Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression

Abstract
1. Introduction
1.1. Non-Alcoholic Fatty Liver Disease (NAFLD) Pathogenesis and Significance
1.2. Bile Acids—from Synthesis over Transport to Recovery
1.3. Relationship of NAFLD and Bile Acids
2. Typical Ligands for Bile Acids
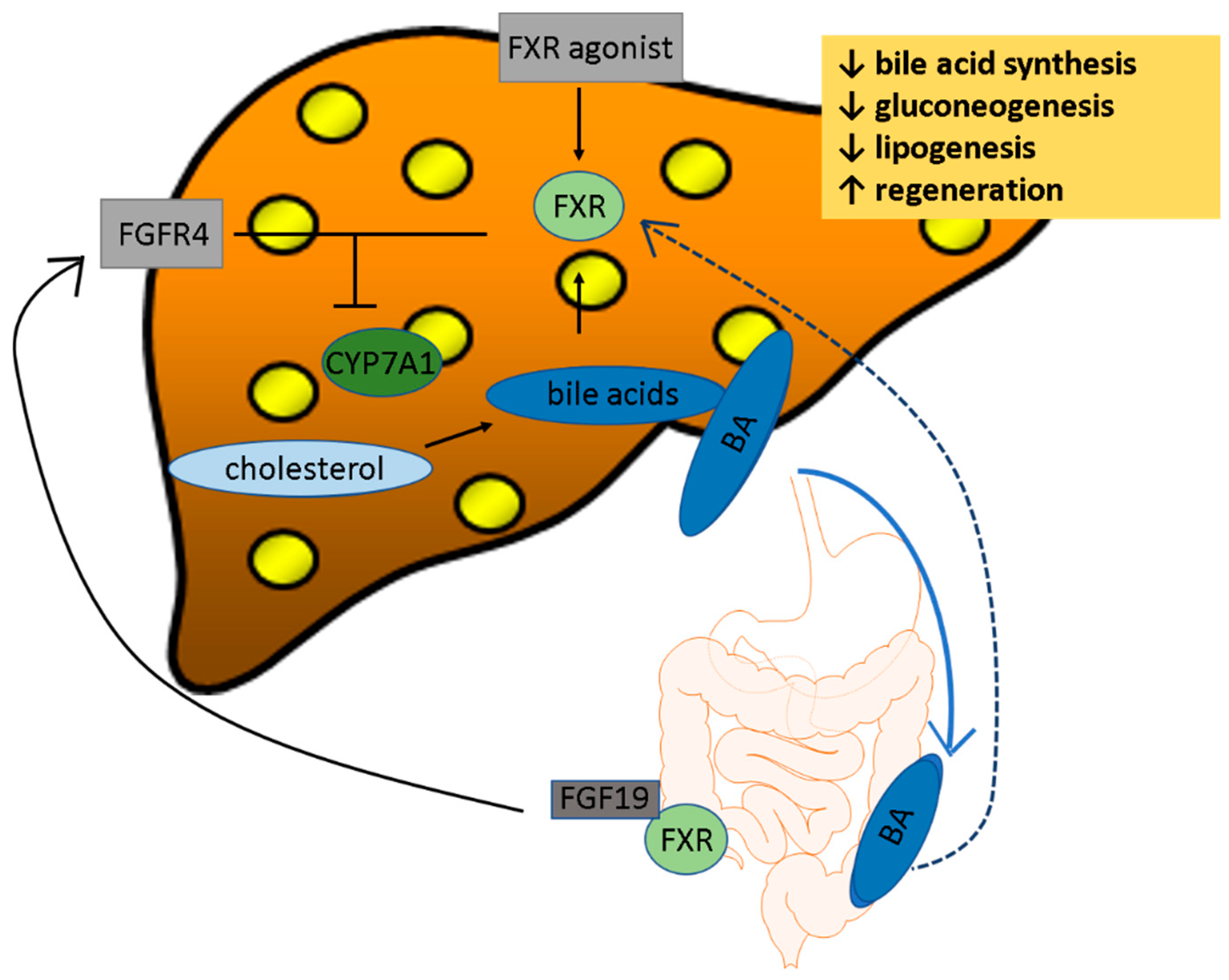
2.1. FXR
2.2. Takeda G-Protein-Coupled Receptor 5 (TGR5)
3. Effects from Bile Acids on Different Metabolic Functions in the Body
3.1. Clinical Manifestation of Dysregulated BAs in NAFLD
3.2. Bile Acid Effects on Glucose Metabolism
3.3. Role of Bile in Lipid Metabolism
3.4. Role of Bile in Cholesterol Metabolism
3.5. Role of Bile in the Intestine, and with Microbiota
4. Pharmacotherapies
4.1. FXR Agonists
4.2. OCA
4.3. Ursodeoxycholic Acid (UDCA)
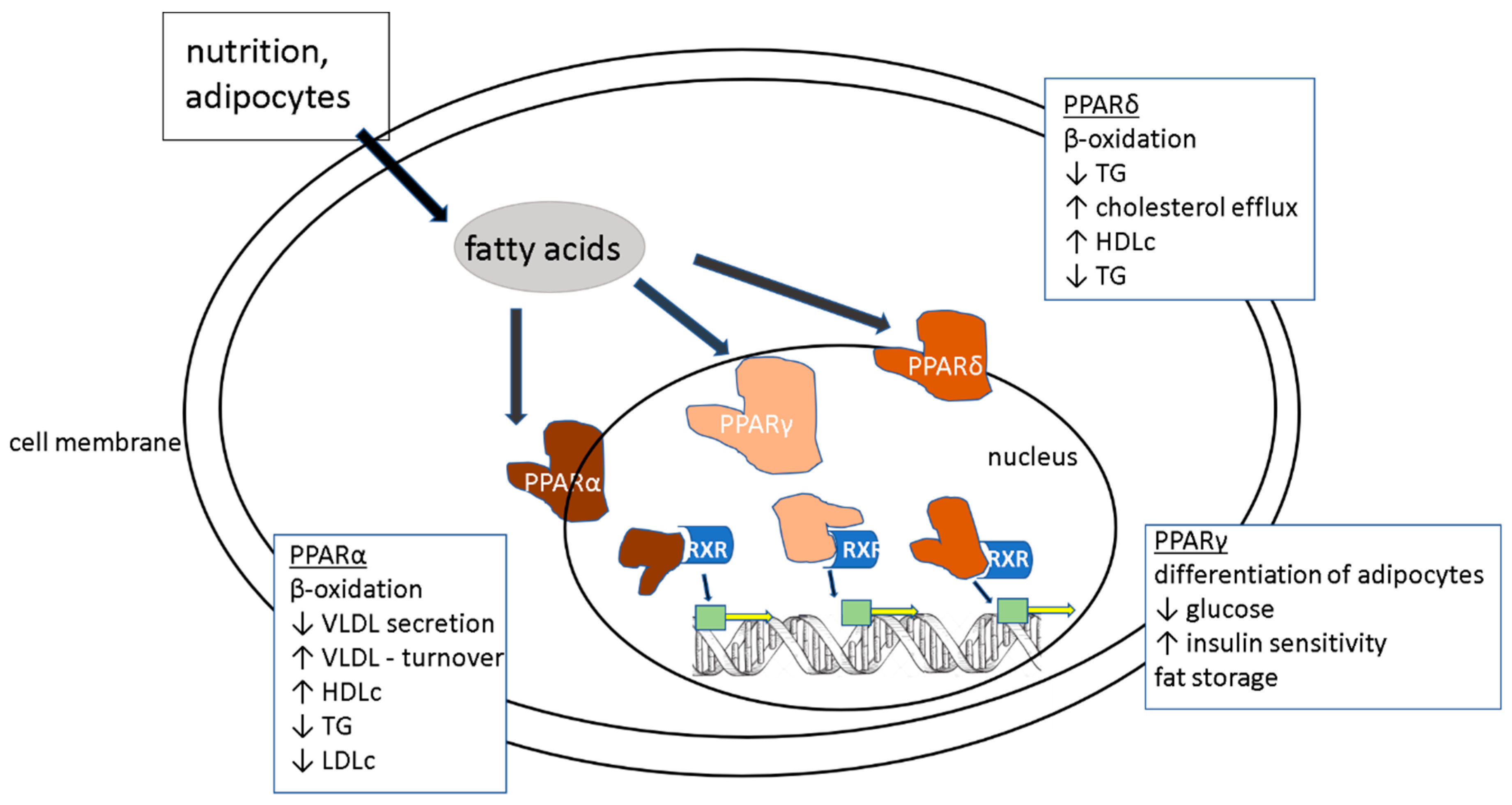
4.4. PPARα Agonists
4.5. TGR5
4.6. Bile Acid Conjugates
4.7. Apical Sodium-Dependent Bile Salt Transporter (ASBT) Inhibitors
5. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Seyda, S.G.; Kucukoglu, O.; Altinbasv, A.; Demir, O.O.; Yilmaz, S.; Akkiz, H.; Otan, E.; Sowa, J.-P.; Canbay, A. Economic growth leads to increase of obesity and associated hepatocellular carcinoma in developing countries. Annu. Hepatol. 2016, 15, 662–672. [Google Scholar]
- Faasse, S.; Braun, H.; Vos, M. The role of NAFLD in cardiometabolic disease: An update. F1000Research 2018, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.; McCullough, A. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Younossi, Z.M. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Implications for liver transplantation. Liver Transpl. 2018, 24, 166–170. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Masuda, N.; Oda, H.; Hirano, S.; Tanaka, H. Enhancement of the 7 alpha-dehydroxylase activity of a gram-positive intestinal anaerobe by flavins. Appl. Environ. Microbiol. 1983, 45, 308–309. [Google Scholar]
- Bhowmik, S.; Chiu, H.-P.; Jones, D.H.; Chiu, H.-J.; Miller, M.D.; Xu, Q.; Farr, C.L.; Ridlon, J.M.; Wells, J.E.; Elsliger, M.-A.; et al. Structure and functional characterization of a bile acid 7α dehydratase BaiE in secondary bile acid synthesis. Proteins Struct. Funct. Bioinform. 2016, 84, 316–331. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.-P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.R.; Barritt, A.S. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Setchell, K.D. Bile acid biosynthesis. Biochemistry 1992, 31, 4737–4749. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Pang, Y.; Wang, X.; Wu, Q.; Liu, H.; Liu, B.; Liu, G.; Ye, M.; Kong, W.; Jiang, C. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm. Sin. B 2019, 9, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol. Appl. Pharm. 2013, 268, 132–140. [Google Scholar] [CrossRef]
