Over-Activated Proteasome Mediates Neuroinflammation on Acute Intracerebral Hemorrhage in Rats

 , , and
, , and 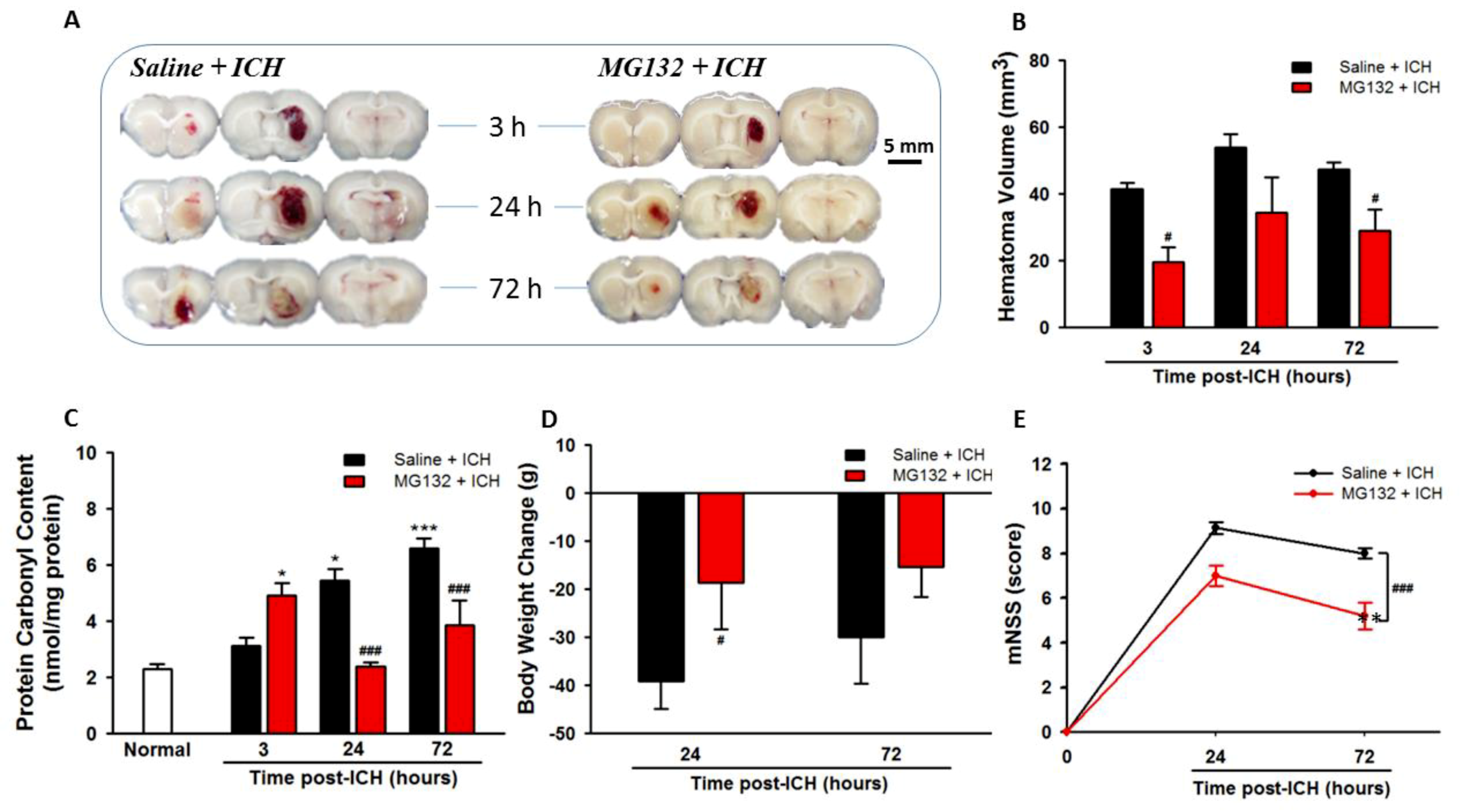{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Methods
2.1.1. Animals
2.1.2. Intracerebral Hemorrhage Induction Model
2.1.3. The Proteasome Inhibitors Administration
2.2. Morphometric Measurements
2.3. Neurobehavioral Assays
2.4. Protein Carbonyl Assay
2.5. Ubiquitin Competitive Enzyme-Linked Immunosorbent Assay
2.6. Cytokines Assay
2.7. Proteasome Activity Assay
2.8. Protein Aggregation Assay
2.9. Immunoprecipitation and Immunoblotting
2.10. Reverse Transcription Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
2.11. Immunohistochemical Staining
2.12. TUNEL Assay
2.13. Transmission Electron Microscopy
2.14. Statistical Analysis
3. Results
3.1. ICH Increases Oxidative Stress, Hematoma Expansion, Body Weight Loss, and Neurological Deficits
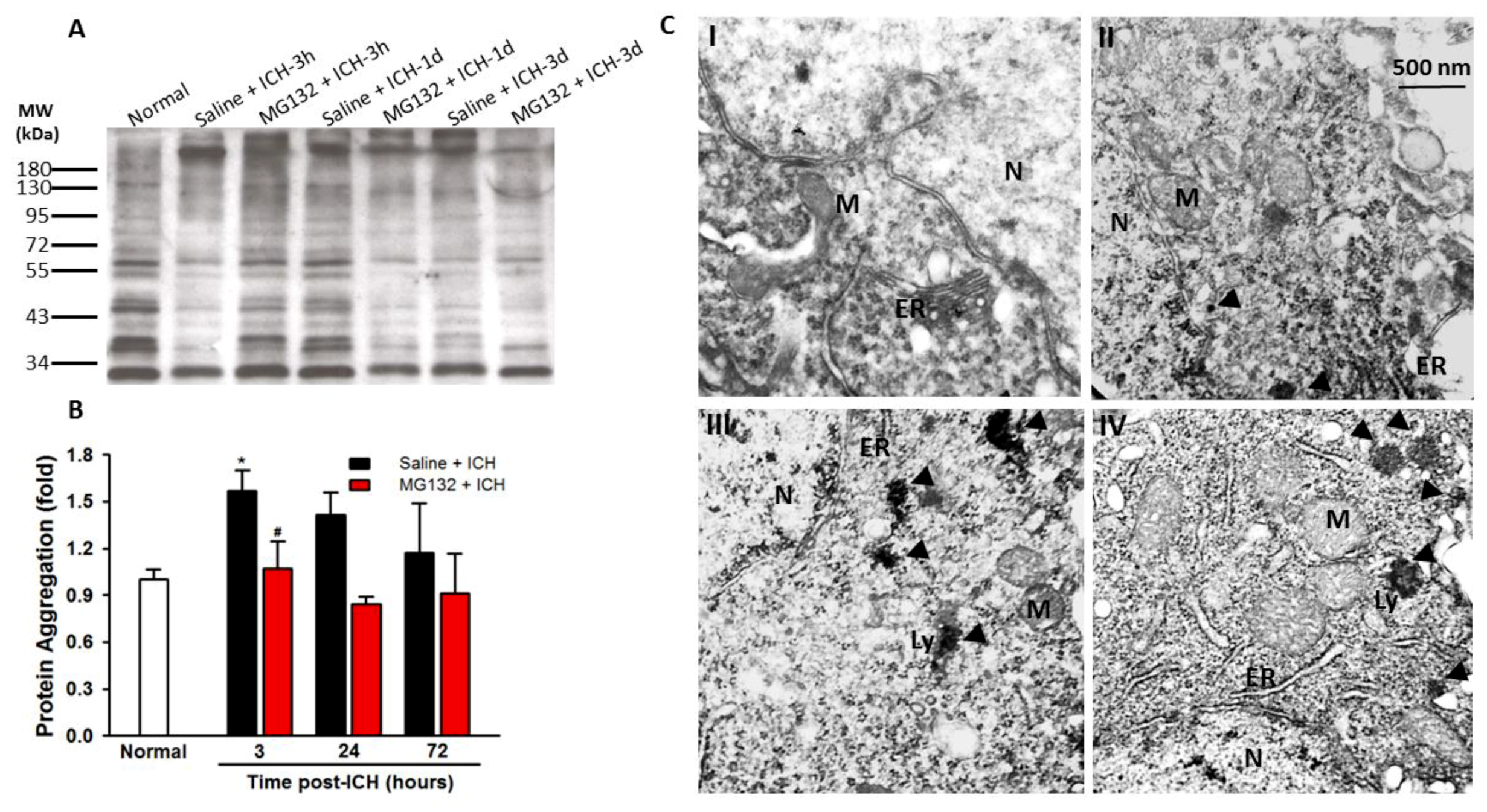
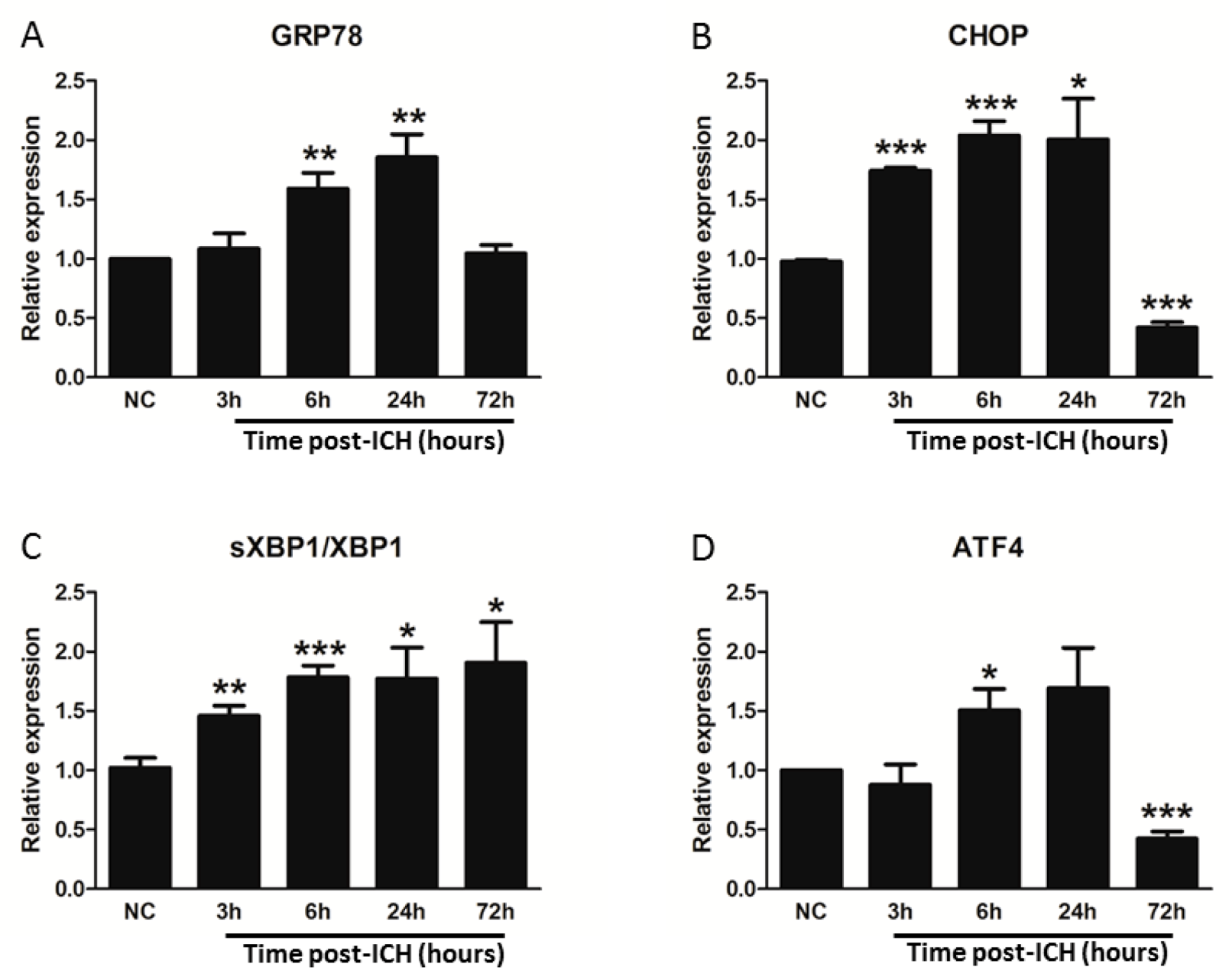
3.2. ICH Induces ER Stress and Proteostasis Disruption
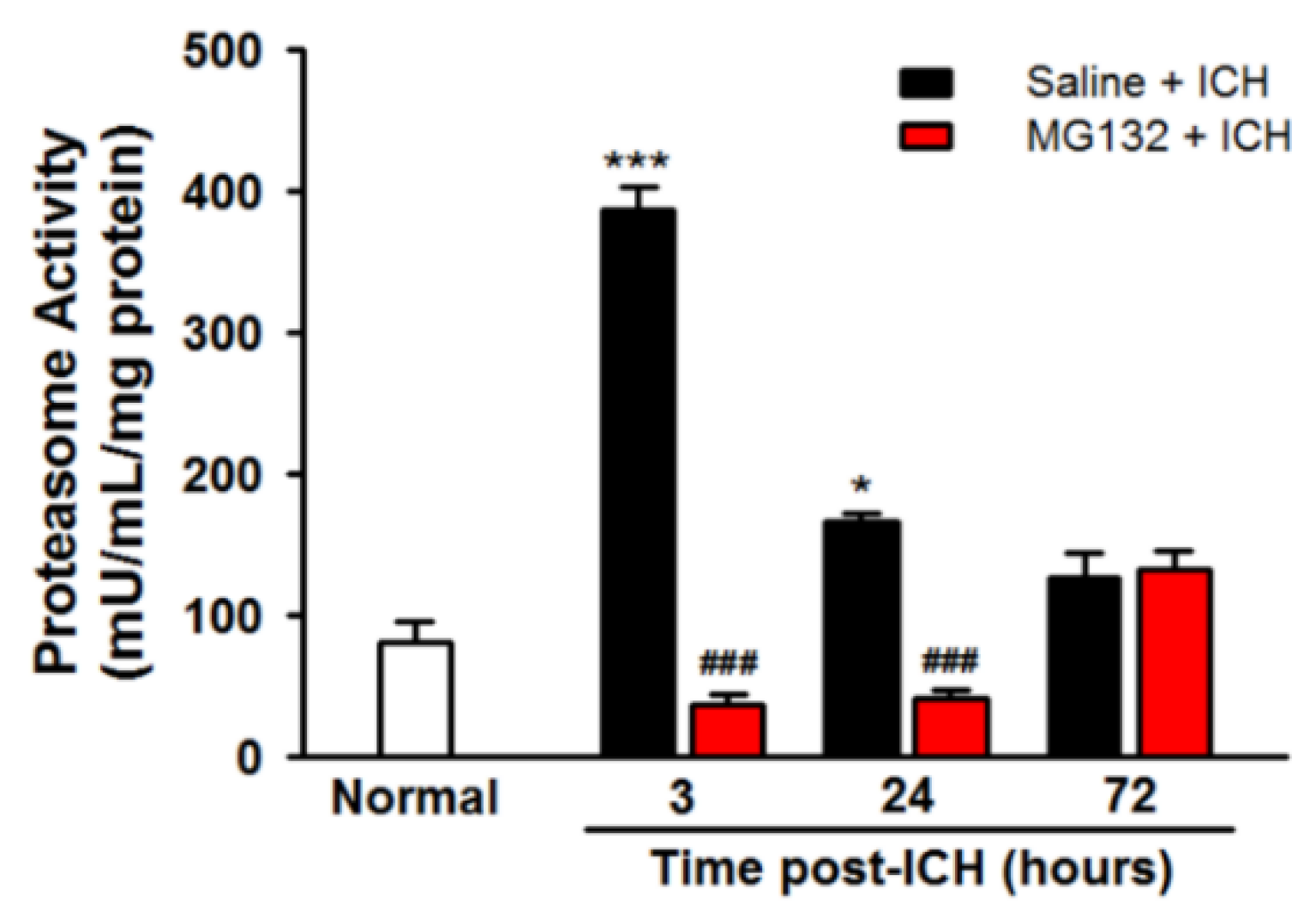
