Mild Iron Overload as Seen in Individuals Homozygous for the Alpha-1 Antitrypsin Pi*Z Variant Does Not Promote Liver Fibrogenesis in HFE Knockout Mice

 ,
, 
Abstract
1. Introduction
2. Material and Methods
2.1. Human Cohort
2.1.1. Cohort of Pi*ZZ and Non-Carrier Subjects
2.1.2. Assessment of Iron Parameters and Exclusion of Concomitant Liver Disease
2.1.3. HFE Mutation Analysis in Pi*ZZ Subjects
2.1.4. Ethical Statement
2.2. Mouse Experiments
2.3. Statistical Analysis
3. Results
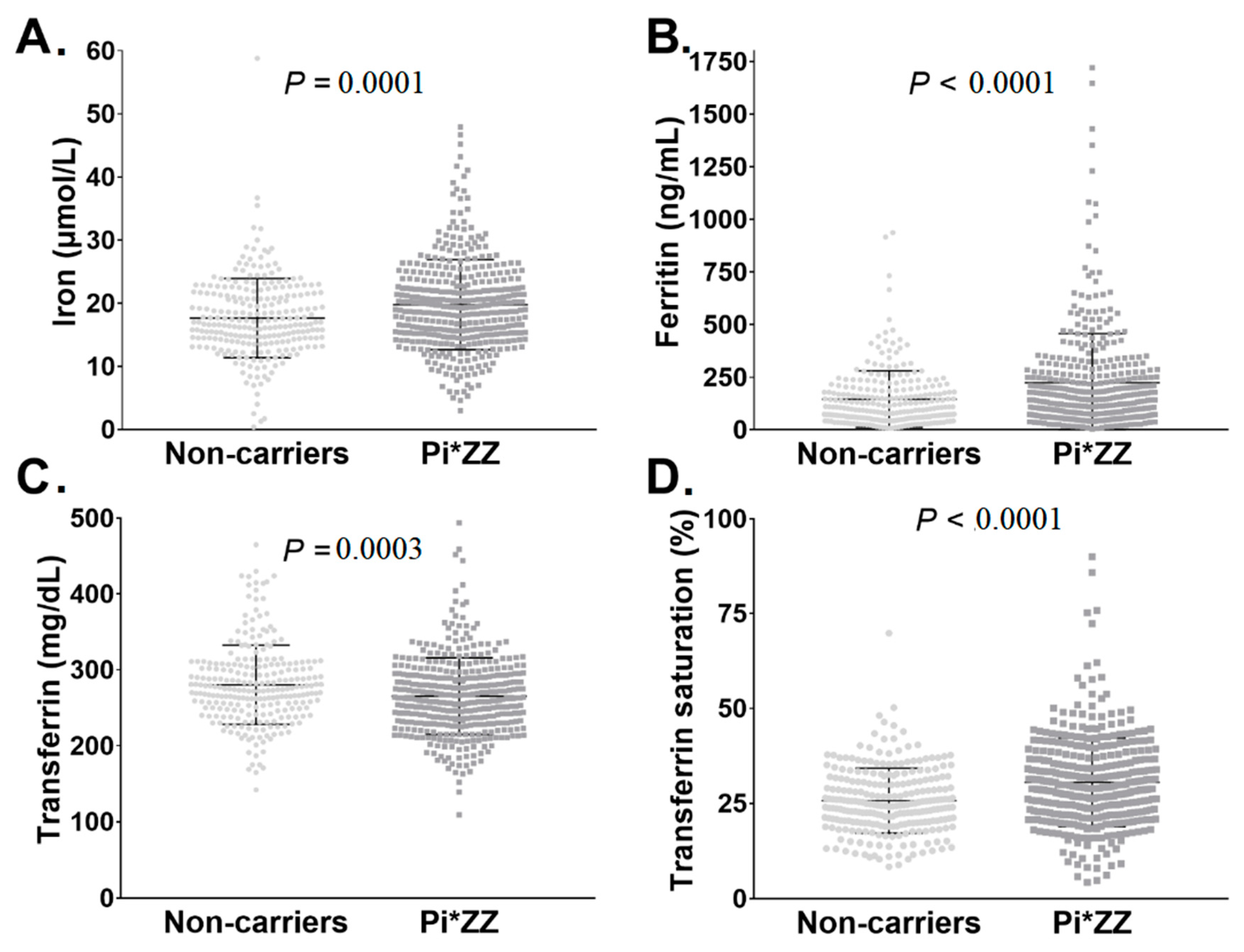
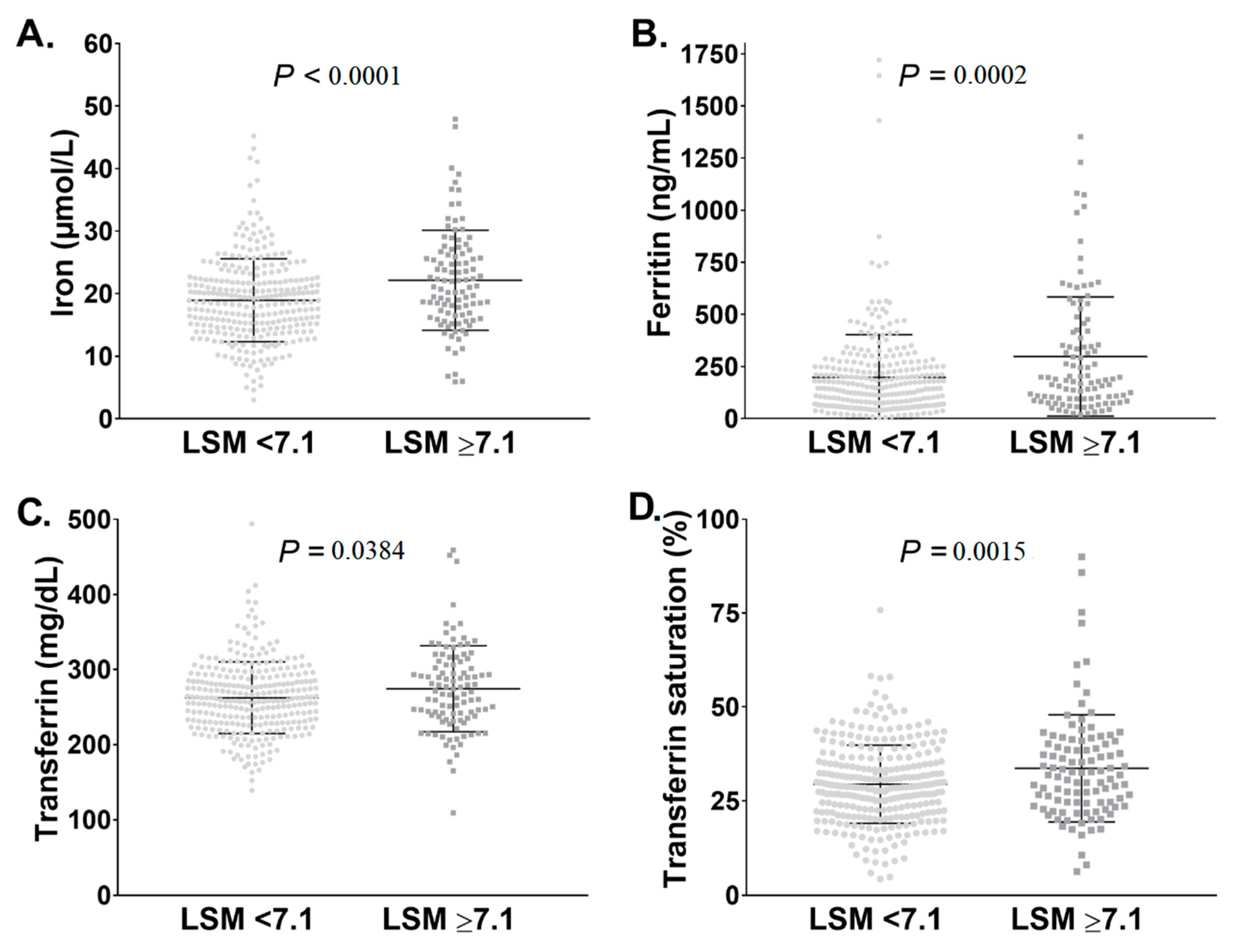
3.1. Pi*ZZ Individuals Displayed Mildly Elevated Iron Parameters
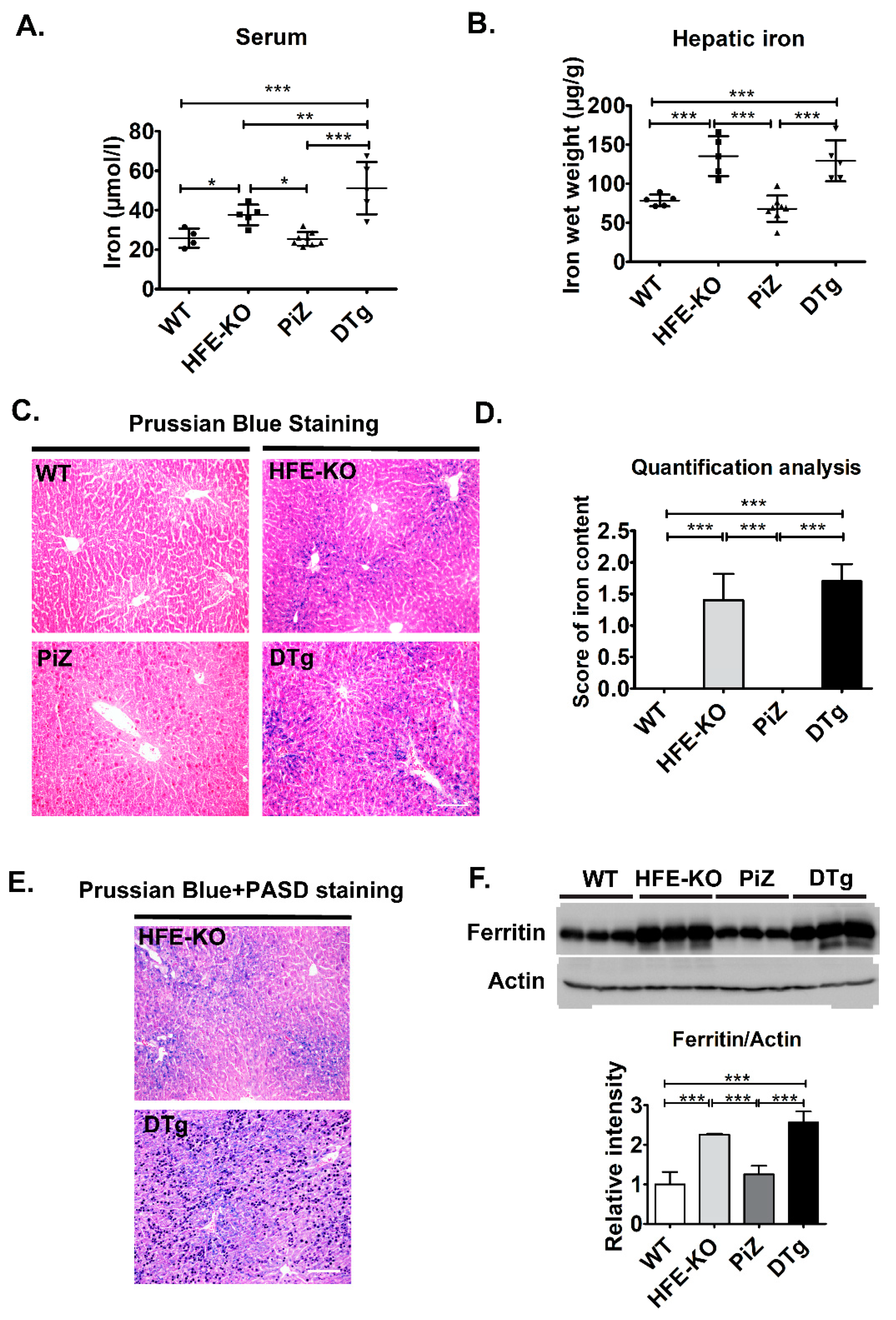
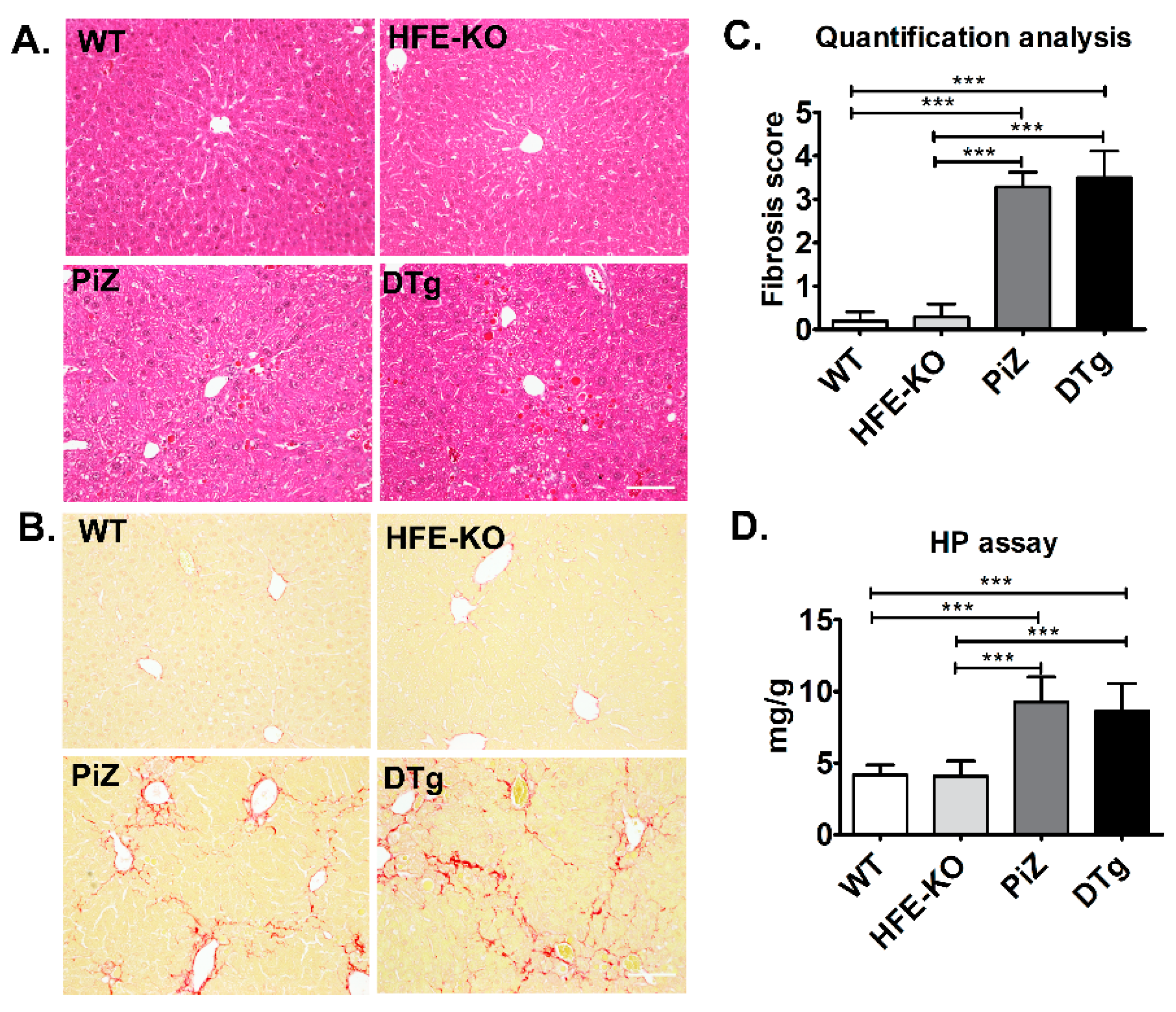
3.2. Mild Iron Overload in Double Transgenicmice Did Not Alter the Course of Pi*Z-Associated Liver Disease
3.3. Presence of HFE Mutations Did Not Associate with Increased Liver Injury in Pi*ZZ Individuals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAT | alpha-1 antitrypsin |
| AATD | Alpha-1 antitrypsin deficiency |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| CAP | Controlled attenuation parameter (via transient elastography (FibroScan®) |
| CAT | COPD assessment test |
| COPD | chronic obstructive pulmonary disease |
| HFE | Hereditary Hemochromatosis Protein |
| LSM | liver stiffness measurement (via transient elastography (FibroScan®) |
| LTOT | long-term oxygen therapy |
| SERPINA1 | serpin family A member 1 (AAT gene) |
| TF | transferrin |
| TSAT | transferrin saturation |
References
- Lomas, D.A.; Hurst, J.R.; Gooptu, B. Update on alpha-1 antitrypsin deficiency: New therapies. J. Hepatol. 2016, 65, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. Alpha1-Antitrypsin deficiency. Nat. Rev. Dis. Primers 2016, 2, 16051. [Google Scholar] [CrossRef] [PubMed]
