A Low-Therapeutic Dose of Lithium Inhibits GSK3 and Enhances Myoblast Fusion in C2C12 Cells



 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. C2C12 Cells
2.2. Western Blotting
2.3. GSK3 Activity Assay
2.4. Generation of GSK3 Double-Knockout Cell-Line
2.5. Myoblast Fusion
2.6. Statistical Analysis
3. Results
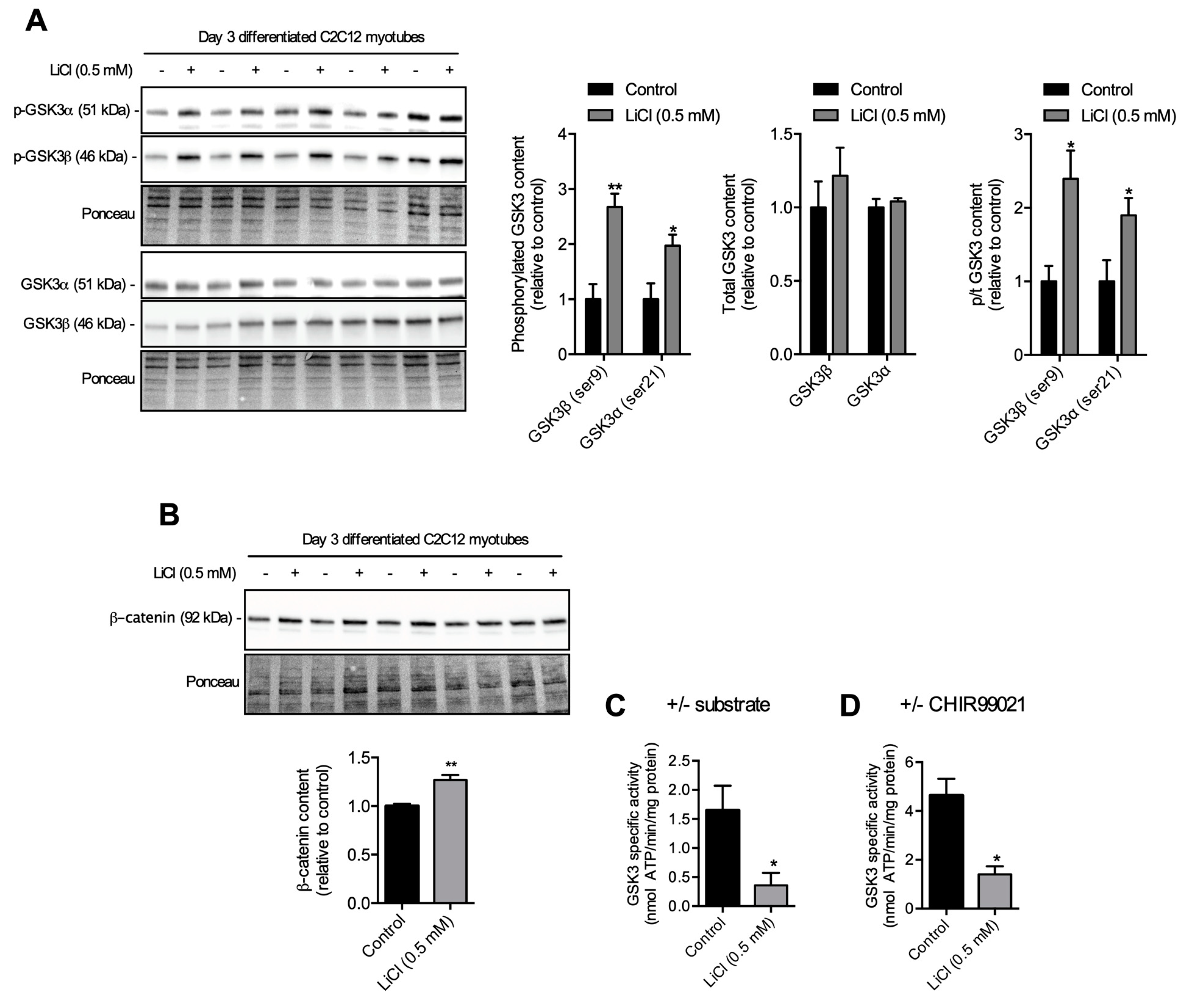
3.1. A Low-Therapeutic Dose of LiCl Inhibits GSK3β and Total GSK3 Activity
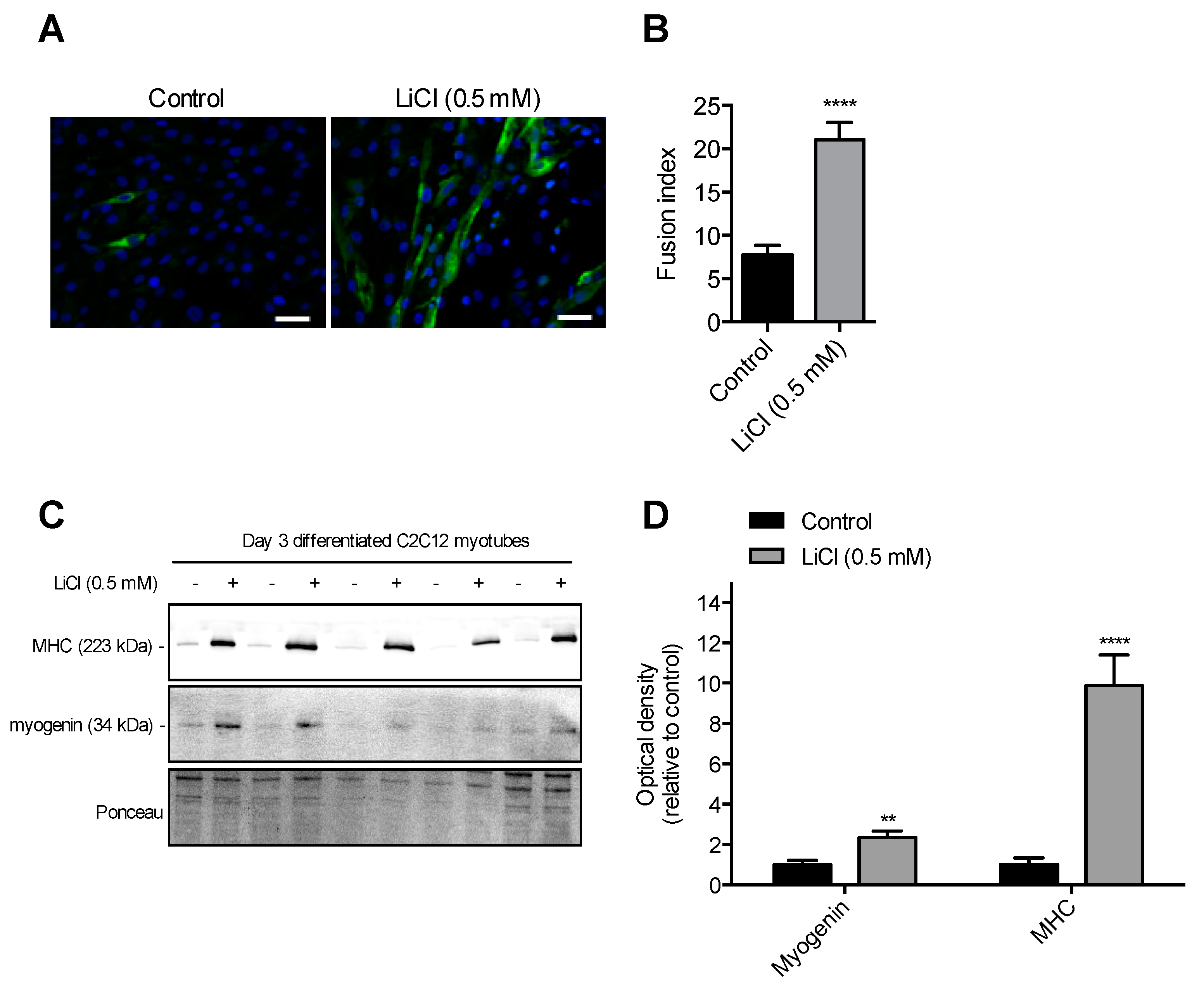
3.2. Sub-Therapeutic Dose of LiCl Augments Myoblast Fusion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boyd, C.M.; Xue, Q.-L.; Simpson, C.F.; Guralnik, J.M.; Fried, L.P. Frailty, hospitalization, and progression of disability in a cohort of disabled older women. Am. J. Med. 2005, 118, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Landi, F.; Cruz-Jentoft, A.J.; Liperoti, R.; Russo, A.; Giovannini, S.; Tosato, M.; Capoluongo, E.; Bernabei, R.; Onder, G. Sarcopenia and mortality risk in frail older persons aged 80 years and older: results from ilSIRENTE study. Age Ageing 2013, 42, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R.; Reginster, J.-Y.; Arnal, J.-F.; Bautmans, I.; Beaudart, C.; Bischoff-Ferrari, H.; Biver, E.; Boonen, S.; Brandi, M.-L.; Chines, A.; et al. Quality of life in sarcopenia and frailty. Calcif. Tissue Int. 2013, 93, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; Williams, J.; Atherton, P.J.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, Dynapenia, and the Impact of Advancing Age on Human Skeletal Muscle Size and Strength; A Quantitative Review. Front. Physiol. 2012, 3, 3. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Dou, Q.; Zhang, W.; Yang, Y.; Xie, X. Sarcopenia as a predictor of all-cause mortality among older nursing home residents: A systematic review and meta-analysis. BMJ Open 2018, 8, e021252. [Google Scholar] [CrossRef]
- McGlory, C.; Vliet, S.; Stokes, T.; Mittendorfer, B.; Phillips, S.M.; Van Vliet, S. The impact of exercise and nutrition on the regulation of skeletal muscle mass. J. Physiol. 2018, 597, 1251–1258. [Google Scholar] [CrossRef]
- McKinlay, B.J.; Wallace, P.; Dotan, R.; Long, D.; Tokuno, C.; Gabriel, D.A.; Falk, B. Effects of Plyometric and Resistance Training on Muscle Strength, Explosiveness, and Neuromuscular Function in Young Adolescent Soccer Players. J. Strength Cond. Res. 2018, 32, 3039–3050. [Google Scholar] [CrossRef]
