Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System

 , , and
, , and
Abstract
1. Introduction
2. Glioblastoma Cancer Stem Cells
3. Circulating Cancer Stem Cells and Epithelial-to-Mesenchymal Transition
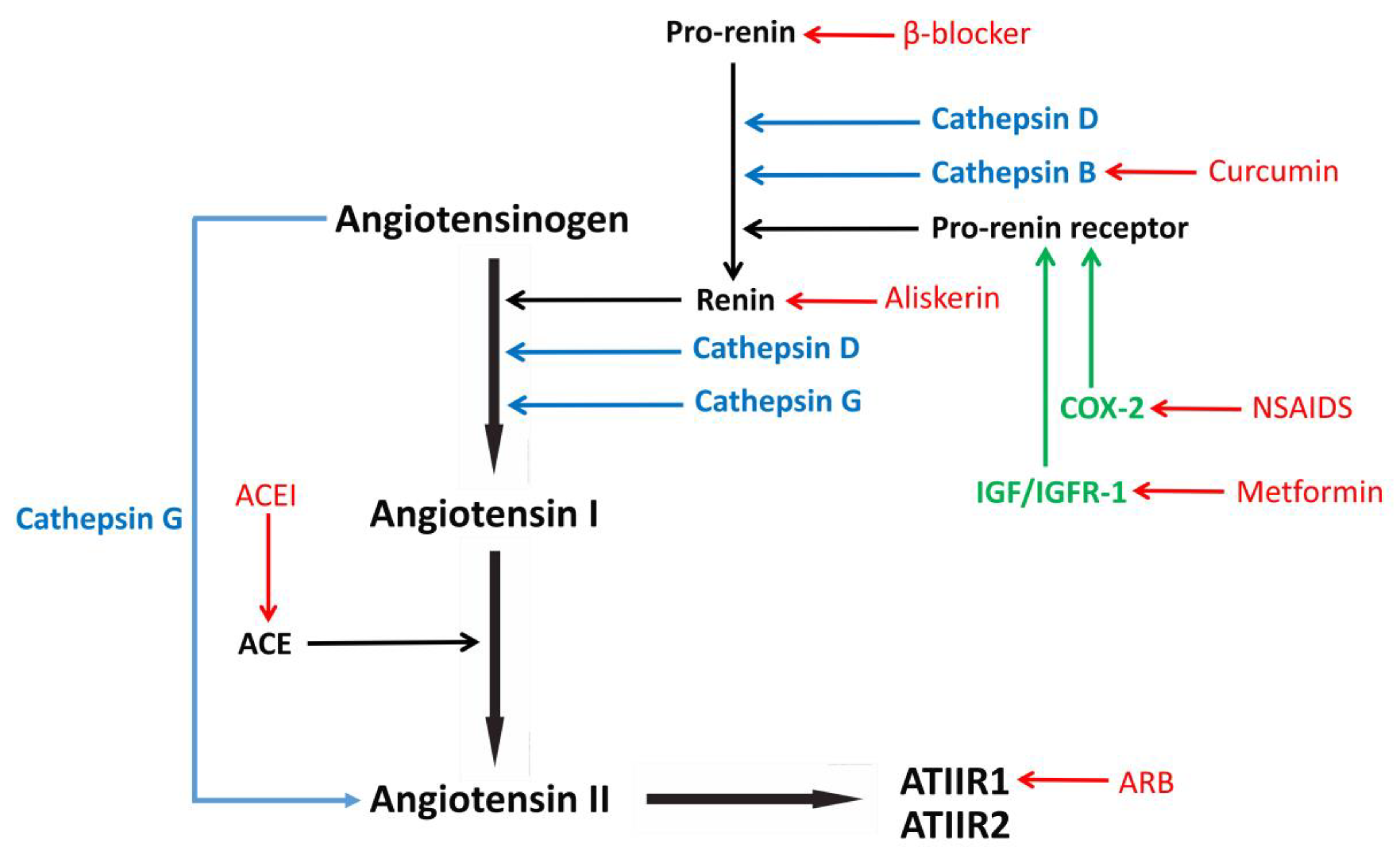
4. The Renin-Angiotensin System
5. Repurposing Drugs that Target the RAS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-C.; Wang, L.-J.; Rajesh, R. Targeting human brain cancer stem cells by curcumin-loaded nanoparticles grafted with anti-aldehyde dehydrogenase and sialic acid: Colocalization of ALDH and CD44. Mater. Sci. Eng. C 2019, 102, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Zentgraf, H.; Balss, J.; Hartmann, C.; Von Deimling, A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009, 118, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [PubMed]
- Paul, Y.; Mondal, B.; Patil, V.; Somasundaram, K. DNA methylation signatures for 2016 WHO classification subtypes of diffuse gliomas. Clin. Epigenetics 2017, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Nahed, B.V.; Wander, S.A.; Iafrate, A.J.; Borger, D.R.; Hu, R.; Thabet, A.; Cahill, D.P.; Perry, A.M.; Joseph, C.P.; et al. Elevation of Urinary 2-Hydroxyglutarate in IDH-Mutant Glioma. Oncologist 2016, 21, 214–219. [Google Scholar] [CrossRef]
- Bent, M.J.V.D.; Weller, M.; Wen, P.Y.; Kros, J.M.; Aldape, K.; Chang, S. A clinical perspective on the 2016 WHO brain tumor classification and routine molecular diagnostics. Neuro-Oncology 2017, 19, 614–624. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2018, 33, 152. [Google Scholar] [CrossRef]
- Shen, R.; Mo, Q.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative Subtype Discovery in Glioblastoma Using iCluster. PLoS ONE 2012, 7, e35236. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Galli, R. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, R. Glioblastoma Stem Cells as a New Therapeutic Target for Glioblastoma. Clin. Med. Insights: Oncol. 2015, 9, CMO.S30271. [Google Scholar] [CrossRef] [PubMed]
- Tejero, R.; Huang, Y.; Katsyv, I.; Kluge, M.; Lin, J.Y.; Tome-Garcia, J.; Daviaud, N.; Wang, Y.; Zhang, B.; Tsankova, N.M.; et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 2019, 42, 252–269. [Google Scholar] [CrossRef]
- Iwadate, Y. Plasticity in Glioma Stem Cell Phenotype and Its Therapeutic Implication. Neurol. Medico-Chirurgica 2018, 58, 61–70. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Chhabra, A. Derivation of Human Induced Pluripotent Stem Cell (iPSC) Lines and Mechanism of Pluripotency: Historical Perspective and Recent Advances. Stem Cell Rev. Rep. 2017, 13, 757–773. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremesekera, A.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer stem cells in glioblastoma multiforme. Front. Surg. 2016, 3, 48. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer stem cell hierarchy in glioblastoma multiforme. Front Surg 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Elsir, T.; Edqvist, P.H.; Carlson, J.; Ribom, D.; Bergqvist, M.; Ekman, S.; Popova, S.N.; Alafuzoff, I.; Ponten, F.; Nistér, M.; et al. A study of embryonic stem cell-related proteins in human astrocytomas: Identification of Nanog as a predictor of survival. Int. J. Cancer 2014, 134, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.P.; Wickremesekera, A.C.; Itinteang, T.; Tan, S.T. Editorial: Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Transl. Cancer Res. 2018, 7, S476–S479. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Berezovsky, A.D.; Poisson, L.M.; Cherba, D.; Webb, C.P.; Transou, A.D.; Lemke, N.W.; Hong, X.; Hasselbach, L.A.; Irtenkauf, S.M.; Mikkelsen, T.; et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16, 193–206.e25. [Google Scholar] [CrossRef]
- Gangemi, R.M.R.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2Silencing in Glioblastoma Tumor-Initiating Cells Causes Stop of Proliferation and Loss of Tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 222. [Google Scholar] [CrossRef]
- Xing, Y.; Ge, Y.; Liu, C.; Zhang, X.; Jiang, J.; Wei, Y. ER stress inducer tunicamycin suppresses the self-renewal of glioma-initiating cell partly through inhibiting Sox2 translation. Oncotarget 2016, 7, 36395–36406. [Google Scholar] [CrossRef]
- Yan, J.-L.; Li, C.; Boonzaier, N.R.; Fountain, D.M.; Larkin, T.J.; Matys, T.; Van Der Hoorn, A.; Price, S.J. Multimodal MRI characteristics of the glioblastoma infiltration beyond contrast enhancement. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419844664. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, S.K.; Shin, J.; Se, Y.B.; Choi, S.H.; Park, S.H.; Choi, S.A.; Lee, J.Y.; Phi, J.H.; Wang, K.C.; et al. A subpopulation of cancer stem cells identifies radiographic characteristics in glioblastoma. Oncol. Lett. 2017, 13, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Fu, J.; Wang, W.; Hofman, F.M.; Chen, T.C.; Chen, L. Distribution of cancer stem cells in two human brain gliomas. Oncol. Lett. 2019, 17, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Stylli, S.S.; Kaye, A.H.; Lock, P. Invadopodia: At the cutting edge of tumour invasion. J. Clin. Neurosci. 2008, 15, 725–773. [Google Scholar] [CrossRef]
- Senft, C.; Priester, M.; Polacin, M.; Schröder, K.; Seifert, V.; Kögel, D.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neurooncol. 2011, 101, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Umper, S.L.H.-J.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.; Van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J.; et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251–41264. [Google Scholar] [CrossRef] [PubMed]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kögel, D. Dietary Curcumin Attenuates Glioma Growth in a Syngeneic Mouse Model by Inhibition of the JAK1,2/STAT3 Signaling Pathway. Clin. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef] [PubMed]
- Seyithanoğlu, M.H.; Abdallah, A.; Kitiş, S.; Güler, E.M.; Koçyiğit, A.; Dündar, T.T.; Gündağ, M.P. Investigation of cytotoxic, genotoxic, and apoptotic effects of curcumin on glioma cells. Cell. Mol. Biol. (Noisy-le-Grand) 2019, 65, 101–108. [Google Scholar] [CrossRef]
- Whitehead, C.A.; Nguyen, H.P.; Morokoff, A.P.; Luwor, R.B.; Paradiso, L.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S. Inhibition of Radiation and Temozolomide-Induced Invadopodia Activity in Glioma Cells Using FDA-Approved Drugs. Transl. Oncol. 2018, 11, 1406–1418. [Google Scholar] [CrossRef]