- Kuscuoglu, D.; Janciauskiene, S.; Hamesch, K.; Haybaeck, J.; Trautwein, C.; Strnad, P. Liver—Master and servant of serum proteome. J. Hepatol. 2018, 69, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, P.; Attanasio, S.; Brunetti-Pierri, N. Mechanisms of liver disease in AATD. In α1-Antitrypsin Deficiency; ERS Monograph: London, UK, 2019. [Google Scholar]
- Patel, D.; Teckman, J.H. Alpha-1-Antitrypsin Deficiency Liver Disease. Clin. Liver Dis. 2018, 22, 643–655. [Google Scholar] [CrossRef]
- Clark, V.C.; Marek, G.; Liu, C.; Collinsworth, A.; Shuster, J.; Kurtz, T.; Nolte, J.; Brantly, M. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J. Hepatol. 2018, 69, 1357–1364. [Google Scholar] [CrossRef]
- Hamesch, K.; Mandorfer, M.; Pereira, V.M.; Moeller, L.S.; Pons, M.; Dolman, G.E.; Reichert, M.C.; Schneider, C.V.; Woditsch, V.; Voss, J.; et al. Liver Fibrosis and Metabolic Alterations in Adults with alpha-1-antitrypsin Deficiency Caused by the Pi* ZZ Mutation. Gastroenterology 2019, 157, 705–719.e18. [Google Scholar] [CrossRef]
- Strnad, P.; Brantly, M.; Bals, R. α1-Antitrypsin Deficiency; ERS Monograph: London, UK, 2019; Volume 85. [Google Scholar]
- Pereira, V.; Hamesch, H.; Strnad, P. Liver fibrosis assessment in adults with alpha1-antitrypsin deficiency. In Liver Elastography: Clinical Use and Interpretation; Springer Nature: Berlin, Germany, 2019; in press. [Google Scholar]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019, 68, 1099–1107. [Google Scholar] [CrossRef]
- Townsend, S.A.; Edgar, R.G.; Ellis, P.R.; Kantas, D.; Newsome, P.N.; Turner, A.M. Systematic review: The natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment. Pharmacol. Ther. 2018, 47, 877–885. [Google Scholar] [CrossRef]
- Lam, M.; Torbenson, M.; Yeh, M.M.; Vivekanandan, P.; Ferrell, L. HFE mutations in alpha-1-antitrypsin deficiency: An examination of cirrhotic explants. Mod. Pathol. 2010, 23, 637–643. [Google Scholar] [CrossRef]