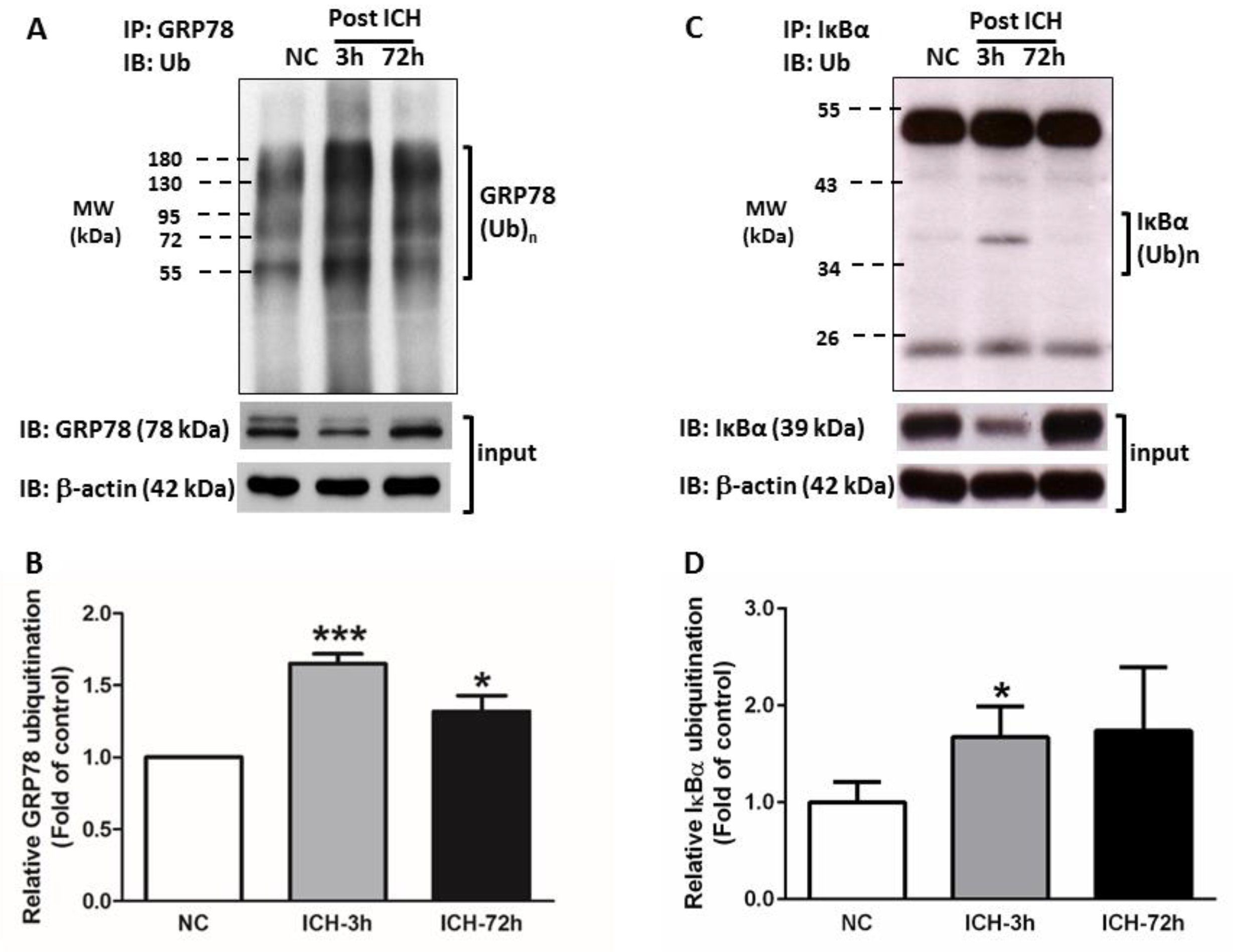
3.3. ICH Induces Proteasome Over-Activation
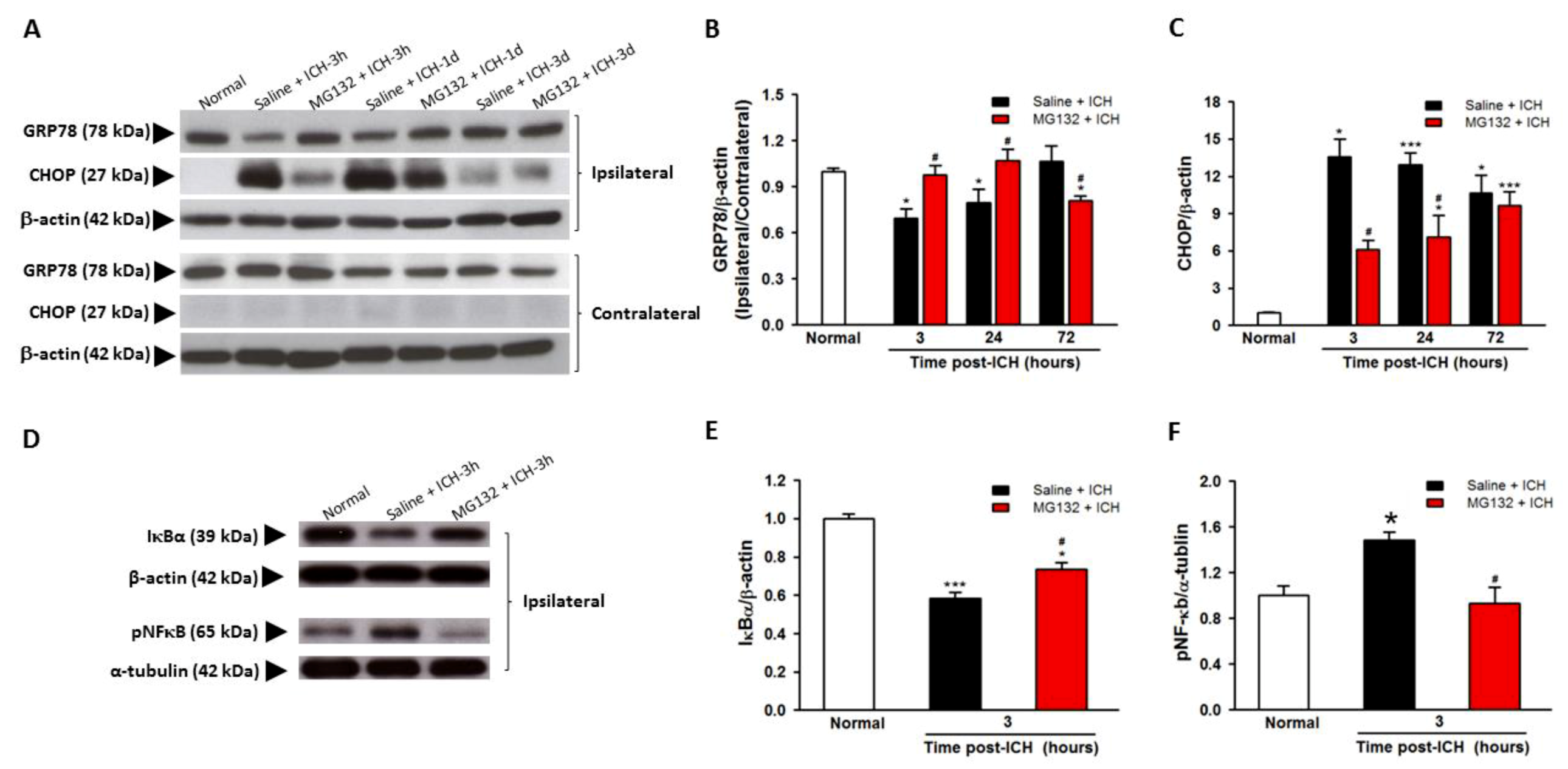
3.4. GRP78 Protein Degradation Occurs at the Early Phase Of ICH and Coincides with the Activation Of Pro-Apoptotic Transcriptional Factor CHOP
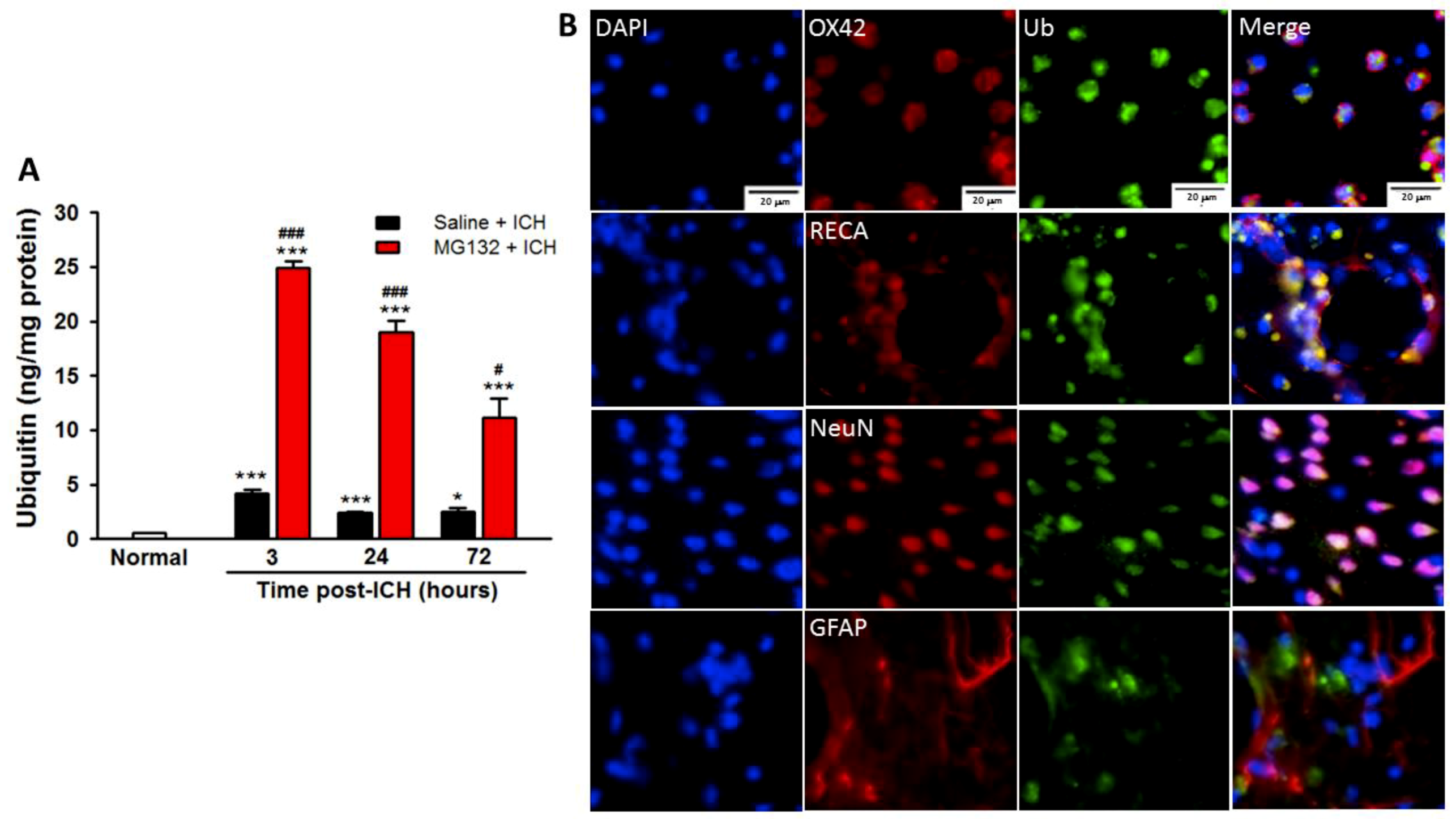
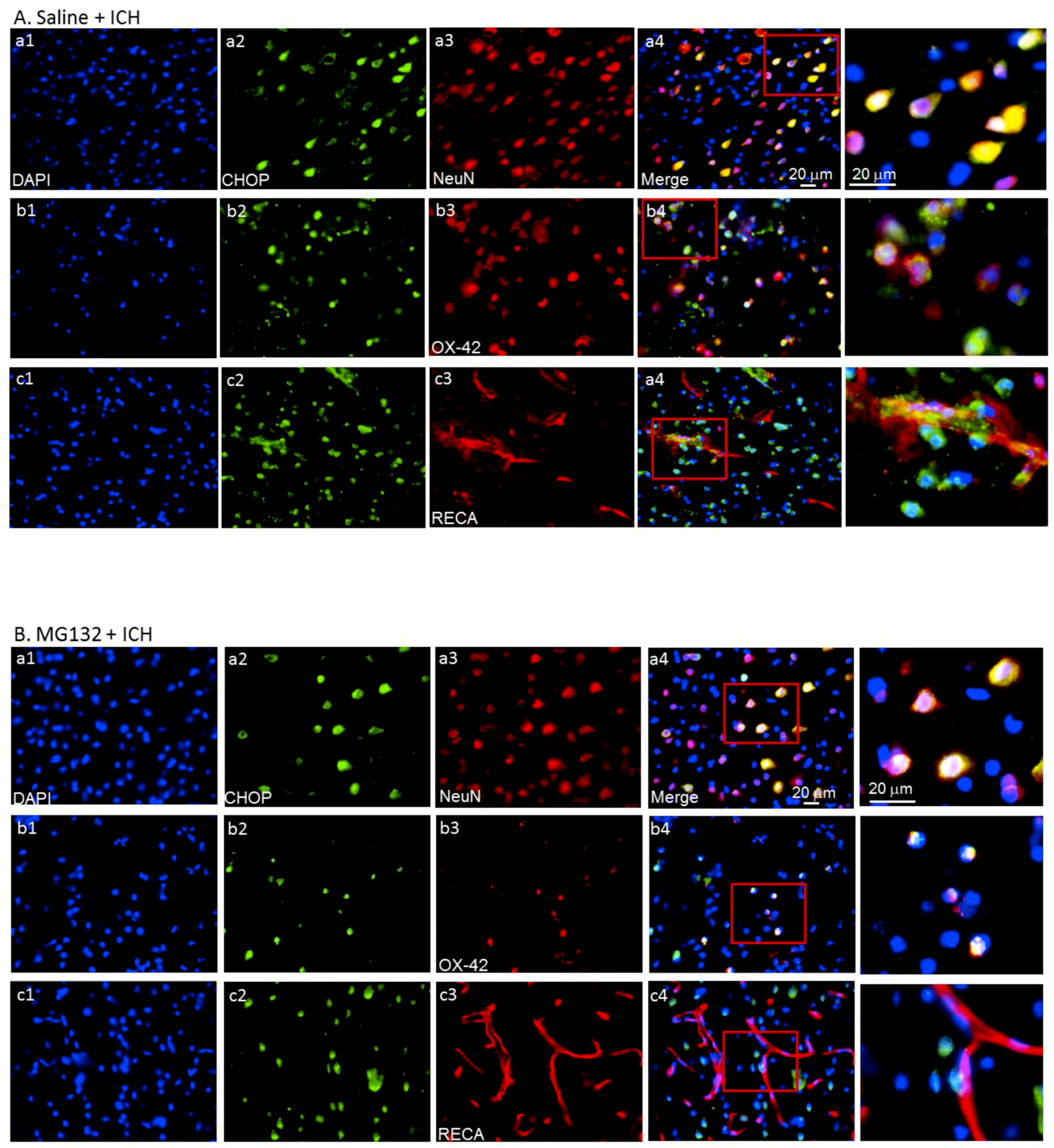
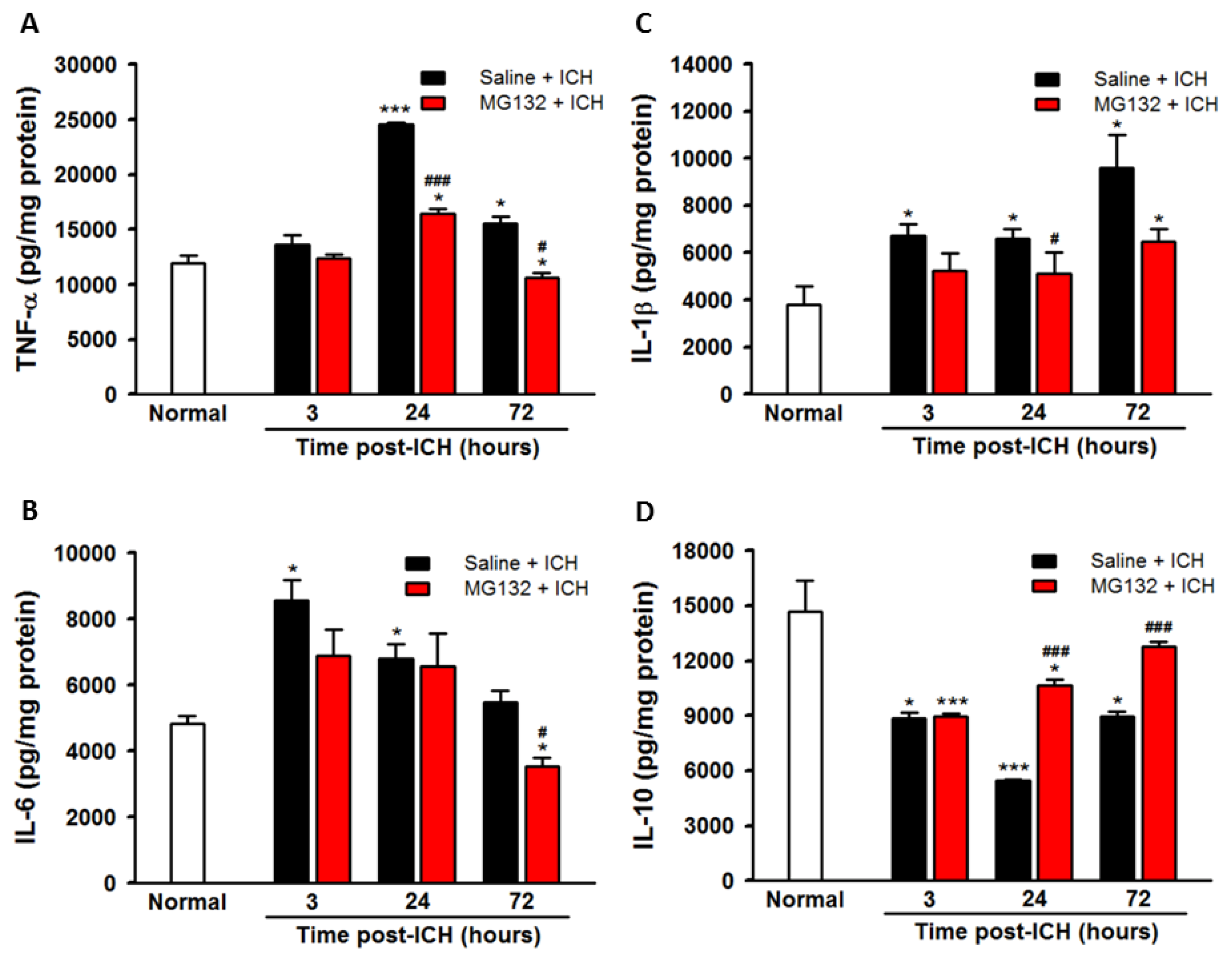
3.5. ICH-Induced Proteasome Over-Activation Exacerbates Neuroinflammation
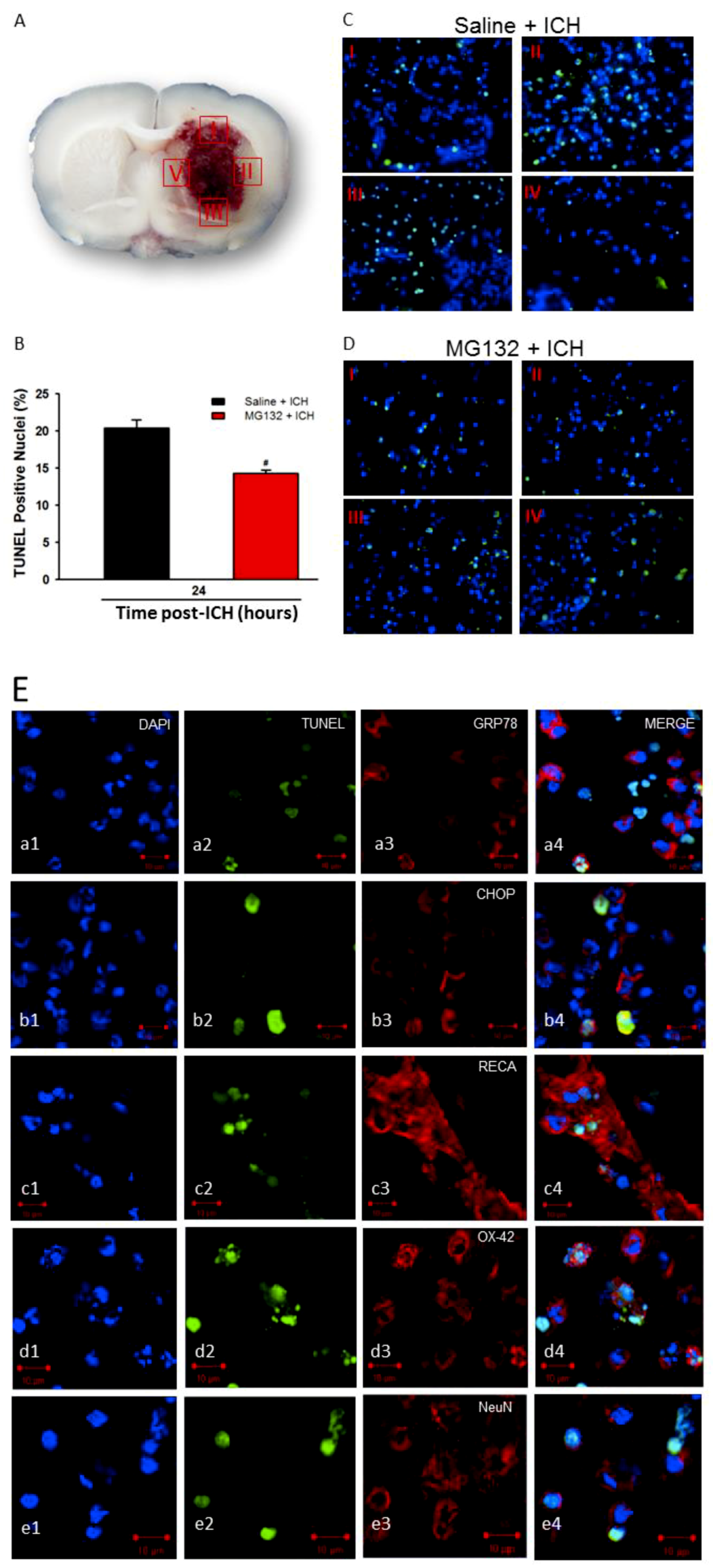
3.6. Apoptosis and TUNEL
3.7. Proteasomal Inhibition Exerts Neuroprotective Effects in Alleviated Hematoma Expansion, Oxidative Stress, Body Weight Loss, and Neurological Deficits
3.8. Proteasomal Inhibition Prevented ICH-Induced Proteasome Over-Activation, Exerts Anti-Neuroinflammation and Anti-Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11, 720–731. [Google Scholar] [CrossRef]
- Duan, X.; Wen, Z.; Shen, H.; Shen, M.; Chen, G. Intracerebral Hemorrhage, Oxidative Stress, and Antioxidant Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 1203285. [Google Scholar] [CrossRef] [PubMed]