- Van Schaijik, B.; Wickremesekera, A.C.; Mantamadiotis, T.; Kaye, A.H.; Tan, S.T.; Stylli, S.S.; Itinteang, T. Circulating tumor stem cells and glioblastoma: A review. J. Clin. Neurosci. 2019, 61, 5–9. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhong, D.; Zhao, Z.; Wang, W.; Li, J.; Zhang, W. Postoperative extracranial metastasis from glioblastoma: A case report and review of the literature. World J. Surg. Oncol. 2017, 15, 231. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Rochat, P.; Law, I.; Scheie, D.; Poulsen, H.S.; Muhic, A. Presentation of Two Cases with Early Extracranial Metastases from Glioblastoma and Review of the Literature. Case Rep. Oncol. Med. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.; Blau, T.; Grau, S.J.; Barbe, M.T.; Fink, G.R.; Galldiks, N. Extracranial Metastases of a Cerebral Glioblastoma: A Case Report and Review of the Literature. Case Rep. Oncol. 2018, 11, 591–600. [Google Scholar] [CrossRef]
- Lewis, G.D.; Rivera, A.L.; Tremont-Lukats, I.W.; Ballester-Fuentes, L.Y.; Zhang, Y.J.; Teh, B.S. GBM skin metastasis: A case report and review of the literature. CNS Oncol. 2017, 6, 203–209. [Google Scholar] [CrossRef]
- Collignon, F.P.; Holland, E.C.; Feng, S. Organ donors with malignant gliomas: An update. Arab. Archaeol. Epigr. 2004, 4, 15–21. [Google Scholar] [CrossRef]
- Pasquier, B.; Pasquier, D.; N’golet, A.; Panh, M.H.; Couderc, P. Extraneural metastases of astrocytomas and glioblastomas: Clinicopathological study of two cases and review of literature. Cancer 1980, 45, 112–125. [Google Scholar] [CrossRef]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous dissemination of glioblastoma multiforme. Sci. Transl. Med. 2014, 6, 247ra101. [Google Scholar] [CrossRef]
- Pilkington, G.J. The paradox of neoplastic glial cell invasion of the brain and apparent metastatic failure. Anticancer Res. 1997, 17, 4103–4105. [Google Scholar]
- Vitiani, L.R.; Pallini, R.; LaRocca, L.M.; Lombardi, D.G.; Signore, M.; Pierconti, F.; Petrucci, G.; Montano, N.; Maira, G.; De Maria, R. Mesenchymal differentiation of glioblastoma stem cells. Cell Death Differ. 2008, 15, 1491–1498. [Google Scholar] [CrossRef]
- Du, L.; Tang, J.-H.; Huang, G.-H.; Xiang, Y.; Lv, S.-Q. The progression of epithelial-mesenchymal transformation in gliomas. Chin. Neurosurg. J. 2017, 3, 23. [Google Scholar] [CrossRef]
- Clément, V.; Sánchez, P.; de Tribolet, N.; Radovanovic, I.; i Altaba, A.R. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Boil. 2007, 17, 192. [Google Scholar] [CrossRef][Green Version]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zhao, L.; Zhao, G. Metformin inhibits TGF-β1-induced epithelial-to-mesenchymal transition-like process and stem-like properties in GBM. Oncotarget 2018, 9, 7023–7035. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, C.; D’Alessio, A.; Lama, G.; Binda, E.; Mangiola, A.; Vescovi, A.L.; Proietti, G.; Masuelli, L.; Bei, R.; Fazi, B.; et al. Cancer stem cells from peritumoral tissue of glioblastoma multiforme: The possible missing link between tumor development and progression. Oncotarget 2018, 9, 28116–28130. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Y.; An, T.; Liu, P.; Zhu, J.; Yang, H.; Zhang, W.; Dong, T.; Jiang, J.; Zhang, Y.; et al. Nuciferine inhibits the progression of glioblastoma by suppressing the SOX2-AKT/STAT3-Slug signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 139. [Google Scholar] [CrossRef] [PubMed]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog and PI3K/Akt/mTOR pathways cooperate in suppressing survival, self-renewal and tumorigenic potential of glioblastoma-initiating cells. Mol. Cell. Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zheng, T.; Zhao, L.; Zhao, G. Resveratrol Suppresses Epithelial-Mesenchymal Transition in GBM by Regulating Smad-Dependent Signaling. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Nguyen, L.; Ager, E.I.; Neo, J.; Christophi, C. Regulation of colorectal cancer cell epithelial to mesenchymal transition by the renin angiotensin system. J. Gastroenterol. Hepatol. 2016, 31, 1773–1782. [Google Scholar] [CrossRef]
- Perdomo-Pantoja, A.; Mejía-Pérez, S.I.; Gómez-Flores-Ramos, L.; Lara-Velazquez, M.; Orillac, C.; Gómez-Amador, J.L.; Wegman-Ostrosky, T. Renin angiotensin system and its role in biomarkers and treatment in gliomas. J. Neurooncol. 2018, 138, 1–15. [Google Scholar] [CrossRef]
- Cousin, C.; Bracquart, D.; Contrepas, A.; Nguyen, G. Potential role of the (pro)renin receptor in cardiovascular and kidney diseases. J. Nephrol. 2010, 23, 508–513. [Google Scholar] [PubMed]
- Neves, F.A.; Duncan, K.G.; Baxter, J.D. Cathepsin B Is a Prorenin Processing Enzyme. Hypertension 1996, 27, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lu, X.; Peng, K.; Zhou, L.; Li, C.; Wang, W.; Yu, X.; Kohan, D.E.; Zhu, S.-F.; Yang, T. COX-2 mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Am. J. Physiol. Physiol. 2014, 307, F25–F32. [Google Scholar] [CrossRef] [PubMed]
- Holmer, S.R.; Hengstenberg, C.; Mayer, B.; Engel, S.; Lowel, H.; Riegger, G.A.J.; Schunkert, H. Marked suppression of renin levels by beta-receptor blocker in patients treated with standard heart failure therapy: A potential mechanism of benefit from beta-blockade. J. Intern. Med. 2001, 249, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Standen, P.; Sferruzzi-Perri, A.N.; Taylor, R.; Heinemann, G.; Zhang, J.V.; Highet, A.R.; Pringle, K.G.; Owens, J.A.; Kumarasamy, V.; Lumbers, E.R.; et al. Maternal insulin-like growth factor 1 and 2 differentially affect the renin–angiotensin system during pregnancy in the guinea pig. Growth Horm. IGF Res. 2015, 25, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef] [PubMed]
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millan, S.; Sanchez-Corona, J. The renin-angiotensin system meets the hallmarks of cancer. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 227–233. [Google Scholar] [CrossRef]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin–angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef]
- Nallaiah, S.; Lee, V.M.Y.; Brasch, H.D.; De Jongh, J.; Van Schaijik, B.; Marsh, R.; Tan, S.T.; Itinteang, T. Cancer stem cells within moderately differentiated head and neck cutaneous squamous cell carcinoma express components of the renin-angiotensin system. J. Plast. Reconstr. Aesthetic Surg. 2019, 72, 1484–1493. [Google Scholar] [CrossRef]