- Schaefer, B.; Haschka, D.; Finkenstedt, A.; Petersen, B.S.; Theurl, I.; Henninger, B.; Janecke, A.R.; Wang, C.Y.; Lin, H.Y.; Veits, L.; et al. Impaired hepcidin expression in alpha-1-antitrypsin deficiency associated with iron overload and progressive liver disease. Hum. Mol. Genet. 2015, 24, 6254–6263. [Google Scholar] [CrossRef] [PubMed]
- Pandur, E.; Nagy, J.; Poor, V.S.; Sarnyai, A.; Huszar, A.; Miseta, A.; Sipos, K. Alpha-1 antitrypsin binds preprohepcidin intracellularly and prohepcidin in the serum. FEBS J. 2009, 276, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitz, M.; Gavaler, J.S.; Kelly, R.H.; Van Thiel, D.H. Association between heterozygous alpha 1-antitrypsin deficiency and genetic hemochromatosis. Hepatology 1992, 16, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Elzouki, A.N.; Hultcrantz, R.; Stal, P.; Befrits, R.; Eriksson, S. Increased PiZ gene frequency for alpha 1 antitrypsin in patients with genetic haemochromatosis. Gut 1995, 36, 922–926. [Google Scholar] [CrossRef]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Lawless, M.W.; Mankan, A.K.; White, M.; O’Dwyer, M.J.; Norris, S. Expression of hereditary hemochromatosis C282Y HFE protein in HEK293 cells activates specific endoplasmic reticulum stress responses. BMC Cell Biol. 2007, 8, 30. [Google Scholar] [CrossRef]
- Joly, P.; Vignaud, H.; Di Martino, J.; Ruiz, M.; Garin, R.; Restier, L.; Belmalih, A.; Marchal, C.; Cullin, C.; Arveiler, B.; et al. ERAD defects and the HFE-H63D variant are associated with increased risk of liver damages in Alpha 1-Antitrypsin Deficiency. PLoS ONE 2017, 12, e0179369. [Google Scholar] [CrossRef]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Neves, J.; Haider, T.; Gassmann, M.; Muckenthaler, M.U. Iron Homeostasis in the Lungs-A Balance between Health and Disease. Pharmaceuticals 2019, 12, 5. [Google Scholar] [CrossRef]