- Hoff, J.T.; Xi, G. Brain edema from intracerebral hemorrhage. Acta Neurochir. Suppl. 2003, 86, 11–15. [Google Scholar] [PubMed]
- Hu, X.; Tao, C.; Gan, Q.; Zheng, J.; Li, H.; You, C. Oxidative Stress in Intracerebral Hemorrhage: Sources, Mechanisms, and Therapeutic Targets. Oxid. Med. Cell. Longev. 2016, 2016, 3215391. [Google Scholar] [CrossRef] [PubMed]
- Kazui, S.; Naritomi, H.; Yamamoto, H.; Sawada, T.; Yamaguchi, T. Enlargement of spontaneous intracerebral hemorrhage. Incidence and time course. Stroke 1996, 27, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.M.; Wu, W.G. Mechanism of hemolysis of red blood cell mediated by ethanol. Biochim. et Biophys. Acta 1991, 1062, 46–50. [Google Scholar] [CrossRef]
- Wang, J.; Doré, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 894–908. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J. Neurosur. 1998, 89, 991–996. [Google Scholar] [CrossRef]
- Huang, F.P.; Xi, G.; Keep, R.F.; Hua, Y.; Nemoianu, A.; Hoff, J.T. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J. Neurosur. 2002, 96, 287–293. [Google Scholar] [CrossRef]