- Johnson, M.R.; Barnes, S.; Kwakye, J.B.; Diasio, R.B. Purification and characterization of bile acid-CoA: amino acid N-acyltransferase from human liver. J. Boil. Chem. 1991, 266, 10227–10233. [Google Scholar]
- Shneider, B.L.; Dawson, P.; Christie, D.M.; Hardikar, W.; Wong, M.H.; Suchy, F.J. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J. Clin. Investig. 1995, 95, 745–754. [Google Scholar] [CrossRef]
- Corbett, C.L.; Bartholomew, T.C.; Billing, B.H.; Summerfield, J.A. Urinary Excretion of Bile Acids in Cholestasis: Evidence for Renal Tubular Secretion in Man. Clin. Sci. 1981, 61, 773–780. [Google Scholar] [CrossRef]
- Li, T.; Apte, U. Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer. Adv. Pharm. 2015, 74, 263–302. [Google Scholar]
- Chen, J.; Deng, W.; Wang, J.; Shao, Y.; Ou, M.; Ding, M. Primary bile acids as potential biomarkers for the clinical grading of intrahepatic cholestasis of pregnancy. Int. J. Gynecol. Obs. 2013, 122, 5–8. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metab. Clin. Exp. 2011, 60, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Legry, V.; Francque, S.; Haas, J.T.; Verrijken, A.; Caron, S.; Chávez-Talavera, O.; Vallez, E.; Vonghia, L.; Dirinck, E.; Verhaegen, A.; et al. Bile Acid Alterations Are Associated with Insulin Resistance, but Not with NASH, in Obese Subjects. J. Clin. Endocrinol. Metab. 2017, 102, 3783–3794. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Singh, S.; Bassirian, S.; Kolar, J.; Faulkner, C.; Sinha, N.; Bettencourt, R.; Gara, N.; Valasek, M.A.; et al. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment. Pharm. Ther. 2019, 49, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Jiang, Z.; Zhang, L. Bile acid regulation: A novel therapeutic strategy in non-alcoholic fatty liver disease. Pharm. Ther. 2018, 190, 81–90. [Google Scholar] [CrossRef]
- Horvatits, T.; Trauner, M.; Fuhrmann, V. Hypoxic liver injury and cholestasis in critically ill patients. Curr. Opin. Crit. Care 2013, 19, 128–132. [Google Scholar] [CrossRef]
- Soroka, C.J.; Boyer, J.L. Biosynthesis and trafficking of the bile salt export pump, BSEP: Therapeutic implications of BSEP mutations. Mol. Asp. Med. 2014, 37, 3–14. [Google Scholar] [CrossRef]
- Okushin, K.; Tsutsumi, T.; Enooku, K.; Fujinaga, H.; Kado, A.; Shibahara, J.; Fukayama, M.; Moriya, K.; Yotsuyanagi, H.; Koike, K. The intrahepatic expression levels of bile acid transporters are inversely correlated with the histological progression of nonalcoholic fatty liver disease. J. Gastroenterol. 2016, 51, 808–818. [Google Scholar] [CrossRef]
- Figge, A.; Lammert, F.; Paigen, B.; Henkel, A.; Matern, S.; Korstanje, R.; Shneider, B.L.; Chen, F.; Stoltenberg, E.; Spatz, K.; et al. Hepatic overexpression of murine Abcb11 increases hepatobiliary lipid secretion and reduces hepatic steatosis. J. Biol. Chem. 2004, 279, 2790–2799. [Google Scholar] [CrossRef]
