
SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 during Serum-Starvation-Induced Ciliogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Cell Culture, Transfection, Ciliogenesis, and Drug Treatment
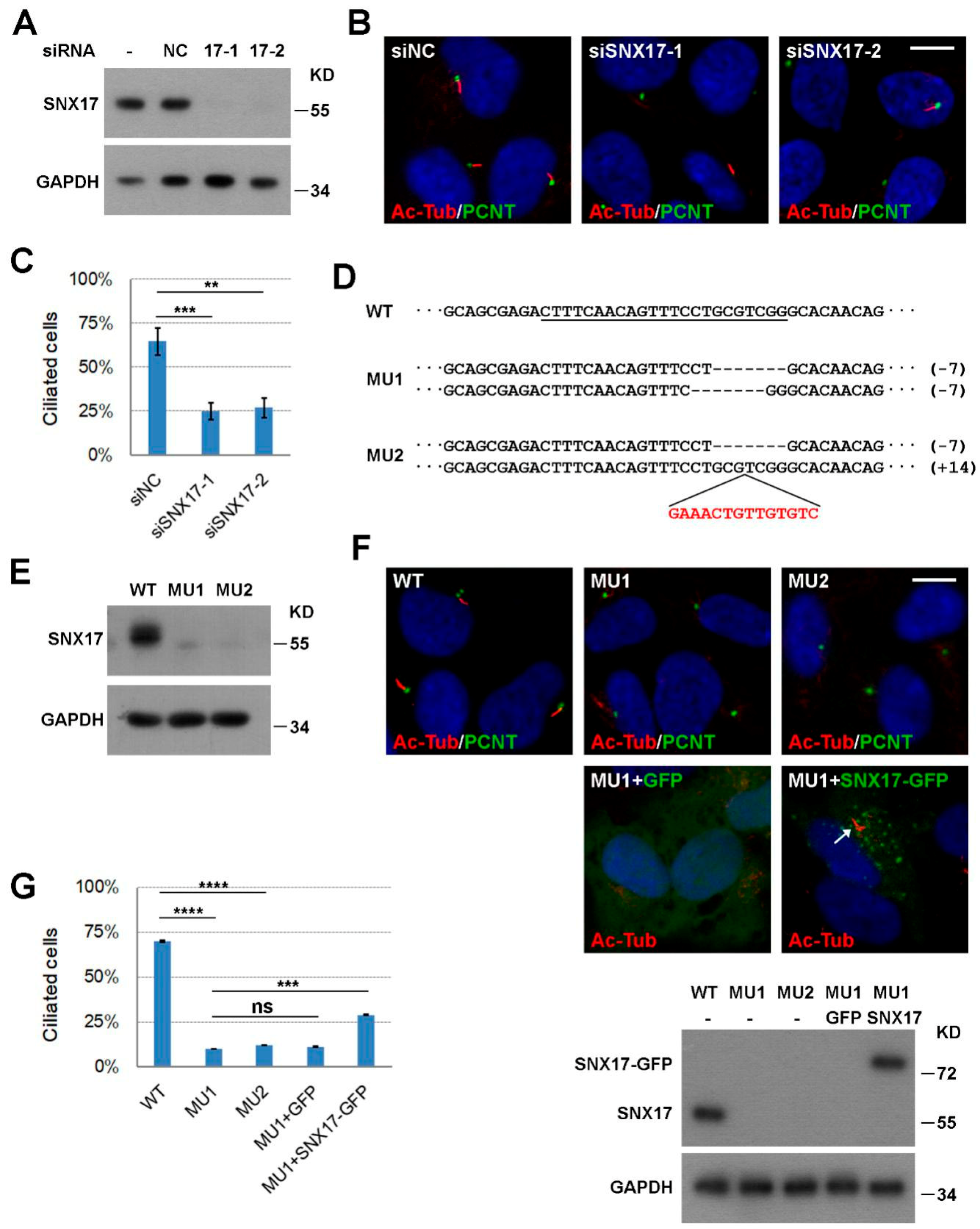
2.3. SNX17 Knockout
2.4. Immunofluorescence Staining
2.5. Immunoprecipitation and Western Blot
2.6. Ubiquitination Assay
2.7. Statistical Analyses
3. Results
3.1. SNX17 Is Required for Serum-Starvation-Induced Ciliogenesis
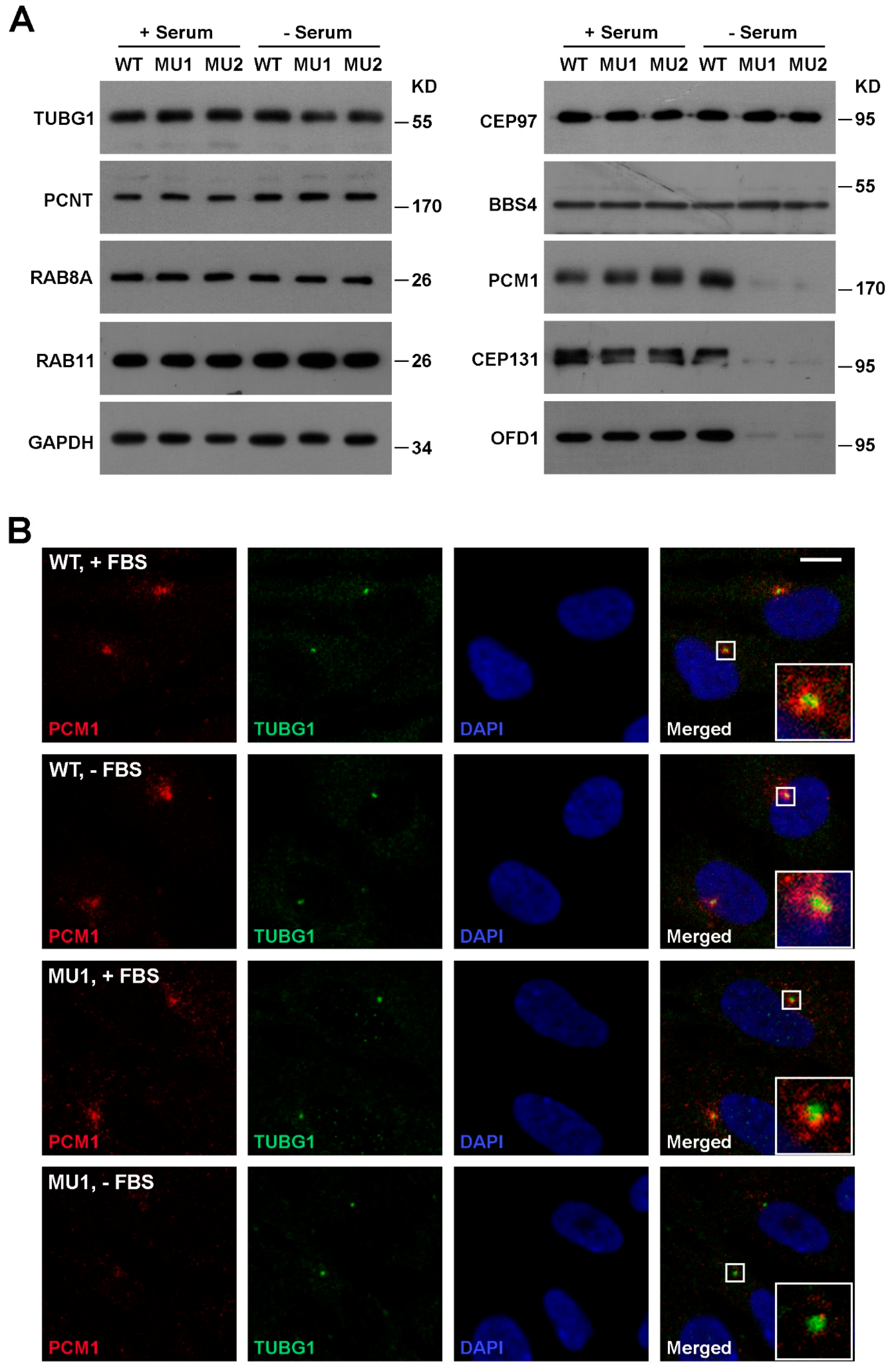
3.2. SNX17 Regulates Homeostasis of Multiple Centriolar Satellite Proteins During Ciliogenesis
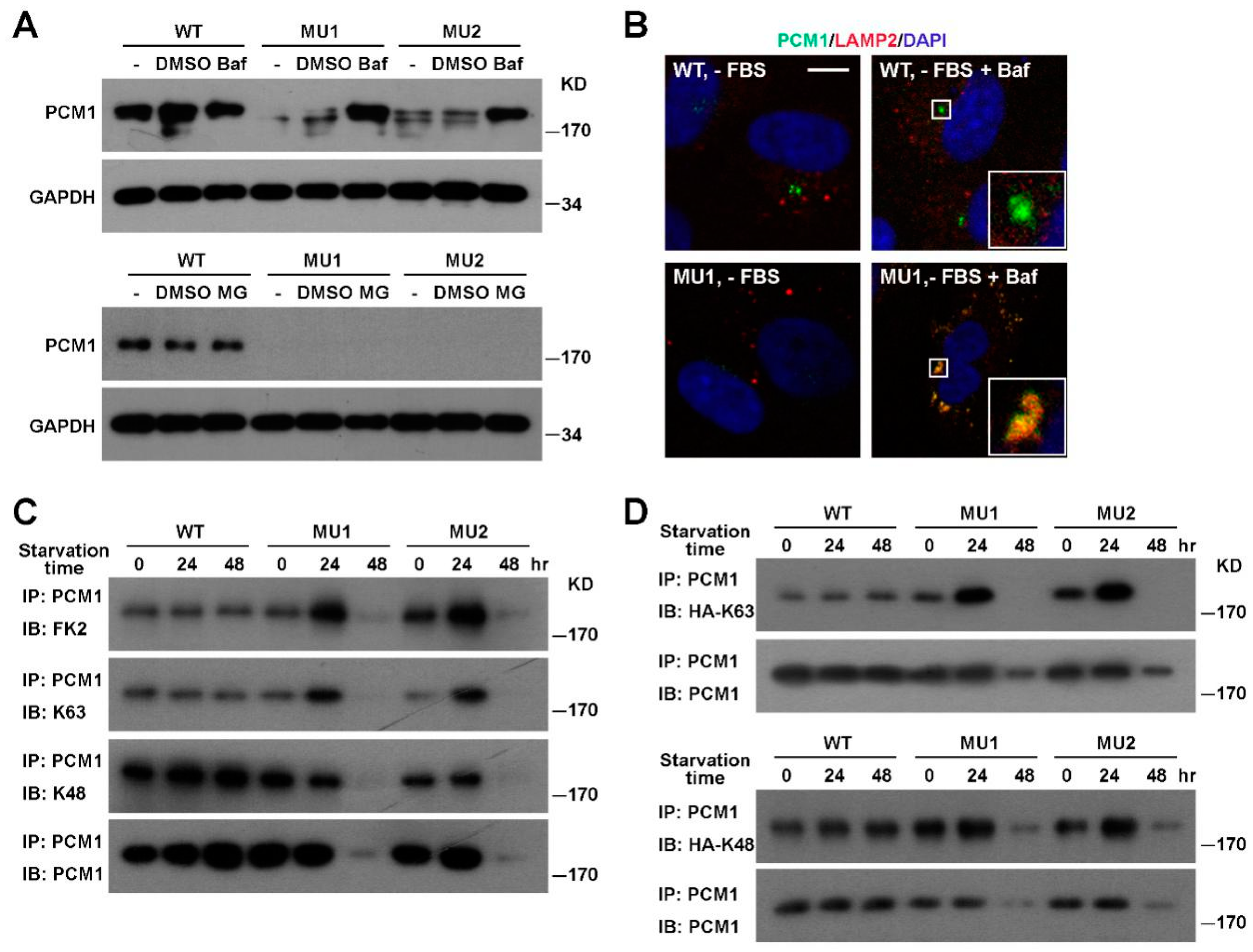
3.3. PCM1 Is Degraded Via the Lysosomal Pathway in SNX17 Knockout Cells
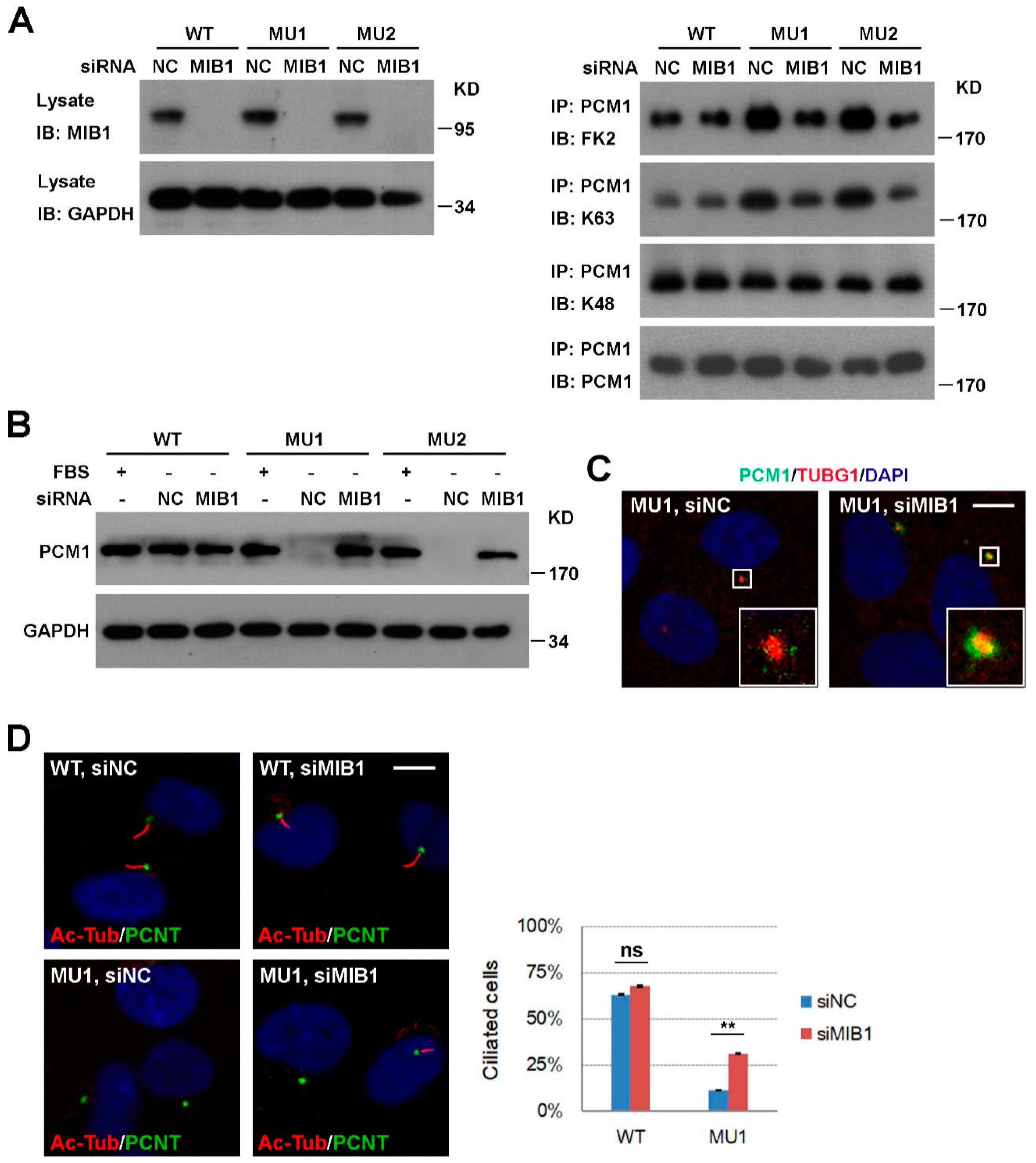
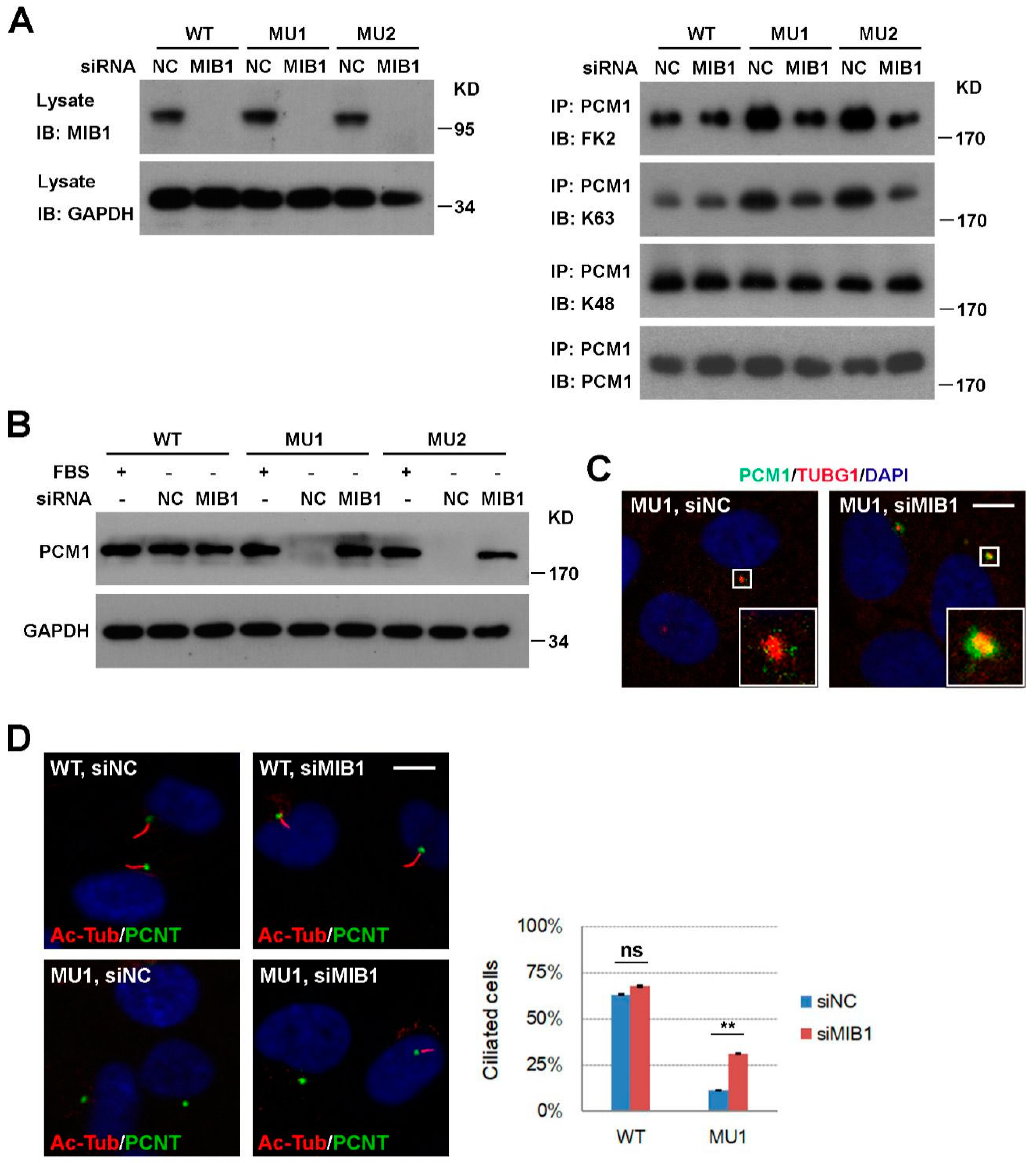
3.4. Knockdown of MIB1 Rescues PCM1 and Ciliogenesis Defects in SNX17 Knockout Cells
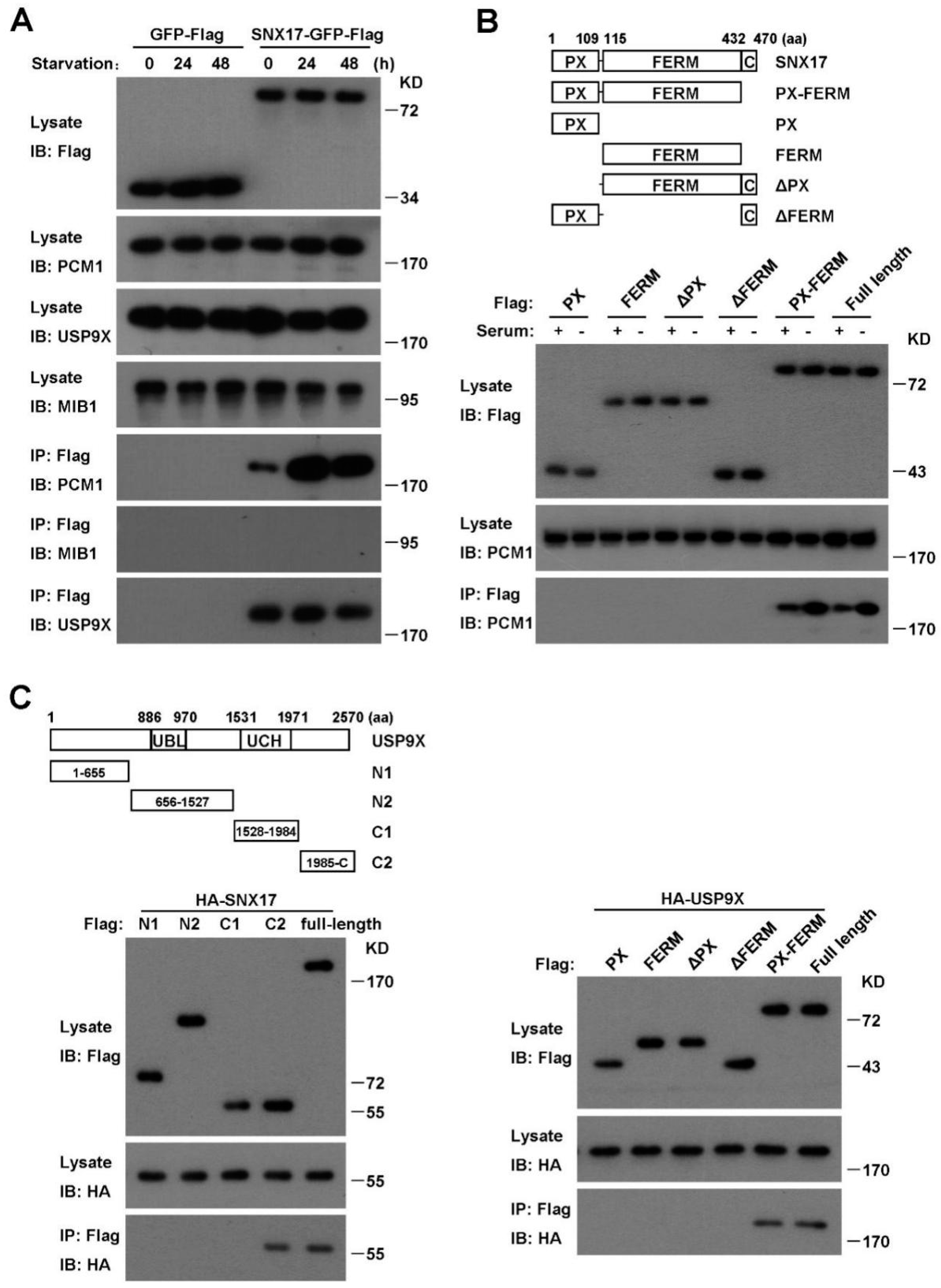
3.5. SNX17 Recruits USP9X to Antagonize the MIB1-Induced Degradation of PCM1
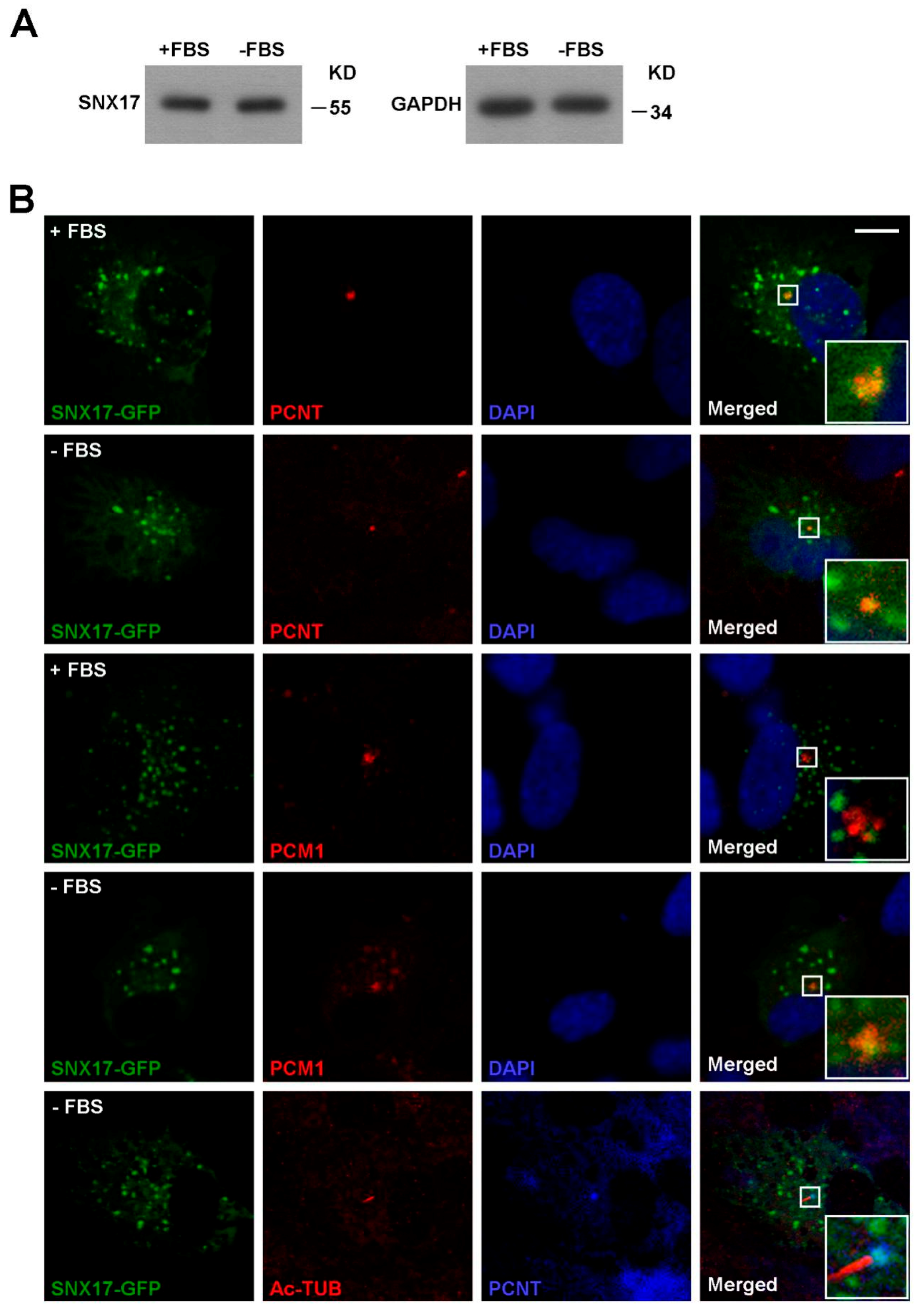
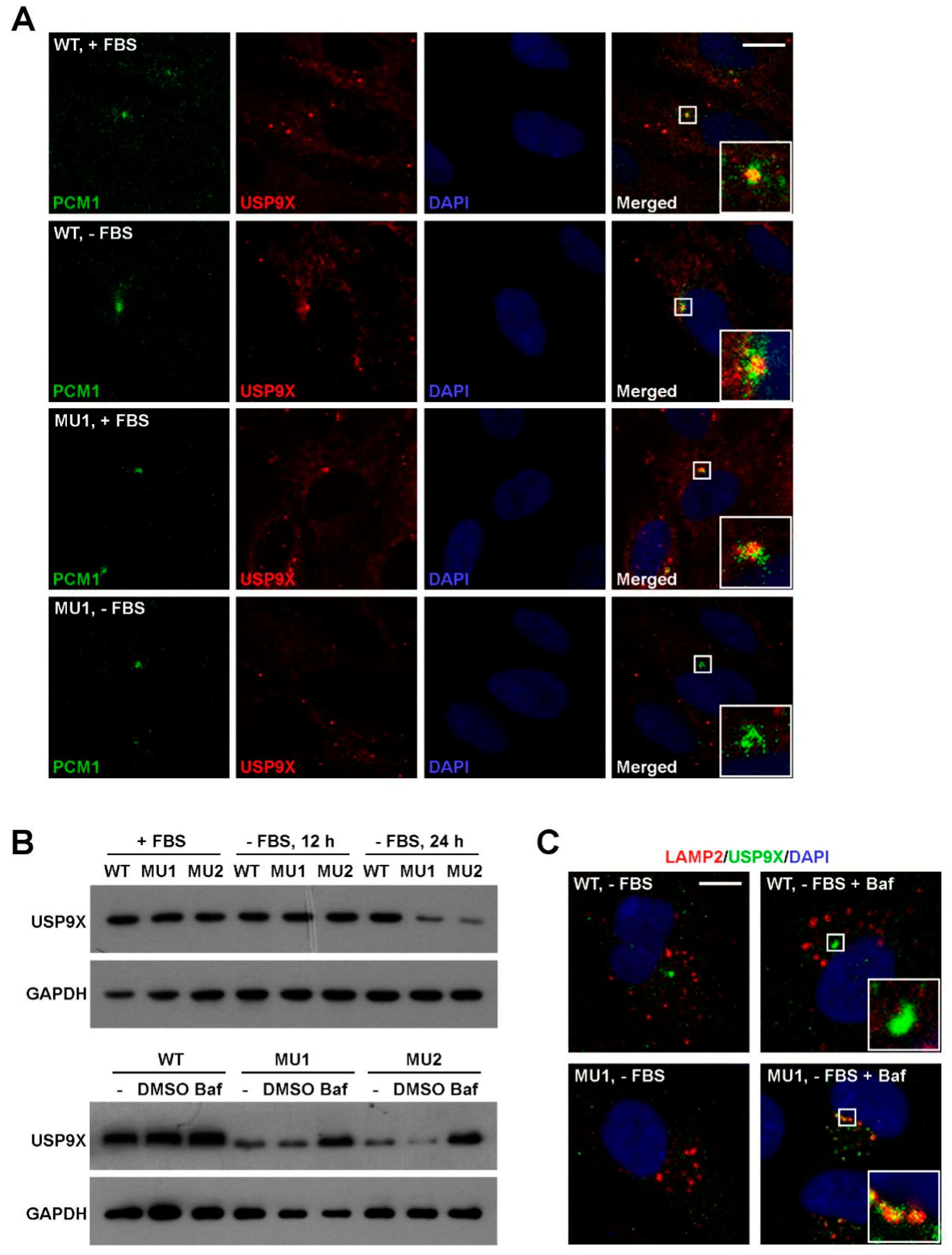
3.6. SNX17 Regulates USP9X Homeostasis Upon Serum Starvation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vertii, A.; Hehnly, H.; Doxsey, S. The centrosome, a multitalented renaissance organelle. Cold Spring Harb. Perspect. Biol. 2016, 8, a025049. [Google Scholar] [CrossRef] [PubMed]