- Zhang, V.; Nemeth, E.; Kim, A. Iron in Lung Pathology. Pharmaceuticals 2019, 12, 30. [Google Scholar] [CrossRef]
- Khiroya, H.; Turner, A.M. The role of iron in pulmonary pathology. Multidiscip. Respir. Med. 2015, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Mumby, S.; Adcock, I.M.; Choi, A.M.K.; Chung, K.F.; Quinlan, G.J. The “Iron”-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 196, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Hilborn, E.D. Indices of iron homeostasis correlate with airway obstruction in an NHANES III cohort. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Wood, A.M.; Barker, A.F.; Brantly, M.L.; Campbell, E.J.; Eden, E.; McElvaney, G.; Rennard, S.I.; Sandhaus, R.A.; Stocks, J.M.; et al. Association of IREB2 and CHRNA3 polymorphisms with airflow obstruction in severe alpha-1 antitrypsin deficiency. Respir. Res. 2012, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Lohr, B.; El-Samalouti, V.; Junge, W.; Maatouk, H.; Halabi, A.; Fahle, A.; Bossert-Reuther, S.; Jung, M.; Berding, C.; Domke, I. Reference range study for various parameters on Roche clinical chemistry analyzers. Clin. Lab. 2009, 55, 465–471. [Google Scholar]
- Lotz, J.; Hafner, G.; Prellwitz, W. Reference study for ferritin assays. Kurzmitt. Clin. Lab. 1997, 43, 993–994. [Google Scholar]
- Schumann, G.; Dati, F. Vorläufige Referenzbereiche für 14 Proteine im Serum (für Erwachsene) nach Standardisierung immunchemischer Methoden unter Bezug auf das internationale Referenzmaterial CRM. Lab. Med. 1995, 19, 401–403. [Google Scholar]
- Friedrich-Rust, M.; Poynard, T.; Castera, L. Critical comparison of elastography methods to assess chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 402–411. [Google Scholar] [CrossRef]