- Makishima, M. Vitamin D Receptor as an Intestinal Bile Acid Sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Guo, G.L.; Lambert, G.; Negishi, M.; Ward, J.M.; Brewer, H.B.; Kliewer, S.A.; Gonzalez, F.J.; Sinal, C.J. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 2003, 278, 45062–45071. [Google Scholar] [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Edwards, P.A.; Kast, H.R.; Anisfeld, A.M. BAREing it all: The adoption of LXR and FXR and their roles in lipid homeostasis. J. Lipid Res. 2002, 43, 2–12. [Google Scholar] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Pircher, P.C.; Kitto, J.L.; Petrowski, M.L.; Tangirala, R.K.; Bischoff, E.D.; Schulman, I.G.; Westin, S.K. Farnesoid X Receptor Regulates Bile Acid-Amino Acid Conjugation. J. Boil. Chem. 2003, 278, 27703–27711. [Google Scholar] [CrossRef]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jiménez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 2017, 66, 1818–1828. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, S.-H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, H.; Zhang, M.; Guo, G.L. Fatty liver diseases, bile acids, and FXR. Acta Pharm. Sin. B 2016, 6, 409–412. [Google Scholar] [CrossRef]
- Li, Y.; Jadhav, K.; Zhang, Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem. Pharm. 2013, 86, 1517–1524. [Google Scholar] [CrossRef]
- Keitel, V.; Ullmer, C.; Häussinger, D. The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Boil. Chem. 2010, 391, 785–789. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. Metabolic interplay between white, beige, brown adipocytes and the liver. J. Hepatol. 2016, 64, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Donepudi, A.C.; Boehme, S.; Li, F.; Chiang, J.Y.L. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology 2017, 65, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-D.; Chen, W.-D.; Yu, D.; Forman, B.M.; Huang, W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Genet, C.; Strehle, A.; Schmidt, C.; Boudjelal, G.; Lobstein, A.; Schoonjans, K.; Souchet, M.; Auwerx, J.; Saladin, R.; Wagner, A. Structure—Activity Relationship Study of Betulinic Acid, A Novel and Selective TGR5 Agonist, and Its Synthetic Derivatives: Potential Impact in Diabetes. J. Med. Chem. 2010, 53, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Sousa, K.M.; Jin, L.; Dong, B.; Kim, B.-W.; Ramirez, R.; Xiao, Z.; Gu, Y.; Yang, Q.; Wang, J.; et al. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 2016, 64, 760–773. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef]