- Hori, A.; Toda, T. Regulation of centriolar satellite integrity and its physiology. Cell Mol. Life Sci. 2017, 74, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Krishnaswami, S.R.; Gleeson, J.G. Cep290 interacts with the centriolar satellite component pcm-1 and is required for rab8 localization to the primary cilium. Hum. Mol. Genet. 2008, 17, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lee, K.; Malonis, R.; Sanchez, I.; Dynlacht, B.D. Tethering of an E3 ligase by Pcm1 regulates the abundance of centrosomal kiaa0586/talpid3 and promotes ciliogenesis. Elife 2016, 5, e12950. [Google Scholar] [CrossRef] [PubMed]
- Villumsen, B.H.; Danielsen, J.R.; Povlsen, L.; Sylvestersen, K.B.; Merdes, A.; Beli, P.; Yang, Y.G.; Choudhary, C.; Nielsen, M.L.; Mailand, N.; et al. A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. Embo J. 2013, 32, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, N.; Liu, L.; Liu, X.; Ding, X.; Song, X.; Yang, S.; Shan, L.; Zhou, X. Usp9x regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun. 2017, 8, 14866. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, Y.; Xu, Y.; Xu, S.; Jiang, Y.; Dong, Q.; Zhou, Y.; Ge, W. The X-Linked deubiquitinase Usp9x is an integral component of centrosome. J. Biol. Chem. 2017, 292, 12874–12884. [Google Scholar] [CrossRef]
- Han, K.J.; Wu, Z.; Pearson, C.G.; Peng, J.; Song, K.; Liu, C.W. Deubiquitylase Usp9x maintains centriolar satellite integrity by stabilizing pericentriolar material 1 protein. J. Cell Sci. 2019, 132, jcs221663. [Google Scholar] [CrossRef]
- Florian, V.; Schluter, T.; Bohnensack, R. A new member of the sorting nexin family interacts with the c-terminus of p-selectin. Biochem. Biophys Res. Commun. 2001, 281, 1045–1050. [Google Scholar] [CrossRef]
- Ghai, R.; Bugarcic, A.; Liu, H.; Norwood, S.J.; Skeldal, S.; Coulson, E.J.; Li, S.S.; Teasdale, R.D.; Collins, B.M. Structural basis for endosomal trafficking of diverse transmembrane cargos by px-ferm proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E643–E652. [Google Scholar] [CrossRef]
- Williams, R.; Schluter, T.; Roberts, M.S.; Knauth, P.; Bohnensack, R.; Cutler, D.F. sorting nexin 17 accelerates internalization yet retards degradation of p-selectin. Mol. Biol. Cell 2004, 15, 3095–3105. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, F.; Heesom, K.J.; Bass, M.D.; Cullen, P.J. Snx17 protects integrins from degradation by sorting between lysosomal and recycling pathways. J. Cell Biol. 2012, 197, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, R.T.; Stremmel, C.; Meves, A.; Meyer, H.; Widmaier, M.; Tseng, H.Y.; Fassler, R. Sorting nexin 17 prevents lysosomal degradation of beta1 integrins by binding to the beta1-integrin tail. Nat. Cell Biol. 2012, 14, 584–592. [Google Scholar] [CrossRef] [PubMed]
- McNally, K.E.; Faulkner, R.; Steinberg, F.; Gallon, M.; Ghai, R.; Pim, D.; Langton, P.; Pearson, N.; Danson, C.M. Retriever is a multiprotein complex for retromer-independent endosomal cargo recycling. Nat. Cell Biol. 2017, 19, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for crispr screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Liu, D.; Liu, N.; Xu, L.; Li, S.; Lin, S.; Pei, D. SNX17 regulates Notch pathway and pancreas development through the retromer-dependent recycling of Jag1. Cell Regen. 2012, 1, 4. [Google Scholar] [CrossRef]
- Das, A.; Qian, J.; Tsang, W.Y. Usp9x counteracts differential ubiquitination of nphp5 by march7 and bbs11 to regulate ciliogenesis. PLoS Genet. 2017, 13, e1006791. [Google Scholar] [CrossRef]