- Tapper, E.B.; Lok, A.S.F. Use of Liver Imaging and Biopsy in Clinical Practice. N. Engl. J. Med. 2017, 377, 2296–2297. [Google Scholar] [CrossRef]
- Kümpers, J.; Fromme, M.; Schneider, C.V.; Trautwein, C.; Denk, H.; Hamesch, K.; Strnad, P. Assessment of liver phenotype in adults with severe alpha1-antitrypsin deficiency (Pi*ZZ genotype). J. Hepatol. 2019, in press. [Google Scholar] [CrossRef]
- Jeffrey, G.P.; Chakrabarti, S.; Hegele, R.A.; Adams, P.C. Polymorphism in intron 4 of HFE may cause overestimation of C282Y homozygote prevalence in haemochromatosis. Nat. Genet. 1999, 22, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Best, L.G.; Harris, P.E.; Spriggs, E.L. Hemochromatosis mutations C282Y and H63D in ‘cis’ phase. Clin. Genet. 2001, 60, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Finegold, M.J.; Clift, S.M.; DeMayo, F.J.; Bullock, D.W.; Woo, S.L. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Investig. 1989, 83, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Tomatsu, S.; Fleming, R.E.; Parkkila, S.; Waheed, A.; Jiang, J.; Fei, Y.; Brunt, E.M.; Ruddy, D.A.; Prass, C.E.; et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA 1998, 95, 2492–2497. [Google Scholar] [CrossRef]
- Levy, J.E.; Montross, L.K.; Andrews, N.C. Genes that modify the hemochromatosis phenotype in mice. J. Clin. Investig. 2000, 105, 1209–1216. [Google Scholar] [CrossRef]
- Czaja, A.J. Review article: Iron disturbances in chronic liver diseases other than haemochromatosis—Pathogenic, prognostic, and therapeutic implications. Aliment. Pharmacol. Ther. 2019, 49, 681–701. [Google Scholar] [CrossRef]
- Karlas, T.; Petroff, D.; Sasso, M.; Fan, J.G.; Mi, Y.Q.; de Ledinghen, V.; Kumar, M.; Lupsor-Platon, M.; Han, K.H.; Cardoso, A.C.; et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J. Hepatol. 2017, 66, 1022–1030. [Google Scholar] [CrossRef]
