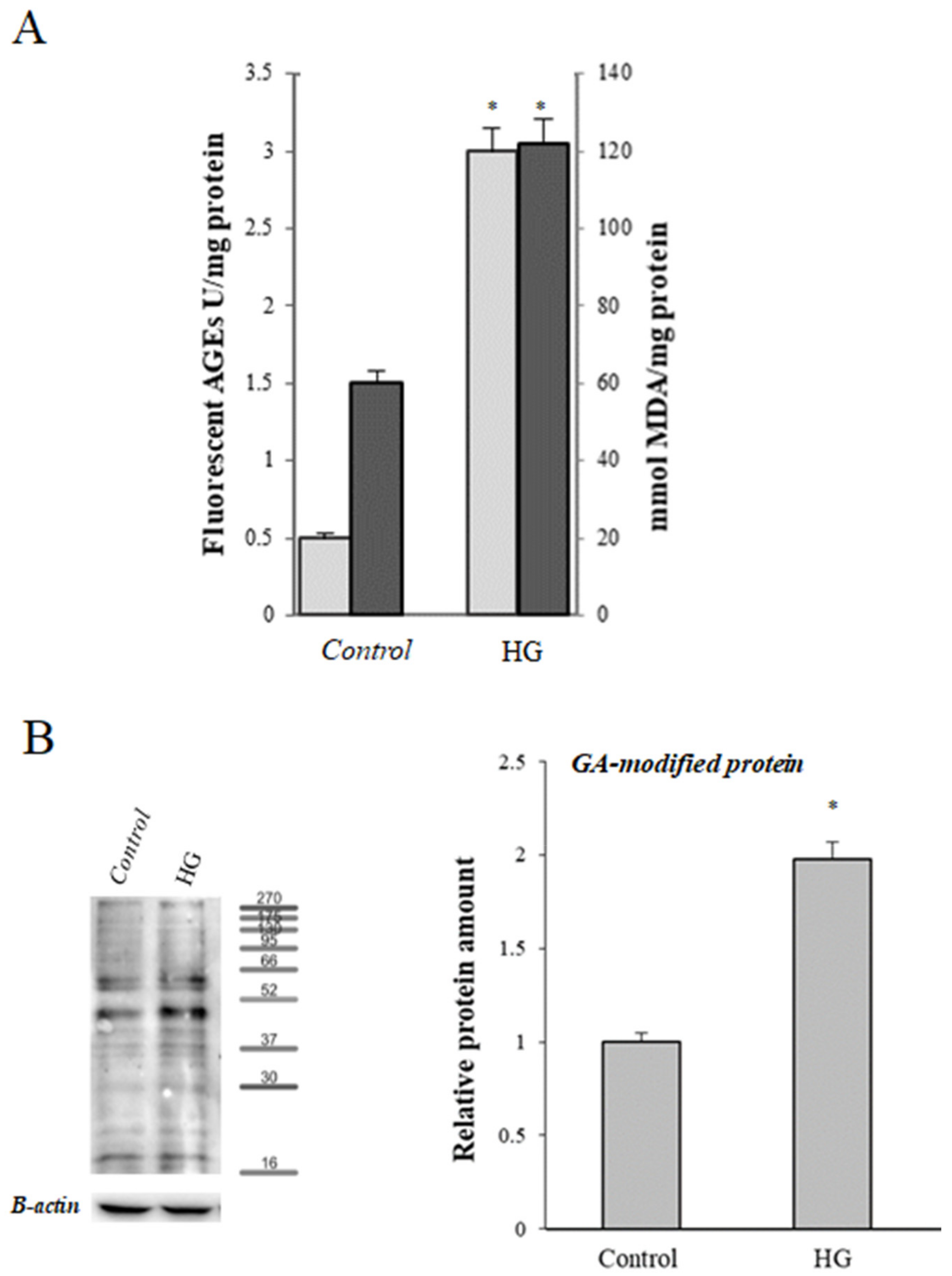
 ) and levels of malondialdehyde (MDA) (
) and levels of malondialdehyde (MDA) ( ) in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. (B) Representative Western blot and relative densitometric analysis of total GA-modified proteins in control or high glucose (HG) cells. Data are normalized on β-actin. Values reported are expressed as mean ± standard deviation of three independent experiments carried out in triplicate (* p < 0.05 vs. control cells).