- Lopez, P.; Pinto, R.S.; Radaelli, R.; Rech, A.; Grazioli, R.; Izquierdo, M.; Cadore, E.L. Benefits of resistance training in physically frail elderly: A systematic review. Aging Clin. Exp. Res. 2018, 30, 889–899. [Google Scholar] [CrossRef]
- Chen, E.H.; Olson, E.N. Unveiling the Mechanisms of Cell-Cell Fusion. Science 2005, 308, 369–373. [Google Scholar] [CrossRef]
- Bruusgaard, J.C.; Johansen, I.B.; Egner, I.M.; Rana, Z.A.; Gundersen, K. Myonuclei acquired by overload exercise precede hypertrophy and are not lost on detraining. Proc. Natl. Acad. Sci. USA 2010, 107, 15111–15116. [Google Scholar] [CrossRef]
- Guerci, A.; Lahoute, C.; Hébrard, S.; Collard, L.; Graindorge, D.; Favier, M.; Cagnard, N.; Batonnet-Pichon, S.; Précigout, G.; García, L.; et al. Srf-Dependent Paracrine Signals Produced by Myofibers Control Satellite Cell-Mediated Skeletal Muscle Hypertrophy. Cell Metab. 2012, 15, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Cheek, D.B.; Powell, G.K.; Scott, R.E. Growth of muscle mass and skeletal collagen in the rat. I. Normal growth. Johns Hopkins Med. J. 1965, 116, 378–387. [Google Scholar]
- Allen, D.L.; Roy, R.R.; Edgerton, V.R. Myonuclear domains in muscle adaptation and disease. Muscle Nerve 1999, 22, 1350–1360. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Biersmith, B.H.; McNally, E.M. Membrane fusion in muscle development and repair. Semin. Cell Dev. Biol. 2015, 45, 48–56. [Google Scholar] [CrossRef]
- McGeachie, J.K.; Grounds, M.D. The timing between skeletal muscle myoblast replication and fusion into myotubes, and the stability of regenerated dystrophic myofibres: An autoradiographic study in mdx mice. J. Anat. 1999, 194, 287–295. [Google Scholar] [CrossRef]
- Anderson, J.E.; Weber, M.; Vargas, C. Deflazacort increases laminin expression and myogenic repair, and induces early persistent functional gain in mdx mouse muscular dystrophy. Cell Transplant. 2000, 9, 551–564. [Google Scholar] [CrossRef]