- Cariou, B.; Van Harmelen, K.; Duran-Sandoval, D.; Van Dijk, T.H.; Grefhorst, A.; Abdelkarim, M.; Caron, S.; Torpier, G.; Fruchart, J.-C.; Gonzalez, F.J.; et al. The Farnesoid X Receptor Modulates Adiposity and Peripheral Insulin Sensitivity in Mice. J. Boil. Chem. 2006, 281, 11039–11049. [Google Scholar] [CrossRef]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet α Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef]
- Conde, J.; Scotece, M.; Gómez, R.; Lopez, V.; Gómez-Reino, J.J.; Lago, F.; Gualillo, O.; Gómez-Reino, J.J.; Gómez-Reino, J.J. Adipokines: Biofactors from white adipose tissue. A complex hub among inflammation, metabolism, and immunity. BioFactors 2011, 37, 413–420. [Google Scholar] [CrossRef]
- Lehr, S.; Hartwig, S.; Sell, H. Adipokines: A treasure trove for the discovery of biomarkers for metabolic disorders. Proteom. Clin. Appl. 2012, 6, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Colombo, G.; Schiavon, L.; Schiavon, L. Adiponectin: A multitasking player in the field of liver diseases. Diabetes Metab. 2014, 40, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Balmer, M.L.; Joneli, J.; Schoepfer, A.; Stickel, F.; Thormann, W.; Dufour, J. Significance of serum adiponectin levels in patients with chronic liver disease. Clin. Sci. 2010, 119, 431–436. [Google Scholar] [CrossRef]
- Wree, A.; Schlattjan, M.; Bechmann, L.P.; Claudel, T.; Sowa, J.-P.; Stojakovic, T.; Scharnagl, H.; Köfeler, H.; Baba, H.A.; Gerken, G.; et al. Adipocyte cell size, free fatty acids and apolipoproteins are associated with non-alcoholic liver injury progression in severely obese patients. Metabolism 2014, 63, 1542–1552. [Google Scholar] [CrossRef]
- Ikejima, K.; Takei, Y.; Honda, H.; Hirose, M.; Yoshikawa, M.; Zhang, Y.-J.; Lang, T.; Fukuda, T.; Yamashina, S.; Kitamura, T.; et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002, 122, 1399–1410. [Google Scholar] [CrossRef]
- Huang, H.-H.; Lee, W.-J.; Chen, S.-C.; Chen, T.-F.; Lee, S.-D.; Chen, C.-Y. Bile Acid and Fibroblast Growth Factor 19 Regulation in Obese Diabetics, and Non-Alcoholic Fatty Liver Disease after Sleeve Gastrectomy. J. Clin. Med. 2019, 8, 815. [Google Scholar] [CrossRef]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef]
- Kwong, E.; Li, Y.; Hylemon, P.B.; Zhou, H. Bile acids and sphingosine-1-phosphate receptor 2 in hepatic lipid metabolism. Acta Pharm. Sin. B 2015, 5, 151–157. [Google Scholar] [CrossRef]
- Krattinger, R.; Boström, A.; Lee, S.M.; Thasler, W.E.; Schiöth, H.B.; Kullak-Ublick, G.A.; Mwinyi, J. Chenodeoxycholic acid significantly impacts the expression of miRNAs and genes involved in lipid, bile acid and drug metabolism in human hepatocytes. Life Sci. 2016, 156, 47–56. [Google Scholar] [CrossRef]