- Ram, R.S.; Brasch, H.D.; Dunne, J.C.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Moderately Differentiated Lip Squamous Cell Carcinoma Express Components of the Renin–Angiotensin System. Front. Surg. 2017, 4, 30. [Google Scholar] [CrossRef]
- Featherston, T.; Yu, H.H.; Dunne, J.C.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Moderately Differentiated Buccal Mucosal Squamous Cell Carcinoma Express Components of the Renin–Angiotensin System. Front. Surg. 2016, 3, 394. [Google Scholar] [CrossRef] [PubMed]
- Itinteang, T.; Dunne, J.C.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma express components of the renin–angiotensin system. J. Clin. Pathol. 2016, 69, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Wickremesekera, S.K.; Van Schaijik, B.; Marsh, R.W.; Brasch, H.D.; Tan, S.T.; Itinteang, T. Cancer stem cells in liver metastasis from colon adenocarcinoma express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- Wickremesekera, A.C.; Brasch, H.D.; Lee, V.M.; Davis, P.F.; Parker, A.; Koeck, H.; Itinteang, T.; Tan, S.T. Cancer stem cell subpopulations in metastatic melanoma to the brain express components of the renin-angiotensin system. J. Cancer Metastasis Treat. 2019, 5, 62. [Google Scholar] [CrossRef]
- Bradshaw, A.R.; Wickremesekera, A.C.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Glioblastoma Multiforme Cancer Stem Cells Express Components of the Renin–Angiotensin System. Front. Surg. 2016, 3, 1985. [Google Scholar] [CrossRef]
- Roth, I.M.; Wickremesekera, A.C.; Wickremesekera, S.K.; Davis, P.F.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells via Modulation of the Renin-Angiotensin System. Front. Oncol. 2019, 9, 745. [Google Scholar] [CrossRef]
- Rempel, S.A.; Rosenblum, M.L.; Mikkelsen, T.; Yan, P.S.; Ellis, K.D.; A Golembieski, W.; Sameni, M.; Rozhin, J.; Ziegler, G.; Sloane, B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994, 54, 6027–6031. [Google Scholar]
- Sivaparvathi, M.; Sawaya, R.; Wang, S.W.; Rayford, A.; Yamamoto, M.; Liottat, L.A.; Nicolson, G.L.; Rao, J.S. Overexpression and localization of cathepsin B during the progression of human gliomas. Clin. Exp. Metastasis 1995, 13, 49–56. [Google Scholar] [CrossRef]
- Konduri, S.; Lakka, S.S.; Tasiou, A.; Yanamandra, N.; Gondi, C.S.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Elevated levels of cathepsin B in human glioblastoma cell lines. Int. J. Oncol. 2001, 19, 519–524. [Google Scholar] [CrossRef]
- Strojnik, T.; Kos, J.; Zidanik, B.; Golouh, R.; Lah, T. Cathepsin B immunohistochemical staining in tumor and endothelial cells is a new prognostic factor for survival in patients with brain tumors. Clin. Cancer Res. 1999, 5, 559–567. [Google Scholar]
- Colin, C.; Voutsinos-Porche, B.; Nanni, I.; Fina, F.; Metellus, P.; Intagliata, D.; Baeza, N.; Bouvier, C.; Delfino, C.; Loundou, A.; et al. High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta Neuropathol. 2009, 118, 745. [Google Scholar] [CrossRef] [PubMed]
- Levicar, N.; Strojnik, T.; Kos, J.; Dewey, R.A.; Pilkington, G.J.; Lah, T.T. Lysosomal enzymes, cathepsins in brain tumour invasion. J. Neuro-Oncology 2002, 58, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Thermo Fisher Scientific. Oncomine. Available online: http://www.oncomine.org (accessed on 20 October 2019).