- Vyas, D.R.; Spangenburg, E.E.; Abraha, T.W.; Childs, T.E.; Booth, F.W. GSK-3β negatively regulates skeletal myotube hypertrophy. Am. J. Physiol. Physiol. 2002, 283, C545–C551. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of igf-1-induced skeletal myotube hypertrophy by pi(3)k/akt/mtor and pi(3)k/akt/gsk3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Van der Velden, J.L.; Schols, A.M.; Willems, J.; Kelders, M.C.; Langen, R.C. Glycogen synthase kinase 3 suppresses myogenic differentiation through negative regulation of nfatc3. J. Biol. Chem. 2008, 283, 358–366. [Google Scholar] [CrossRef]
- Pansters, N.A.M.; Schols, A.M.W.J.; Verhees, K.J.P.; de Theije, C.C.; Snepvangers, F.J.; Kelders, M.C.J.M.; Ubags, N.D.J.; Haegens, A.; Langen, R.C.J. Muscle-specific gsk-3β ablation accelerates regeneration of disuse-atrophied skeletal muscle. Biochim. Biophys. Acta 2015, 1852, 490–506. [Google Scholar] [CrossRef]
- Suzuki, A.; Pelikan, R.C.; Iwata, J. WNT/β-Catenin Signaling Regulates Multiple Steps of Myogenesis by Regulating Step-Specific Targets. Mol. Cell. Boil. 2015, 35, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Malhi, G.S.; Outhred, T. Therapeutic Mechanisms of Lithium in Bipolar Disorder: Recent Advances and Current Understanding. CNS Drugs 2016, 30, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr. Boil. 1996, 6, 1664–1669. [Google Scholar] [CrossRef]
- Hedya, S.A.; Swoboda, H.D. Lithium Toxicity. StatPearls Publishing. January 2018. Treasure Island (FL), [Updated 2018 Dec 28]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499992/ (accessed on 18 February 2019).
- Malhi, G.S.; Berk, M. Is the safety of lithium no longer in the balance? Lancet 2012, 379, 690–692. [Google Scholar] [CrossRef]
- Maddalena, L.A.; Selim, S.M.; Fonseca, J.; Messner, H.; McGowan, S.; Stuart, J.A. Hydrogen peroxide production is affected by oxygen levels in mammalian cell culture. Biochem. Biophys. Res. Commun. 2017, 493, 246–251. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Watson, C.J.; Bott, K.N.; Moradi, F.; A Maddalena, L.; Bellissimo, C.A.; Turner, K.D.; Peters, S.J.; Leblanc, P.J.; MacNeil, A.J.; et al. Neurogranin is expressed in mammalian skeletal muscle and inhibits calcineurin signaling and myoblast fusion. Am. J. Physiol. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Maddala, R.N.M.; Ashwal, A.J.; Rao, M.S.; Padmakumar, R. Chronic lithium intoxication: Varying electrocardiogram manifestations. Indian J. Pharmacol. 2017, 49, 127–129. [Google Scholar]