- Gallon, M.; Cullen, P.J. Retromer and sorting nexins in endosomal sorting. Biochem Soc Trans. 2015, 43, 33–47. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Wang, J.; Tseng, I.C.; Huang, T.; Zhao, Y.; Zheng, Q.; Gao, Y.; Luo, H. Snx27 deletion causes hydrocephalus by impairing ependymal cell differentiation and ciliogenesis. J. Neurosci. 2016, 36, 12586–12597. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, B.; Xu, L.; Li, H.; Xia, J.; Yin, W.; Li, Z.; Shi, D.; Li, S.; Lin, S.; et al. A Snx10/V-atpase pathway regulates ciliogenesis in vitro and in vivo. Cell Res. 2012, 22, 333–345. [Google Scholar] [CrossRef]
- Han, K.J.; Foster, D.G.; Zhang, N.Y.; Kanisha, K.; Dzieciatkowska, M.; Sclafani, R.A.; Hansen, K.C.; Peng, J.; Liu, C.W. Ubiquitin-specific protease 9x deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J. Biol. Chem. 2012, 287, 43741–43752. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Dimitriadi, M.; Terzic, B.; Cable, C.; Hart, A.C.; Chitnis, A.; Fischbeck, K.H.; Burnett, B.G. The E3 ubiquitin ligase mind bomb 1 ubiquitinates and promotes the degradation of survival of motor neuron protein. Mol. Biol. Cell 2013, 24, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Cajanek, L.; Glatter, T.; Nigg, E.A. The