Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents

 ,
,  , and
, and
Abstract

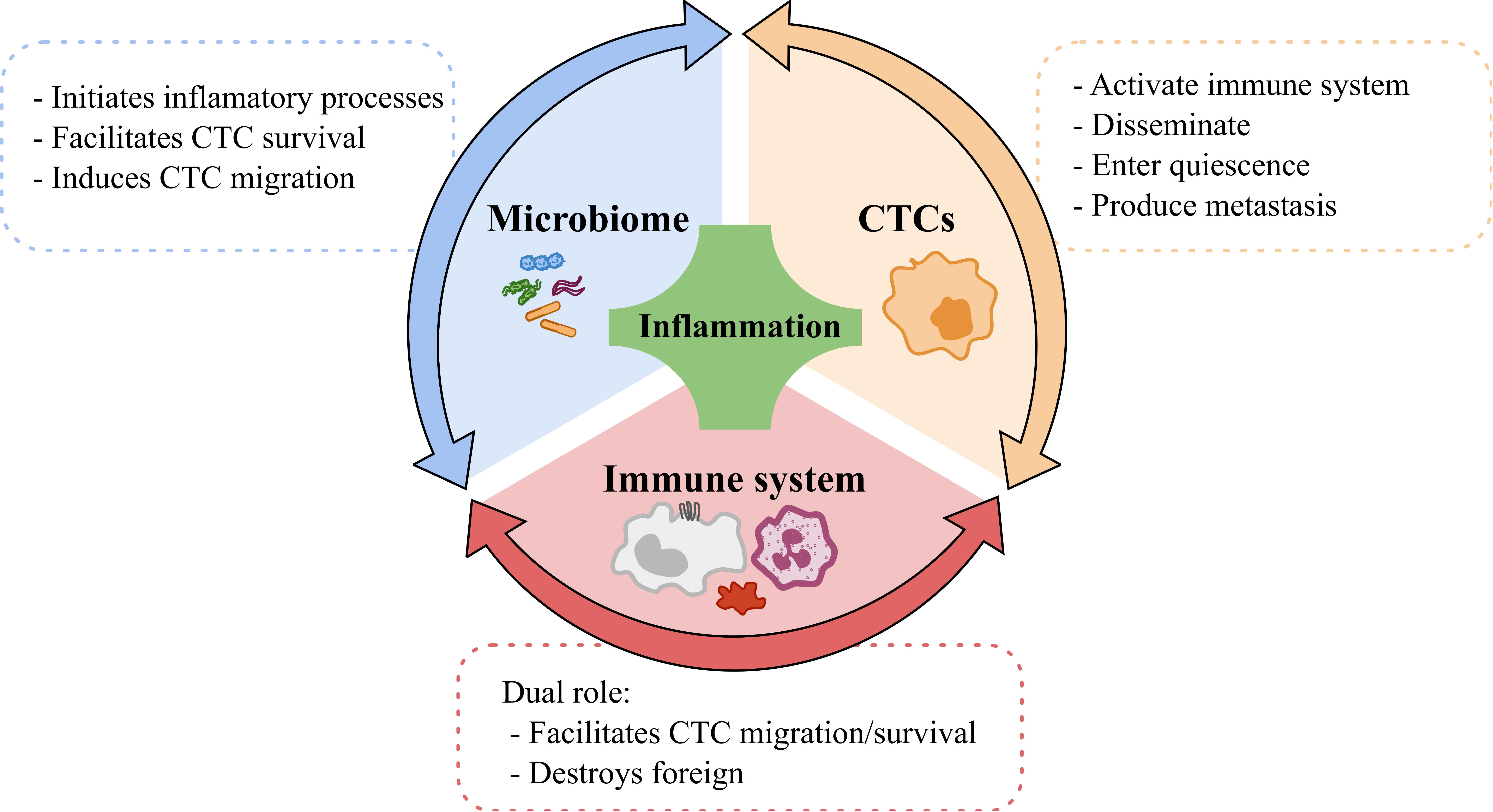{kind=link}
{kind=link}
1. Introduction
2. Promotion of Circulating Tumor Cells through the Immune System
3. Survival of Circulating Tumor Cells Through the Interaction of Microbiota with the Immune System
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bayarri-Lara, C.; Ortega, F.G.; De Guevara, A.C.L.; Puche, J.L.; Zafra, J.R.; De Miguel-Pérez, D.; Ramos, A.S.-P.; Giraldo-Ospina, C.F.; Gómez, J.A.N.; Delgado-Rodríguez, M.; et al. Circulating Tumor Cells Identify Early Recurrence in Patients with Non-Small Cell Lung Cancer Undergoing Radical Resection. PLoS ONE 2016, 11, e0148659. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Ureña, M.; Ortega, F.G.; De Miguel-Pérez, D.; Rodriguez-Martínez, A.; García-Puche, J.L.; Ilyine, H.; Lorente, J.A.; Exposito-Hernandez, J.; Garrido-Navas, M.C.; Delgado-Ramirez, M.; et al. Circulating tumor cells criteria (CyCAR) versus standard RECIST criteria for treatment response assessment in metastatic colorectal cancer patients. J. Transl. Med. 2018, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Mamdouhi, T.; Twomey, J.D.; McSweeney, K.M.; Zhang, B. Fugitives on the run: Circulating tumor cells (CTCs) in metastatic diseases. Cancer Metastasis Rev. 2019, 38, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Rejniak, K.A. Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv. Exp. Med. Biol. 2016, 936, 93–106. [Google Scholar] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Tsai, W.-S.; Chen, J.-S.; Shao, H.-J.; Wu, J.-C.; Lai, J.-M.; Lu, S.-H.; Hung, T.-F.; Chiu, Y.-C.; You, J.-F.; Hsieh, P.-S.; et al. Circulating Tumor Cell Count Correlates with Colorectal Neoplasm Progression and Is a Prognostic Marker for Distant Metastasis in Non-Metastatic Patients. Sci. Rep. 2016, 6, 24517. [Google Scholar] [CrossRef]
- Al-Mehdi, A.; Tozawa, K.; Fisher, A.; Shientag, L.; Lee, A.; Muschel, R. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: A new model for metastasis. Nat. Med. 2000, 6, 100–102. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Abdallah, E.A.; Braun, A.C.; Flores, B.C.; Senda, L.; Urvanegia, A.C.; Calsavara, V.; De Jesus, V.H.F.; Almeida, M.F.A.; Begnami, M.D.; Coimbra, F.J.; et al. The Potential Clinical Implications of Circulating Tumor Cells and Circulating Tumor Microemboli in Gastric Cancer. Oncologist 2019, 24, e854–e863. [Google Scholar] [CrossRef]
- Whisner, C.M.; Athena Aktipis, C. The Role of the Microbiome in Cancer Initiation and Progression: How Microbes and Cancer Cells Utilize Excess Energy and Promote One Another’s Growth. Curr. Nutr. Rep. 2019, 8, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.V.; Cocom-Chan, B.; Hernandez-Montes, G.; Portillo-Bobadilla, T.; Resendis-Antonio, O. Host-Microbiome Interaction and Cancer: Potential Application in Precision Medicine. Front. Physiol. 2016, 7, 606. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Mukherjee, P. Role of Microbiome in Modulating Immune Responses in Cancer. Mediat. Inflamm. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.M.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Blomberg, O.S.; Spagnuolo, L.; de Visser, K.E. Immune regulation of metastasis: Mechanistic insights and therapeutic opportunities. Dis. Model. Mech. 2018, 11, dmm036236. [Google Scholar] [CrossRef]