- Nakahara, M.; Fujii, H.; Maloney, P.R.; Shimizu, M.; Sato, R. Bile Acids Enhance Low Density Lipoprotein Receptor Gene Expression via a MAPK Cascade-mediated Stabilization of mRNA. J. Boil. Chem. 2002, 277, 37229–37234. [Google Scholar] [CrossRef]
- Fuchs, C.; Claudel, T.; Trauner, M. Bile Acid-Mediated Control of Liver Triglycerides. Semin. Liver Dis. 2013, 33, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Lawn, R.M. ABCAThe gatekeeper for eliminating excess tissue cholesterol. J. Lipid Res. 2001, 42, 1173–1179. [Google Scholar] [PubMed]
- Eckardstein, A.V.; Kardassis, D. High Density Lipoproteins: From Biological Understanding to Clinical Exploitation; Handbook of Experimental Pharmacology; Cham, S.L., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 224. [Google Scholar]
- Li, G.; Thomas, A.M.; Williams, J.A.; Kong, B.; Liu, J.; Inaba, Y.; Xie, W.; Guo, G.L. Farnesoid X Receptor Induces Murine Scavenger Receptor Class B Type I via Intron Binding. PLoS ONE 2012, 7, e35895. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Kong, B.; Stieger, B.; Tschopp, O.; Schultze, S.M.; Rau, M.; Weber, A.; Müllhaupt, B.; Guo, G.L.; Geier, A. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 2015, 35, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef]
- Janssen, A.W.F.; Houben, T.; Katiraei, S.; Dijk, W.; Boutens, L.; Van Der Bolt, N.; Wang, Z.; Brown, J.M.; Hazen, S.L.; Mandard, S.; et al. Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: A potential role for bile acids. J. Lipid Res. 2017, 58, 1399–1416. [Google Scholar] [CrossRef]
- Ushiroda, C.; Naito, Y.; Takagi, T.; Uchiyama, K.; Mizushima, K.; Higashimura, Y.; Yasukawa, Z.; Okubo, T.; Inoue, R.; Honda, A.; et al. Green tea polyphenol (epigallocatechin-3-gallate) improves gut dysbiosis and serum bile acids dysregulation in high-fat diet-fed mice. J. Clin. Biochem. Nutr. 2019, 65, 34–46. [Google Scholar] [CrossRef]
- Appleby, R.N.; Moghul, I.; Khan, S.; Yee, M.; Manousou, P.; Neal, T.D.; Walters, J.R.F. Non-alcoholic fatty liver disease is associated with dysregulated bile acid synthesis and diarrhea: A prospective observational study. PLoS ONE 2019, 14, e0211348. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Mueller, M.; Thorell, A.; Claudel, T.; Jha, P.; Koefeler, H.; Lackner, C.; Hoesel, B.; Fauler, G.; Stojakovic, T.; Einarsson, C.; et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J. Hepatol. 2015, 62, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Rautiainen, H.; Kärkkäinen, P.; Karvonen, A.-L.; Nurmi, H.; Pikkarainen, P.; Nuutinen, H.; Färkkilä, M. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: A three-year randomized trial. Hepatology 2005, 41, 747–752. [Google Scholar] [CrossRef]