- Thompson, K.J.; Shoham, S.; Connor, J.R. Iron and neurodegenerative disorders. Brain Res. Bull. 2001, 55, 155–164. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- Siesjo, B.K.; Agardh, C.D.; Bengtsson, F. Free radicals and brain damage. Cerebrovasc. Brain Metab. Rev. 1989, 1, 165–211. [Google Scholar] [PubMed]
- Wu, J.; Hua, Y.; Keep, R.F.; Schallert, T.; Hoff, J.T.; Xi, G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2002, 953, 45–52. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Pathophysiology of brain edema formation. Neurosur. Clin. N. Am. 2002, 13, 371–383. [Google Scholar] [CrossRef]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef]
- Niu, M.; Dai, X.; Zou, W.; Yu, X.; Teng, W.; Chen, Q.; Sun, X.; Yu, W.; Ma, H.; Liu, P. Autophagy, Endoplasmic Reticulum Stress and the Unfolded Protein Response in Intracerebral Hemorrhage. Transl. Neurosci. 2017, 8, 37–48. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel) 2013, 4, 306–333. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, K.; Kaufman, R.J. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, C.; Di Napoli, M. Ubiquitin-proteasome system and proteasome inhibition: New strategies in stroke therapy. Stroke 2004, 35, 1506–1518. [Google Scholar] [CrossRef]