E3 ubiquitin ligase mib1 regulates plk4 and centriole biogenesis. J. Cell Sci. 2015, 128, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, R.; Hussain, M.; Attri, P.; Kaur, E.; Tripathi, V.; Priya, S.; Dhapola, P.; Saha, D.; Madhavan, V.; Chowdhury, S.; et al. Blm potentiates c-jun degradation and alters its function as an oncogenic transcription factor. Cell Rep. 2018, 24, 947–961. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Berman, M.; Kong, Y.Y.; Dorf, M.E. Mapping a dynamic innate immunity protein interaction network regulating type i interferon production. Immunity 2011, 35, 426–440. [Google Scholar] [CrossRef]
- Douanne, T.; Andre-Gregoire, G.; Thys, A.; Trillet, K.; Gavard, J.; Bidere, N. Cyld regulates centriolar satellites proteostasis by counteracting the e3 ligase Mib1. Cell Rep. 2019, 27, 1657–1665. [Google Scholar] [CrossRef]
- Vong, Q.P.; Cao, K.; Li, H.Y.; Iglesias, P.A.; Zheng, Y. Chromosome alignment and segregation regulated by ubiquitination of survivin. Science 2005, 310, 1499–1504. [Google Scholar] [CrossRef]
- Skowyra, A.; Allan, L.A.; Saurin, A.T.; Clarke, P.R. Usp9x limits mitotic checkpoint complex turnover to strengthen the spindle assembly checkpoint and guard against chromosomal instability. Cell Rep. 2018, 23, 852–865. [Google Scholar] [CrossRef]