- Namisaki, T.; Noguchi, R.; Moriya, K.; Kitade, M.; Aihara, Y.; Douhara, A.; Nishimura, N.; Takeda, K.; Okura, Y.; Kawaratani, H.; et al. Beneficial effects of combined ursodeoxycholic acid and angiotensin-II type 1 receptor blocker on hepatic fibrogenesis in a rat model of nonalcoholic steatohepatitis. J. Gastroenterol. 2016, 51, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Ghaleh, H.E.G.; Motlagh, B.M.; Azarbayjani, A.F.; Zarei, L. Therapeutic effects of curcumin and ursodexycholic acid on non-alcoholic fatty liver disease. Biomed. Pharm. 2019, 115, 108938. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. Fibrates and cholestasis. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef]
- Li, F.; Patterson, A.D.; Krausz, K.W.; Tanaka, N.; Gonzalez, F.J. Metabolomics reveals an essential role for peroxisome proliferator-activated receptor α in bile acid homeostasis. J. Lipid Res. 2012, 53, 1625–1635. [Google Scholar] [CrossRef]
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558. [Google Scholar] [CrossRef]
- Kim, M.S.; Kung, S.; Grewal, T.; Roufogalis, B.D. Methodologies for investigating natural medicines for the treatment of nonalcoholic fatty liver disease (NAFLD). Curr. Pharm. Biotechnol. 2012, 13, 278–291. [Google Scholar]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef]
- Ullmer, C.; Sanchez, R.A.; Sprecher, U.; Raab, S.; Mattei, P.; Dehmlow, H.; Sewing, S.; Iglesias, A.; Beauchamp, J.; Conde-Knape, K. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br. J. Pharm. 2013, 169, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.D.; Rodriguez, D.; Kohler, J.; Jiang, Z.; Wan, S.; Blanco, E.; King, A.J.; Chen, T.; Bell, N.; Dragoli, D.; et al. Intestinal TGR5 agonism improves hepatic steatosis and insulin sensitivity in Western diet-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Comeglio, P.; Cellai, I.; Mello, T.; Filippi, S.; Maneschi, E.; Corcetto, F.; Corno, C.; Sarchielli, E.; Morelli, A.; Rapizzi, E.; et al. INT-767 prevents NASH and promotes visceral fat brown adipogenesis and mitochondrial function. J. Endocrinol. 2018, 238, 107–127. [Google Scholar] [CrossRef]
- Safadi, R.; Konikoff, F.M.; Hershkovitz, A.; Gilat, T.; Halpern, M.; Rosenthal-Galili, Z.; Zuckerman, E.; Abu-Mouch, S.; Fich, A.; Sikuler, E.; et al. The Fatty Acid–Bile Acid Conjugate Aramchol Reduces Liver Fat Content in Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091. [Google Scholar] [CrossRef]
- Pellicciari, R.; Passeri, D.; Franco, F.; de Mostarda, S.; Filipponi, P.; Colliva, C.; Gadaleta, R.M.; Franco, P.; Carotti, A.; Macchiarulo, A.; et al. Discovery of 3α, 7α, 11β-Trihydroxy-6α-ethyl-5β-cholan-24-oic Acid (TC-100), a Novel Bile Acid as Potent and Highly Selective FXR Agonist for Enterohepatic Disorders. J. Med. Chem. 2016, 59, 9201–9214. [Google Scholar] [CrossRef]