) and levels of malondialdehyde (MDA) () in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. (B) Representative Western blot and relative densitometric analysis of total GA-modified proteins in control or high glucose (HG) cells. Data are normalized on β-actin. Values reported are expressed as mean ± standard deviation of three independent experiments carried out in triplicate (* p < 0.05 vs. control cells).
) in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. (B) Representative Western blot and relative densitometric analysis of total GA-modified proteins in control or high glucose (HG) cells. Data are normalized on β-actin. Values reported are expressed as mean ± standard deviation of three independent experiments carried out in triplicate (* p < 0.05 vs. control cells).
) and levels of malondialdehyde (MDA) () in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells. (B) Representative Western blot and relative densitometric analysis of total GA-modified proteins in control or high glucose (HG) cells. Data are normalized on β-actin. Values reported are expressed as mean ± standard deviation of three independent experiments carried out in triplicate (* p < 0.05 vs. control cells).
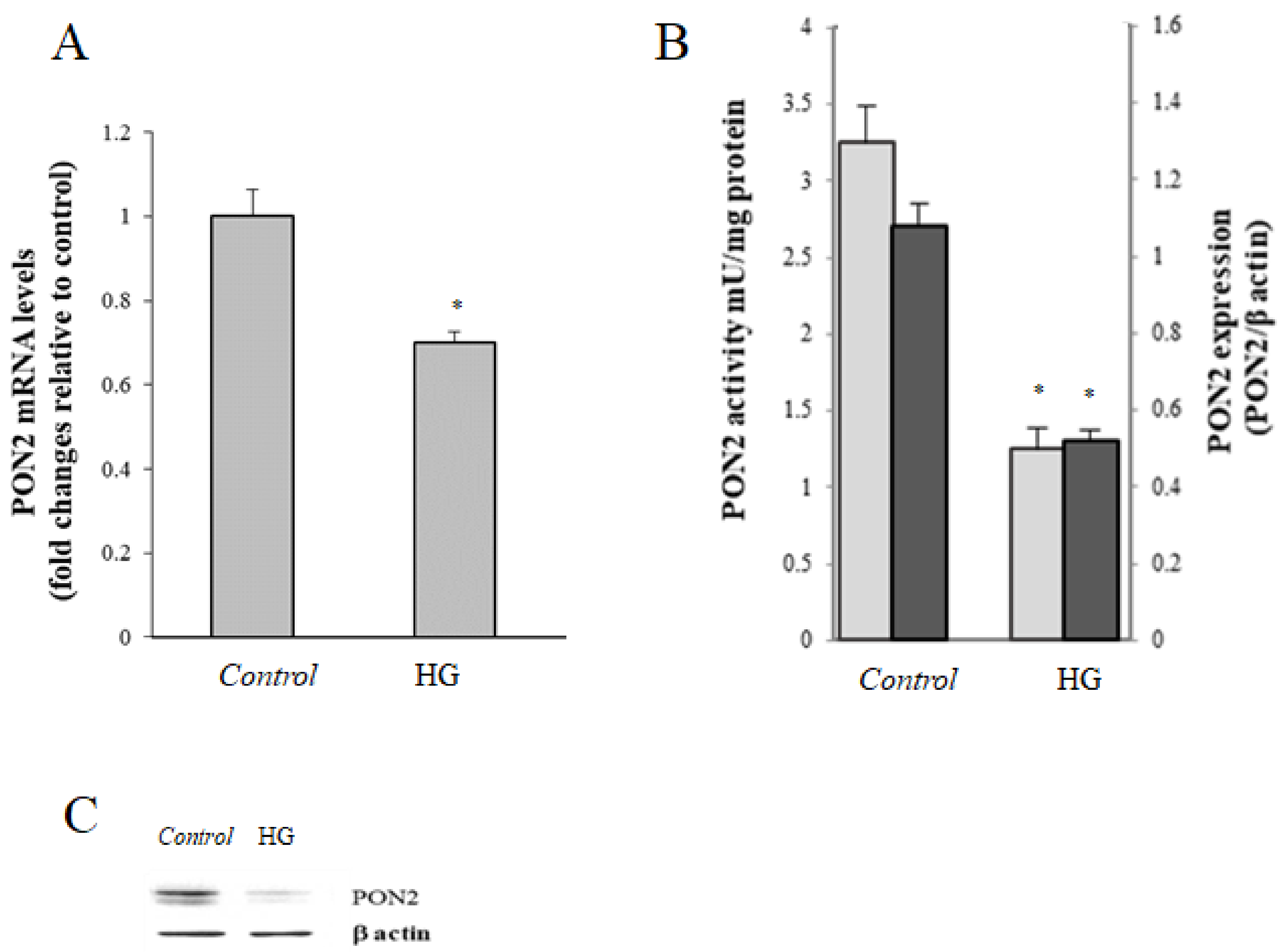
 ) and activity (
) and activity ( ) in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells; data are normalized on β-actin. (C) Representative Western blot of PON2 expression. Values reported are expressed as mean ± standard deviation (* p ≤ 0.05 vs. control cells).
) and activity () in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells; data are normalized on β-actin. (C) Representative Western blot of PON2 expression. Values reported are expressed as mean ± standard deviation (* p ≤ 0.05 vs. control cells).