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pander, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Gopinath, S.; Malla, R.; Alapati, K.; Gorantla, B.; Gujrati, M.; Dinh, D.H.; Rao, J.S. Cathepsin B and uPAR regulate self-renewal of glioma-initiating cells through GLI-regulated Sox2 and Bmi1 expression. Carcinogenesis 2013, 34, 550–559. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Ding, Y.-M.; Hueng, D.-Y.; Chen, J.-Y.; Chen, Y. Caffeine suppresses the progression of human glioblastoma via cathepsin B and MAPK signaling pathway. J. Nutr. Biochem. 2016, 33, 63–72. [Google Scholar] [CrossRef]
- Alhajala, H.S.; Nguyen, H.S.; Shabani, S.; Best, B.; Kaushal, M.; Al-Gizawiy, M.M.; Ahn, E.-Y.E.; Knipstein, J.A.; Mirza, S.; Schmainda, K.M.; et al. Irradiation of pediatric glioblastoma cells promotes radioresistance and enhances glioma malignancy via genome-wide transcriptome changes. Oncotarget 2018, 9, 34122–34131. [Google Scholar] [CrossRef]
- Koh, S.P.; Wickremesekera, A.C.; Brasch, H.D.; Marsh, R.; Tan, S.T.; Itinteang, T. Expression of Cathepsins B, D, and G in Isocitrate Dehydrogenase-Wildtype Glioblastoma. Front. Surg. 2017, 4, 28. [Google Scholar] [CrossRef]
- Ramírez-Expósito, M.J.; Martínez-Martos, J.M. Differential Effects of Doxazosin on Renin-Angiotensin-System- Regulating Aminopeptidase Activities in Neuroblastoma and Glioma Tumoral Cells. CNS Neurol. Disord. Drug Targets 2019, 18, 29–36. [Google Scholar] [CrossRef]
- Rivera, E.; Arrieta, O.; Guevara, P.; Duarte-Rojo, A.; Sotelo, J. AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br. J. Cancer 2001, 85, 1396–1399. [Google Scholar] [CrossRef][Green Version]
- Arrieta, O.; Guevara, P.; Escobar, E.; García-Navarrete, R.; Pineda, B.; Sotelo, J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer 2005, 92, 1247–1252. [Google Scholar] [CrossRef]
- Ursu, R.; Thomas, L.; Psimaras, D.; Chinot, O.; Le Rhun, E.; Ricard, D.; Charissoux, M.; Cuzzubbo, S.; Sejalon, F.; Quillien, V.; et al. Angiotensin II receptor blockers, steroids and radiotherapy in glioblastoma—A randomised multicentre trial (ASTER trial). An ANOCEF study. Eur. J. Cancer 2019, 109, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Juillerat-Jeanneret, L.; Celerier, J.; Bernasconi, C.C.; Nguyen, G.; Wostl, W.; Maerki, H.P.; Janzer, R.-C.; Corvol, P.; Gasc, J.-M. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br. J. Cancer 2004, 90, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Januel, E.; Ursu, R.; Alkhafaji, A.; Marantidou, A.; Doridam, J.; Belin, C.; Lévy-Piedbois, C.; Carpentier, A.F.; Levy-Piedbois, C. Impact of renin-angiotensin system blockade on clinical outcome in glioblastoma. Eur. J. Neurol. 2015, 22, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.-E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef]
- Skaga, E.; Skaga, I.Ø.; Grieg, Z.; Sandberg, C.J.; Langmoen, I.A.; Vik-Mo, E.O. The efficacy of a coordinated pharmacological blockade in glioblastoma stem cells with nine repurposed drugs using the CUSP9 strategy. J. Cancer Res. Clin. Oncol. 2019, 145, 1495–1507. [Google Scholar] [CrossRef]
- Christian, J.B.; Lapane, K.L.; Hume, A.L.; Eaton, C.B.; Weinstock, M.A. Association of ACE Inhibitors and Angiotensin Receptor Blockers with Keratinocyte Cancer Prevention in the Randomized VATTC Trial. J. Natl. Cancer Inst. 2008, 100, 1223–1232. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol Reduces Cancer Risk: A Population-Based Cohort Study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef]