- Liu, H.; Pang, G.; Ren, J.; Zhao, Y.; Wang, J. A novel class of apical sodium--dependent bile salt transporter inhibitors: 1-(2,4-bifluorophenyl)-7-dialkylamino-1,8-naphthyridine-3-carboxamides. Acta Pharm. Sin. B 2017, 7, 223–229. [Google Scholar] [CrossRef]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; McNulty, J.; Verschuren, L.; Palmer, M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr-/-.Leiden mice. PLoS ONE 2019, 14, e0218459. [Google Scholar] [CrossRef]
- Ge, M.-X.; Niu, W.-X.; Ren, J.-F.; Cai, S.-Y.; Yu, D.-K.; Liu, H.-T.; Zhang, N.; Zhang, Y.-X.; Wang, Y.-C.; Shao, R.-G.; et al. A novel ASBT inhibitor, IMB17-15, repressed nonalcoholic fatty liver disease development in high-fat diet-fed Syrian golden hamsters. Acta Pharm. Sin. 2019, 40, 895–907. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Name/Dosage | Name of Study | Clinicaltrials.gov ID | Phase | Status |
|---|---|---|---|---|
| FXR agonists | ||||
| OCA (Obeticholic Acid) | The Farnesoid X Receptor (FXR) Ligand Obeticholic Acid in NASH Treatment Trial (FLINT) | NCT01265498 | 2 | completed |
| Study of INT-747 in Patients With Diabetes and Presumed NAFLD | NCT00501592 | 2 | completed | |
| Combination Obeticholic Acid (OCA) and Statins for Monitoring of Lipids (CONTROL) | NCT02633956 | 2 | completed | |
| 10, 25mg | Randomized Global Phase 3 Study to Evaluate the Impact on NASH With Fibrosis of Obeticholic Acid Treatment (REGENERATE) | NCT02548351 | 3 | recruiting |
| 10mg, 10 or 25mg | Study Evaluating the Efficacy and Safety of Obeticholic Acid in Subjects With Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis | NCT03439254 | 3 | recruiting |
| PPAR agonists | ||||
| Saroglitazar (PPARα and – γ agonist) | Safety, Tolerability and Efficacy of Saroglitazar Mg 4 mg in Liver Transplant Recipients With NAFLD | NCT03639623 | 2 | recruiting |
| IVA-337 (Lanifibranor) (Pan-PPAR agonist) | Phase 2b Study in NASH to Assess IVA337 (NATIVE) | NCT03008070 | 2b | recruiting |
| Lobeglitazone (PPARγ agonist) | A 24 Week, Multicenter, Prospective, Open-labeled, Single-arm, Exploratory Phase 4 Clinical Trial to Evaluate the Safety and Efficacy of Lobeglitazone in Decreasing Intrahepatic Fat Contents in Type 2 Diabetes With NAFLD | NCT02285205 | 4 | completed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. https://doi.org/10.3390/cells8111358
Gottlieb A, Canbay A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells. 2019; 8(11):1358. https://doi.org/10.3390/cells8111358
Chicago/Turabian StyleGottlieb, Aline, and Ali Canbay. 2019. "Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression" Cells 8, no. 11: 1358. https://doi.org/10.3390/cells8111358
APA StyleGottlieb, A., & Canbay, A. (2019). Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells, 8(11), 1358. https://doi.org/10.3390/cells8111358