- Leone, K.; Poggiana, C.; Zamarchi, R. The Interplay between Circulating Tumor Cells and the Immune System: From Immune Escape to Cancer Immunotherapy. Diagnostics 2018, 8, 59. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Circulating tumor cell interactions with macrophages: Implications for biology and treatment. Transl. Lung Cancer Res. 2017, 6, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Machlus, K.R.; Italiano, J.E. The incredible journey: From megakaryocyte development to platelet formation. J. Cell Biol. 2013, 201, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Li, N. Platelets in cancer metastasis: To help the “villain” to do evil. Int. J. Cancer 2016, 138, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S. Inflammation and haemostasis. Biochem. Med. 2012, 22, 49–62. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Yu, L.-X.; Yan, L.; Yang, W.; Wu, F.-Q.; Ling, Y.; Chen, S.-Z.; Tang, L.; Tan, Y.-X.; Cao, D.; Wu, M.-C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Plantureux, L.; Mège, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers (Basel) 2018, 10, 441. [Google Scholar] [CrossRef]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef]
- Serrano, M.J.; Ortega, F.G.; Alvarez-Cubero, M.J.; Nadal, R.; Sánchez-Rovira, P.; Salido, M.; Rodriguez, M.; García-Puche, J.L.; Delgado-Rodríguez, M.; Sole, F.; et al. EMT and EGFR in CTCs cytokeratin negative non-metastatic breast cancer. Oncotarget 2014, 5, 7486–7497. [Google Scholar] [CrossRef]
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- Gruber, I.; Landenberger, N.; Staebler, A.; Hahn, M.; Wallwiener, D.; Fehm, T. Relationship between circulating tumor cells and peripheral T-cells in patients with primary breast cancer. Anticancer. Res. 2013, 33, 2233–2238. [Google Scholar] [PubMed]
- Tao, L.; Zhang, L.; Peng, Y.; Tao, M.; Li, L.; Xiu, D.; Yuan, C.; Ma, Z.; Jiang, B. Neutrophils assist the metastasis of circulating tumor cells in pancreatic ductal adenocarcinoma: A new hypothesis and a new predictor for distant metastasis. Medicine (Baltimore) 2016, 95, e4932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qiao, X.; Shi, H.; Han, X.; Liu, W.; Tian, X.; Zeng, X. Circulating tumor-associated neutrophils (cTAN) contribute to circulating tumor cell survival by suppressing peripheral leukocyte activation. Tumor Biol. 2016, 37, 5397–5404. [Google Scholar] [CrossRef]