- Piccolo, P.; Annunziata, P.; Soria, L.R.; Attanasio, S.; Barbato, A.; Castello, R.; Carissimo, A.; Quagliata, L.; Terracciano, L.M.; Brunetti-Pierri, N. Down-regulation of hepatocyte nuclear factor-4alpha and defective zonation in livers expressing mutant Z alpha1-antitrypsin. Hepatology 2017, 66, 124–135. [Google Scholar] [CrossRef]
- Matsuo, S.; Ogawa, M.; Muckenthaler, M.U.; Mizui, Y.; Sasaki, S.; Fujimura, T.; Takizawa, M.; Ariga, N.; Ozaki, H.; Sakaguchi, M.; et al. Hepatocyte Nuclear Factor 4alpha Controls Iron Metabolism and Regulates Transferrin Receptor 2 in Mouse Liver. J. Biol. Chem. 2015, 290, 30855–30865. [Google Scholar] [CrossRef]
- Bhagat, C.I.; Fletcher, S.; Joseph, J.; Beilby, J.P. Plasma ferritin in acute hepatocellular damage. Clin. Chem. 2000, 46, 885–886. [Google Scholar]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lee, S.Y.; Neely, E.; Nandar, W.; Moyo, M.; Simmons, Z.; Connor, J.R. Mutant HFE H63D protein is associated with prolonged endoplasmic reticulum stress and increased neuronal vulnerability. J. Biol. Chem. 2011, 286, 13161–13170. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Non-Carriers (n = 254) | Pi*ZZ Carriers (n = 409) | p Value |
|---|---|---|---|
| Age (years) | 51.8 ± 15.4 | 56.1 ± 12.2 | <0.0001 |
| Women (%) | 50.8 | 46.0 | 0.23 |
| BMI (kg/m2) | 25.5 ± 4.6 | 25.0 ± 4.3 | 0.30 |
| BMI ≥ 30 kg/m2 (%) | 17.8 | 15.7 | 0.48 |
| Diabetes (%) | 5.1 | 3.4 | 0.28 |
| Mean alcohol consumption (g/d) | 7.5 ± 10.3 | 6.0 ± 9.5 | 0.15 |
| COPD assessment test (points) | 7.6 ± 6.4 | 17.3 ± 8.8 | <0.0001 |
| Long-term oxygen therapy (%) | 0.4 | 21.1 | <0.0001 |
| AAT augmentation therapy (%) | 0 | 60.4 | <0.0001 |
| Liver stiffness (kPa) * | 4.7 ± 2.8 | 7.1 ± 7.3 | <0.0001 |
| Controlled attenuation parameter (dB/m) | 245 ± 59 | 268 ± 60 | <0.0001 |