- Qiao, Y.; Yang, T.; Gan, Y.; Li, W.; Wang, C.; Gong, Y.; Lu, Z. Associations between aspirin use and the risk of cancers: A meta-analysis of observational studies. BMC Cancer 2018, 18, 288. [Google Scholar] [CrossRef]
- Viegas, A.; Manso, J.; Corvo, M.C.; Marques, M.M.B.; Cabrita, E.J. Binding of Ibuprofen, Ketorolac, and Diclofenac to COX-1 and COX-2 Studied by Saturation Transfer Difference NMR. J. Med. Chem. 2011, 54, 8555–8562. [Google Scholar] [CrossRef]
- Ferrandez, A.; Prescott, S.; Burt, R.W. COX-2 and colorectal cancer. Curr. Pharm. Des. 2003, 9, 2229–2251. [Google Scholar] [CrossRef]
- Forget, P.; Vandenhende, J.; Berlière, M.; Machiels, J.-P.; Nussbaum, B.; Legrand, C.; De Kock, M. Do Intraoperative Analgesics Influence Breast Cancer Recurrence After Mastectomy? A Retrospective Analysis. Anesthesia Analg. 2010, 110, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Mihajluk, K.; Simms, C.; Reay, M.; Madureira, P.; Howarth, A.; Murray, P.; Nasser, S.; Duckworth, C.; Pritchard, D.; Pilkington, G.; et al. IP1867B suppresses the insulin-like growth factor 1 receptor (IGF1R) ablating epidermal growth factor receptor inhibitor resistance in adult high grade gliomas. Cancer Lett. 2019, 458, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.L.; Thaker, P.H.; Nick, A.M.; Ramondetta, L.M.; Kumar, S.; Urbauer, D.L.; Matsuo, K.; Squires, K.C.; Coleman, R.L.; Lutgendorf, S.K.; et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer 2015, 121, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Hwa, Y.L.; Shi, Q.; Kumar, S.K.; Lacy, M.Q.; Gertz, M.A.; Kapoor, P.; Buadi, F.K.; Leung, N.; Dingli, D.; Go, R.S.; et al. Beta-blockers improve survival outcomes in patients with multiple myeloma: A retrospective evaluation. Am. J. Hematol. 2017, 92, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-E.; Ho, C.-C.; Lin, S.-H.; Yeh, K.-T.; Chen, M.-K. Cathepsin B Expression and the Correlation with Clinical Aspects of Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, 0152165. [Google Scholar] [CrossRef] [PubMed]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Levin, V.A.; Chan, J.; Datta, M.; Yee, J.L.; Jain, R.K. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J. Neuro-Oncol. 2017, 134, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Rundle-Thiele, D.; Head, R.; Cosgrove, L.; Martin, J.H. Repurposing some older drugs that cross the blood-brain barrier and have potential anticancer activity to provide new treatment options for glioblastoma. Br. J. Clin. Pharmacol. 2016, 81, 199–209. [Google Scholar] [CrossRef]
- Itinteang, T.; Chudakova, D.A.; Dunne, J.C.; Davis, P.F.; Tan, S.T. Expression of Cathepsins B, D, and G in Infantile Hemangioma. Front. Surg. 2015, 2, 26. [Google Scholar] [CrossRef]
- Al Hassan, M.; Fakhoury, I.; El Masri, Z.; Ghazale, N.; Dennaoui, R.; El Atat, O.; Kanaan, A.; El-Sibai, M. Metformin Treatment Inhibits Motility and Invasion of Glioblastoma Cancer Cells. Anal. Cell. Pathol. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Tan, S.T. Treatment of Patients with Advanced Cancer by Targeting Cancer Stem Cells Using Modulators of the Renin-Angiotensin System; Australian New Zealand Clinical Trials Registry: Wellington, New Zealand; Available online: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=377393 (accessed on 20 October 2019).