- Engel, K.; Rudelius, M.; Slawska, J.; Jacobs, L.; Ahangarian Abhari, B.; Altmann, B.; Kurutz, J.; Rathakrishnan, A.; Fernandez-Saiz, V.; Brunner, A. Usp9x stabilizes xiap to regulate mitotic cell death and chemoresistance in aggressive b-cell lymphoma. Embo Mol. Med. 2016, 8, 851–862. [Google Scholar] [CrossRef]
- Homan, C.C.; Kumar, R.; Nguyen, L.S.; Haan, E.; Raymond, F.L.; Abidi, F.; Raynaud, M.; Schwartz, C.E.; Wood, S.A.; Gecz, J.; et al. Mutations in Usp9x are associated with x-linked intellectual disability and disrupt neuronal cell migration and growth. Am. J. Hum. Genet. 2014, 94, 470–478. [Google Scholar] [CrossRef]
- Reijnders, M.R.; Zachariadis, V.; Latour, B.; Jolly, L.; Mancini, G.M.; Pfundt, R.; Wu, K.M.; van Ravenswaaij-Arts, C.M.; Veenstra-Knol, H.E.; Anderlid, B.M.; et al. De novo loss-of-function mutations in usp9x cause a female-specific recognizable syndrome with developmental delay and congenital malformations. Am. J. Hum. Genet. 2016, 98, 373–381. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: DNA constructs, siRNAs and cell lines used in this study are available from the authors. All small chemicals and antibodies used in this study are commercially available. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Xia, J.; Zhang, L.; Zhao, S.; Li, S.; Wang, H.; Cheng, S.; Li, H.; Yin, W.; Pei, D.; et al. SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 during Serum-Starvation-Induced Ciliogenesis. Cells 2019, 8, 1335. https://doi.org/10.3390/cells8111335
Wang P, Xia J, Zhang L, Zhao S, Li S, Wang H, Cheng S, Li H, Yin W, Pei D, et al. SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 during Serum-Starvation-Induced Ciliogenesis. Cells. 2019; 8(11):1335. https://doi.org/10.3390/cells8111335
Chicago/Turabian StyleWang, Pengtao, Jianhong Xia, Leilei Zhang, Shaoyang Zhao, Shengbiao Li, Haiyun Wang, Shan Cheng, Heying Li, Wenguang Yin, Duanqing Pei, and et al. 2019. "SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 during Serum-Starvation-Induced Ciliogenesis" Cells 8, no. 11: 1335. https://doi.org/10.3390/cells8111335
APA StyleWang, P., Xia, J., Zhang, L., Zhao, S., Li, S., Wang, H., Cheng, S., Li, H., Yin, W., Pei, D., & Shu, X. (2019). SNX17 Recruits USP9X to Antagonize MIB1-Mediated Ubiquitination and Degradation of PCM1 during Serum-Starvation-Induced Ciliogenesis. Cells, 8(11), 1335. https://doi.org/10.3390/cells8111335