- Uppal, A.; Wightman, S.C.; Ganai, S.; Weichselbaum, R.R.; An, G. Investigation of the essential role of platelet-tumor cell interactions in metastasis progression using an agent-based model. Theor. Biol. Med. Model. 2014, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Moore, K.L.; Patel, K.D.; Bruehl, R.E.; Li, F.; Johnson, D.A.; Lichenstein, H.S.; Cummings, R.D.; Bainton, D.F.; McEver, R.P. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 1995, 128, 661–671. [Google Scholar] [CrossRef]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J.; Zhou, X. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-Derived SPARC Bridges Tumor Cell-Extracellular Matrix Interactions toward Metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.R.; Schmid, M.C. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediat. Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-N.; Koo, K.H.; Sung, J.Y.; Yun, U.-J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heeke, S.; Mograbi, B.; Alix-Panabières, C.; Hofman, P. Never Travel Alone: The Crosstalk of Circulating Tumor Cells and the Blood Microenvironment. Cells 2019, 8, 714. [Google Scholar] [CrossRef]
- Abylkassov, R.; Xie, Y. Role of Yes-associated protein in cancer: An update. Oncol. Lett. 2016, 12, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, M.; Shirai, T.; Tamura, S.; Suzuki-Inoue, K.; Ehata, S.; Takahashi, K.; Miyazono, K.; Hayakawa, Y.; Sato, T.; Takeda, K.; et al. ASK1 facilitates tumor metastasis through phosphorylation of an ADP receptor P2Y12 in platelets. Cell Death Differ. 2017, 24, 2066–2076. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef]
- Cianci, R.; Pagliari, D.; Piccirillo, C.A.; Fritz, J.H.; Gambassi, G. The Microbiota and Immune System Crosstalk in Health and Disease. Mediat. Inflamm. 2018, 2018, 1–3. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Chu, Q.; Zhang, P. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019, 447, 41–47. [Google Scholar] [CrossRef]
- Ong, H.S.; Yim, H.C.H. Microbial Factors in Inflammatory Diseases and Cancers. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; pp. 153–174. [Google Scholar]
- Sussman, D.A.; Santaolalla, R.; Strobel, S. Cancer in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2012, 28, 327–333. [Google Scholar] [CrossRef]
- Sieling, P.A.; Chung, W.; Duong, B.T.; Godowski, P.J.; Modlin, R.L. Toll-like receptor 2 ligands as adjuvants for human Th1 responses. J. Immunol. 2003, 170, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Wynendaele, E.; Verbeke, F.; D’Hondt, M.; Hendrix, A.; Van De Wiele, C.; Burvenich, C.; Peremans, K.; De Wever, O.; Bracke, M.; De Spiegeleer, B. Crosstalk between the microbiome and cancer cells by quorum sensing peptides. Peptides 2015, 64, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) 2017, 9, 52. [Google Scholar]
- Gerhauser, C. Impact of dietary gut microbial metabolites on the epigenome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170359. [Google Scholar] [CrossRef]
- Ji, J.; Shu, D.; Zheng, M.; Wang, J.; Luo, C.; Wang, Y.; Guo, F.; Zou, X.; Lv, X.; Li, Y.; et al. Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci. Rep. 2016, 6, 24838. [Google Scholar] [CrossRef] [PubMed]
- Gaines, S.; Williamson, A.J.; Kandel, J. How the microbiome is shaping our understanding of cancer biology and its treatment. Semin. Colon Rectal Surg. 2018, 29, 12–16. [Google Scholar] [CrossRef]