{kind=link}
{kind=link}
| Number of Glioblastoma Samples | Number of Corresponding Normal Brain Samples | Total Number of Measured Genes | Mean Fold Change (Log2) | p-Value | Sample Type | Platform | Study |
|---|---|---|---|---|---|---|---|
| 542 | 10 | 12,624 | 2.0662 | 1.96 × 10-8 | mRNA | Human Genome U133A Array | TCGA Brain |
| 27 | 4 | 14,836 | 1.819 | 1.84 × 10-5 | mRNA | Not Defined | Bredel Brain 2 |
| 81 | 23 | 19,574 | 1.543 | 4.02 × 10-7 | mRNA | Human Genome U133 Plus 2.0 Array | Sun Brain |
| Authors | Year | Subjects | Medications | Effects |
|---|---|---|---|---|
| Rivera, et al. | 2001 | C6 rat glioma | Losartan | Reduction in tumor volume, decreased vascular density, mitotic index, cell proliferation |
| Juillerat-Jeanneret, et al. | 2004 | Human GB cell cultures | Renin inhibitors | Induced apoptosis in human glioblastoma cells |
| Arrieta, et al. | 2005 | C6 rat glioma | Losartan | Decreased tumor volume, induction of apoptosis in dose-dependent manner |
| Januel, et al. | 2015 | GB patients | ACEIs, ARBs | Improved progression-free survival and overall survival in multivariate analysis |
| Levin, et al. | 2017 | GB patients | Angiotensin system inhibitors (not specified) +/− bevacizumab | Improved survival, further survival advantage when renin-angiotensin system inhibitors were combined with low-dose bevacizumab |
| Mihajluk, et al. | 2019 | Human GB cell cultures | Reformulated aspirin (IP1867B) | Reduction in high-grade glioma cell viability, suppressed IL6/STAT3 and NF-κB networks, reduction in IGF1 and EGFR expression, less gastrointestinal side effects compared to conventional aspirin |
| Ramirez-Exposito, et al. | 2019 | Human neuroblastoma NB69, astroglioma U373-MG | Doxazosin | Concentration-dependent inhibition of cell growth, modification of proteolytic regulatory enzymes of RAS cascade |
| Skaga, et al. | 2019 | Human GB stem cell cultures | Aprepitants, auranofin, captopril, celecoxib, disulfiram, itraconazole, minocycline, quetiapine, sertraline | The combination effect of CUSP9 with temozolomide was superior to temozolomide monotherapy in clinical plasma concentrations |
| Ursu, et al. | 2019 | GB patients | Losartan | No difference in steroid requirement to reduce peritumoral edema |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, D.C.H.; Roth, I.M.; Wickremesekera, A.C.; Davis, P.F.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells 2019, 8, 1364. https://doi.org/10.3390/cells8111364
Tan DCH, Roth IM, Wickremesekera AC, Davis PF, Kaye AH, Mantamadiotis T, Stylli SS, Tan ST. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells. 2019; 8(11):1364. https://doi.org/10.3390/cells8111364
Chicago/Turabian StyleTan, David C. H., Imogen M. Roth, Agadha C. Wickremesekera, Paul F. Davis, Andrew H. Kaye, Theo Mantamadiotis, Stanley S. Stylli, and Swee T. Tan. 2019. "Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System" Cells 8, no. 11: 1364. https://doi.org/10.3390/cells8111364
APA StyleTan, D. C. H., Roth, I. M., Wickremesekera, A. C., Davis, P. F., Kaye, A. H., Mantamadiotis, T., Stylli, S. S., & Tan, S. T. (2019). Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells, 8(11), 1364. https://doi.org/10.3390/cells8111364