- Liew, H.K.; Pang, C.Y.; Hsu, C.W.; Wang, M.J.; Li, T.Y.; Peng, H.F.; Kuo, J.S.; Wang, J.Y. Systemic administration of urocortin after intracerebral hemorrhage reduces neurological deficits and neuroinflammation in rats. J. Neuroinflamm. 2012, 9, 13. [Google Scholar] [CrossRef]
- MacLellan, C.L.; Silasi, G.; Poon, C.C.; Edmundson, C.L.; Buist, R.; Peeling, J.; Colbourne, F. Intracerebral hemorrhage models in rat: Comparing collagenase to blood infusion. J. Cereb. Blood Flow Metab. 2008, 28, 516–525. [Google Scholar] [CrossRef]
- Chen, S.F.; Hsu, C.W.; Huang, W.H.; Wang, J.Y. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 2008, 155, 1279–1296. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Goder, V. Roles of ubiquitin in endoplasmic reticulum-associated protein degradation (ERAD). Curr. Protein Pept. Sci. 2012, 13, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yao, J.; Wang, G.; Wang, Y.; Wang, B.; Ge, P. Ischemic postconditioning protects neuronal death caused by cerebral ischemia and reperfusion via attenuating protein aggregation. Int. J. Med. Sci. 2012, 9, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Luo, Y.; Liu, C.L.; Hu, B. Protein aggregation and proteasome dysfunction after brain ischemia. Stroke 2007, 38, 3230–3236. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Tuhrim, S.; Broderick, J.P.; Batjer, H.H.; Hondo, H.; Hanley, D.F. Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 2001, 344, 1450–1460. [Google Scholar] [CrossRef]
- Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 2010, 92, 463–477. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Mayer, R.J. From neurodegeneration to neurohomeostasis: The role of ubiquitin. Drug News Perspect. 2003, 16, 103–108. [Google Scholar] [CrossRef]
- Fietta, P.; Delsante, G. Proteasomes and immunoproteasomes. Riv. Biol. 2010, 103, 29–50. [Google Scholar] [PubMed]
- Fenstermacher, J.D.; Knight, R.A.; Ewing, J.R.; Nagaraja, T.; Nagesh, V.; Yee, J.S.; Arniego, P.A. Estimating blood-brain barrier opening in a rat model of hemorrhagic transformation with Patlak plots of Gd-DTPA contrast-enhanced MRI. Acta Neurochir. Suppl. 