| Experimental Group | WT | HFE-KO | PiZ | DTg |
|---|---|---|---|---|
| Body weight (g) | 22.9 ± 1.04 | 23.4 ± 1.6 | 22.3 ± 1.5 | 22.8 ± 1.7 |
| Liver/Body weight (g) | 0.04 ± 0.002 a,b | 0.04 ± 0.004 c,d | 0.05 ± 0.004 a,c | 0.05 ± 0.005 b,d |
| ALT (U/L) | 21 ± 6.3 | 18.6 ± 5.4 | 25.5 ± 6 | 24.6 ± 3.9 |
| AST (U/L) | 42.6 ± 11.1 | 48.6 ± 6.5 | 57.7 ± 13.5 | 56.4 ± 17.3 |
| ALP (U/L) | 99.6 ± 40.7 | 77.4 ± 9.1 | 84.4 ± 11.3 | 99.6 ± 46.3 |
| Experimental Group | WT | HFE-KO | PiZ | DTg |
|---|---|---|---|---|
| Body weight (g) | 33.2 ± 2.9 a,b | 32.8 ± 3.6 c,d | 27.8 ± 1.5 a,c | 26.4 ± 2.4 b,d |
| Liver/Body weight (g) | 0.04 ± 0.004 | 0.05 ± 0.01 | 0.05 ± 0.004 | 0.05 ± 0.005 |
| ALT (U/L) | 49 ± 18.8 | 75.6 ± 48.6 | 56.6 ± 18.6 | 51.2 ± 20.6 |
| AST (U/L) | 79.8 ± 32.3 | 106 ± 48.6 | 122.7 ± 29 | 118.1 ± 26.5 |
| (U/L) | 105.8 ± 47.2 e,f | 94.2 ± 32 g,h | 204 ± 68.8 e,g | 189.4 ± 76.7 f,h |
| Category | C282Y & H63D Non-Carriers (n = 246) | C282Y Hetero-zygous (n = 24) | H63D Hetero-zygous (n = 99) | H63D Homo-zygous (n = 7) | C282Y/H63D Compound Heterozygous (n = 4) |
|---|---|---|---|---|---|
| Age (years) | 56.5 ± 12.1 | 53.1 ± 13.8 | 56.0 ± 11.7 | 55.6 ± 15.9 | 65.7 ± 11.1 |
| Women (%) | 44.3 | 50.0 | 50.5 | 42.9 | 46.3 |
| BMI (kg/m2) | 24.9 ± 3.8 | 25.8 ± 5.0 | 25.2 ± 5.4 | 23.8 ± 2.3 | 23.5 ± 2.2 |
| BMI ≥ 30 kg/m2 (%) | 14.3 | 20.8 | 20.2 | 0 | 0 |
| Diabetes (%) | 2.8 | 0 | 6.1 | 0 | 25.0 |
| Mean alcohol consumption (g/d) | 6.6 ± 9.6 | 2.6 ± 3.5 | 5.3 ± 10.3 | 2.9 ± 4.4 | 3.0 ± 5.9 |
| COPD assessment test (points) | 17.3 ± 8.9 | 15.8 ± 8.5 | 18.7 ± 8.3 | 14.4 ± 13.2 | 13.2 ± 8.2 |
| Long-term oxygen therapy (%) | 20.8 | 20.8 | 24.5 | 14.3 | 25.0 |
| AAT augmentation therapy (%) | 61.1 | 50.0 | 64.6 | 57.1 | 50.0 |
| Ferritin (ng/mL) | 205.8 ± 203.2 | 160.7 ± 154.6 | 246.9 ± 228.3 | 167.6 ± 93.9 | 238.0 ± 160.8 |