- Findlay, A.R.; Bengoechea, R.; Pittman, S.K.; Chou, T.-F.; True, H.L.; Weihl, C.C. Lithium chloride corrects weakness and myopathology in a preclinical model of LGMD1D. Neurol. Genet. 2019, 5, e318. [Google Scholar] [CrossRef]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of gsk-3α and gsk-3β in wnt/β-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef]
- Patel, S.; Doble, B.W.; MacAulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol. Cell. Biol. 2008, 28, 6314–6328. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhong, Z.; Zheng, Z.; Shi, X.-M.; Zhang, W. Inhibition of Glycogen Synthase Kinase-3β Attenuates Glucocorticoid-Induced Suppression of Myogenic Differentiation In Vitro. PLoS ONE 2014, 9, e105528. [Google Scholar] [CrossRef] [PubMed]
- Villa-Moruzzi, E.; Puntoni, F.; Marin, O. Activation of protein phosphatase-1 isoforms and glycogen synthase kinase-3 beta in muscle from mdx mice. Int. J. Biochem. Cell Biol. 1996, 28, 13–22. [Google Scholar] [CrossRef]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 2012, 122, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Kwon, E.-S.; Kwon, K.-S. Molecular mechanisms and therapeutic interventions in sarcopenia. Osteoporos. Sarcopenia 2017, 3, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.C.; DelBello, M.P.; Bryan, H.S.; Adler, C.M.; Kowatch, R.A.; Stanford, K.; Strakowski, S.M. Open-Label Lithium for the Treatment of Adolescents With Bipolar Depression. J. Am. Acad. Child Adolesc. Psychiatry 2006, 45, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Paré, M.; Baechler, B.; Fajardo, V.; Earl, E.; Wong, E.; Campbell, T.; Tupling, A.R.; Quadrilatero, J. Effect of acute and chronic autophagy deficiency on skeletal muscle apoptotic signaling, morphology, and function. Biochim. Biophys. Acta 2017, 1864, 708–718. [Google Scholar] [CrossRef]
- Risson, V.; Mazelin, L.; Roceri, M.; Sanchez, H.; Moncollin, V.; Corneloup, C.; Richard-Bulteau, H.; Vignaud, A.; Baas, D.; Defour, A.; et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009, 187, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Bombardier, E.; Irvine, T.; Metherel, A.H.; Stark, K.D.; Duhamel, T.; Rush, J.W.; Green, H.J.; Tupling, A.R. Dietary docosahexaenoic acid supplementation reduces SERCA Ca2+ transport efficiency in rat skeletal muscle. Chem. Phys. Lipids 2015, 187, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Bombardier, E.; McMillan, E.; Tran, K.; Wadsworth, B.J.; Gamu, D.; Hopf, A.; Vigna, C.; Smith, I.C.; Bellissimo, C.; et al. Phospholamban overexpression in mice causes a centronuclear myopathy-like phenotype. Dis. Model. Mech. 2015, 8, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Bombardier, E.; Tran, K.; Metherel, A.H.; Irvine, T.; Holloway, G.P.; Green, H.J.; Stark, K.D.; Tupling, A.R. Sarcoplasmic Reticulum Phospholipid Fatty Acid Composition and Sarcolipin Content in Rat Skeletal Muscle. J. Membr. Biol. 2015, 248, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Bombardier, E.; Vigna, C.; Devji, T.; Bloemberg, D.; Gamu, D.; Gramolini, A.O.; Quadrilatero, J.; Tupling, A.R. Co-Expression of SERCA Isoforms, Phospholamban and Sarcolipin in Human Skeletal Muscle Fibers. PLoS ONE 2013, 8, e84304. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Chambers, P.J.; Juracic, E.S.; Rietze, B.A.; Gamu, D.; Bellissimo, C.; Kwon, F.; Quadrilatero, J.; Russell Tupling, A. Sarcolipin deletion in mdx mice impairs calcineurin signalling and worsens dystrophic pathology. Hum. Mol. Genet. 2018, 27, 4094–4102. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Gamu, D.; Mitchell, A.; Bloemberg, D.; Bombardier, E.; Chambers, P.J.; Bellissimo, C.; Quadrilatero, J.; Tupling, A.R. Sarcolipin deletion exacerbates soleus muscle atrophy and weakness in phospholamban overexpressing mice. PLoS ONE 2017, 12, 0173708. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Rietze, B.A.; Chambers, P.J.; Bellissimo, C.; Bombardier, E.; Quadrilatero, J.; Tupling, A.R. Effects of sarcolipin deletion on skeletal muscle adaptive responses to functional overload and unload. Am. J. Physiol. 2017, 313, C154–C161. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Smith, I.C.; Bombardier, E.; Chambers, P.J.; Quadrilatero, J.; Tupling, A.R. Diaphragm assessment in mice overexpressing phospholamban in slow-twitch type I muscle fibers. Brain Behav. 2016, 6, e00470. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Trojanowski, N.; Castelli, L.M.; Miotto, P.M.; Amoye, F.; Ward, W.E.; Tupling, A.R.; LeBlanc, P.J. Saturation of serca’s lipid annulus may protect against its thermal inactivation. Biochem. Biophys. Res. Commun. 2017, 484, 456–460. [Google Scholar] [CrossRef]