) in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells; data are normalized on β-actin. (C) Representative Western blot of PON2 expression. Values reported are expressed as mean ± standard deviation (* p ≤ 0.05 vs. control cells).
) and activity () in control (25 mM glucose) or high glucose (HG) (50 mM glucose) cells; data are normalized on β-actin. (C) Representative Western blot of PON2 expression. Values reported are expressed as mean ± standard deviation (* p ≤ 0.05 vs. control cells).





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morresi, C.; Cianfruglia, L.; Sartini, D.; Cecati, M.; Fumarola, S.; Emanuelli, M.; Armeni, T.; Ferretti, G.; Bacchetti, T. Effect of High Glucose-Induced Oxidative Stress on Paraoxonase 2 Expression and Activity in Caco-2 Cells. Cells 2019, 8, 1616. https://doi.org/10.3390/cells8121616
Morresi C, Cianfruglia L, Sartini D, Cecati M, Fumarola S, Emanuelli M, Armeni T, Ferretti G, Bacchetti T. Effect of High Glucose-Induced Oxidative Stress on Paraoxonase 2 Expression and Activity in Caco-2 Cells. Cells. 2019; 8(12):1616. https://doi.org/10.3390/cells8121616
Chicago/Turabian StyleMorresi, Camilla, Laura Cianfruglia, Davide Sartini, Monia Cecati, Stefania Fumarola, Monica Emanuelli, Tatiana Armeni, Gianna Ferretti, and Tiziana Bacchetti. 2019. "Effect of High Glucose-Induced Oxidative Stress on Paraoxonase 2 Expression and Activity in Caco-2 Cells" Cells 8, no. 12: 1616. https://doi.org/10.3390/cells8121616
APA StyleMorresi, C., Cianfruglia, L., Sartini, D., Cecati, M., Fumarola, S., Emanuelli, M., Armeni, T., Ferretti, G., & Bacchetti, T. (2019). Effect of High Glucose-Induced Oxidative Stress on Paraoxonase 2 Expression and Activity in Caco-2 Cells. Cells, 8(12), 1616. https://doi.org/10.3390/cells8121616