| Transferrin (mg/dL) | 268.8 ± 55.3 | 259.6 ± 37.9 | 261.4 ± 3.6 | 247.7 ± 57.2 | 233.8 ± 43.1 |
| TSAT (%) * | 29.2 ± 10.4 | 33.9 ± 17.1 | 30.3 ± 9.3 | 38.3 ± 8.9 | 57.8 ± 20.1 |
| Liver stiffness (kPa) | 7.1 ± 6.7 | 6.0 ± 2.8 | 5.9 ± 3.1 | 5.1 ± 1.3 | 6.4 ± 3.7 |
| LSM ≥ 7.1 kPa (%) | 25.6 | 22.7 | 20.4 | 14.3 | 25.0 |
| LSM ≥ 10 kPa (%) | 13.0 | 9.1 | 5.1 | 0 | 25.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guldiken, N.; Hamesch, K.; Schuller, S.M.; Aly, M.; Lindhauer, C.; Schneider, C.V.; Fromme, M.; Trautwein, C.; Strnad, P. Mild Iron Overload as Seen in Individuals Homozygous for the Alpha-1 Antitrypsin Pi*Z Variant Does Not Promote Liver Fibrogenesis in HFE Knockout Mice. Cells 2019, 8, 1415. https://doi.org/10.3390/cells8111415
Guldiken N, Hamesch K, Schuller SM, Aly M, Lindhauer C, Schneider CV, Fromme M, Trautwein C, Strnad P. Mild Iron Overload as Seen in Individuals Homozygous for the Alpha-1 Antitrypsin Pi*Z Variant Does Not Promote Liver Fibrogenesis in HFE Knockout Mice. Cells. 2019; 8(11):1415. https://doi.org/10.3390/cells8111415
Chicago/Turabian StyleGuldiken, Nurdan, Karim Hamesch, Shari Malan Schuller, Mahmoud Aly, Cecilia Lindhauer, Carolin V. Schneider, Malin Fromme, Christian Trautwein, and Pavel Strnad. 2019. "Mild Iron Overload as Seen in Individuals Homozygous for the Alpha-1 Antitrypsin Pi*Z Variant Does Not Promote Liver Fibrogenesis in HFE Knockout Mice" Cells 8, no. 11: 1415. https://doi.org/10.3390/cells8111415
APA StyleGuldiken, N., Hamesch, K., Schuller, S. M., Aly, M., Lindhauer, C., Schneider, C. V., Fromme, M., Trautwein, C., & Strnad, P. (2019). Mild Iron Overload as Seen in Individuals Homozygous for the Alpha-1 Antitrypsin Pi*Z Variant Does Not Promote Liver Fibrogenesis in HFE Knockout Mice. Cells, 8(11), 1415. https://doi.org/10.3390/cells8111415