- Sears, C.L.; Geis, A.L.; Housseau, F. Bacteroides fragilis subverts mucosal biology: From symbiont to colon carcinogenesis. J. Clin. Investig. 2014, 124, 4166–4172. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Huycke, M.M. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015, 64, 459–468. [Google Scholar] [CrossRef]
- Chandrakesan, P.; Weygant, N.; May, R.; Qu, D.; Chinthalapally, H.R.; Sureban, S.M.; Ali, N.; Lightfoot, S.A.; Umar, S.; Houchen, C.W. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014, 5, 9269–9280. [Google Scholar] [CrossRef]
- Chandrakesan, P.; Panneerselvam, J.; Qu, D.; Weygant, N.; May, R.; Bronze, M.; Houchen, C. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J. Carcinog. Mutagen. 2016, 7, 1–8. [Google Scholar]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer–initiating cells. J. Clin. Investig. 2014, 124, 1283–1295. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cubero, M.J.; Vázquez-Alonso, F.; Puche-Sanz, I.; Ortega, F.G.; Martin-Prieto, M.; Garcia-Puche, J.L.; Pascual-Geler, M.; Lorente, J.A.; Cozar-Olmo, J.M.; Serrano, M.J. Dormant Circulating Tumor Cells in Prostate Cancer: Therapeutic, Clinical and Biological Implications. Curr. Drug Targets 2016, 17, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Orberg, E.T.; Geis, A.L.; Chan, J.L.; Fu, K.; Shields, C.E.D.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor−κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e24. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Ma, C.; Luo, H.; Gao, F.; Tang, Q.; Chen, W. Fusobacterium nucleatum promotes the progression of colorectal cancer by interacting with E-cadherin. Oncol. Lett. 2018, 16, 2606–2612. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Navas, C.; de Miguel-Pérez, D.; Exposito-Hernandez, J.; Bayarri, C.; Amezcua, V.; Ortigosa, A.; Valdivia, J.; Guerrero, R.; Garcia Puche, J.L.; Lorente, J.A.; et al. Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents. Cells 2019, 8, 1382. https://doi.org/10.3390/cells8111382
Garrido-Navas C, de Miguel-Pérez D, Exposito-Hernandez J, Bayarri C, Amezcua V, Ortigosa A, Valdivia J, Guerrero R, Garcia Puche JL, Lorente JA, et al. Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents. Cells. 2019; 8(11):1382. https://doi.org/10.3390/cells8111382
Chicago/Turabian StyleGarrido-Navas, Carmen, Diego de Miguel-Pérez, Jose Exposito-Hernandez, Clara Bayarri, Victor Amezcua, Alba Ortigosa, Javier Valdivia, Rosa Guerrero, Jose Luis Garcia Puche, Jose Antonio Lorente, and et al. 2019. "Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents" Cells 8, no. 11: 1382. https://doi.org/10.3390/cells8111382
APA StyleGarrido-Navas, C., de Miguel-Pérez, D., Exposito-Hernandez, J., Bayarri, C., Amezcua, V., Ortigosa, A., Valdivia, J., Guerrero, R., Garcia Puche, J. L., Lorente, J. A., & Serrano, M. J. (2019). Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents. Cells, 8(11), 1382. https://doi.org/10.3390/cells8111382