- Fu, M.H.; Tupling, A.R. Protective effects of Hsp70 on the structure and function of SERCA2a expressed in HEK-293 cells during heat stress. Am. J. Physiol. Circ. Physiol. 2009, 296, H1175–H1183. [Google Scholar] [CrossRef] [PubMed]
- Tupling, A.R.; Bombardier, E.; Gupta, S.C.; Hussain, D.; Vigna, C.; Bloemberg, D.; Quadrilatero, J.; Trivieri, M.G.; Babu, G.J.; Backx, P.H.; et al. Enhanced Ca2+ transport and muscle relaxation in skeletal muscle from sarcolipin-null mice. American journal of physiology. Cell Physiol. 2011, 301, C841–C849. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.C.; Bombardier, E.; Vigna, C.; Tupling, A.R. Atp consumption by sarcoplasmic reticulum Ca2+ pumps accounts for 40–50% of resting metabolic rate in mouse fast and slow twitch skeletal muscle. PLoS ONE 2013, 8, e68924. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Vector | Target Site | Targeted Region |
|---|---|---|---|
| GSK3α | U6gRNA-Cas9-2A-RFP | GCGCGGACTAGCTCGTTCGCGG | 168–189 |
| GSK3β | U6gRNA-Cas9-2A-GFP | GGCTTGCAGCTCTCCGCAAAGG | 450–470 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurgan, N.; Whitley, K.C.; Maddalena, L.A.; Moradi, F.; Stoikos, J.; Hamstra, S.I.; Rubie, E.A.; Kumar, M.; Roy, B.D.; Woodgett, J.R.; et al. A Low-Therapeutic Dose of Lithium Inhibits GSK3 and Enhances Myoblast Fusion in C2C12 Cells. Cells 2019, 8, 1340. https://doi.org/10.3390/cells8111340
Kurgan N, Whitley KC, Maddalena LA, Moradi F, Stoikos J, Hamstra SI, Rubie EA, Kumar M, Roy BD, Woodgett JR, et al. A Low-Therapeutic Dose of Lithium Inhibits GSK3 and Enhances Myoblast Fusion in C2C12 Cells. Cells. 2019; 8(11):1340. https://doi.org/10.3390/cells8111340
Chicago/Turabian StyleKurgan, Nigel, Kennedy C. Whitley, Lucas A. Maddalena, Fereshteh Moradi, Joshua Stoikos, Sophie I. Hamstra, Elizabeth A. Rubie, Megha Kumar, Brian D. Roy, James R. Woodgett, and et al. 2019. "A Low-Therapeutic Dose of Lithium Inhibits GSK3 and Enhances Myoblast Fusion in C2C12 Cells" Cells 8, no. 11: 1340. https://doi.org/10.3390/cells8111340
APA StyleKurgan, N., Whitley, K. C., Maddalena, L. A., Moradi, F., Stoikos, J., Hamstra, S. I., Rubie, E. A., Kumar, M., Roy, B. D., Woodgett, J. R., Stuart, J. A., & Fajardo, V. A. (2019). A Low-Therapeutic Dose of Lithium Inhibits GSK3 and Enhances Myoblast Fusion in C2C12 Cells. Cells, 8(11), 1340. https://doi.org/10.3390/cells8111340