2003, 86, 35–37. [Google Scholar] [PubMed]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Modi, J.P.; Wu, J.Y. Mechanisms of Neuronal Protection against Excitotoxicity, Endoplasmic Reticulum Stress, and Mitochondrial Dysfunction in Stroke and Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [PubMed]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Schroder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Sokka, A.L.; Putkonen, N.; Mudo, G.; Pryazhnikov, E.; Reijonen, S.; Khiroug, L.; Belluardo, N.; Lindholm, D.; Korhonen, L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J. Neurosci. 2007, 27, 901–908. [Google Scholar] [CrossRef]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Tanahashi, N.; Qiu, J.H.; Saito, N.; Chi, S.; Kawahara, N.; Tanaka, K.; Kirino, T. Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Canu, N.; Atlante, A.; Petragallo, V.; Calissano, P.; Marra, E. Proteasome inhibitors prevent cytochrome c release during apoptosis but not in excitotoxic death of cerebellar granule neurons. FEBS Lett. 2002, 515, 8–12. [Google Scholar] [CrossRef]
- Keller, J.N.; Markesbery, W.R. Proteasome inhibition results in increased poly-ADP-ribosylation: Implications for neuron death. J. Neurosci. Res. 2000, 61, 436–442. [Google Scholar] [CrossRef]
- Qiu, J.H.; Asai, A.; Chi, S.; Saito, N.; Hamada, H.; Kirino, T. Proteasome inhibitors induce cytochrome c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J. Neurosci. 2000, 20, 259–265. [Google Scholar] [CrossRef]
- Taglialatela, G.; Kaufmann, J.A.; Trevino, A.; Perez-Polo, J.R. Central nervous system DNA fragmentation induced by the inhibition of nuclear factor kappa B. Neuroreport 1998, 9, 489–493. [Google Scholar] [CrossRef]
- Berti, R.; Williams, A.J.; Velarde, L.C.; Moffett, J.R.; Elliott, P.J.; Adams, J.; Yao, C.; Dave, J.R.; Tortella, F.C. Effect of the proteasome inhibitor MLN519 on the expression of inflammatory molecules following middle cerebral artery occlusion and reperfusion in the rat. Neurotox. Res. 2003, 5, 505–514. [Google Scholar] [CrossRef]
- Williams, A.J.; Hale, S.L.; Moffett, J.R.; Dave, J.R.; Elliott, P.J.; Adams, J.; Tortella, F.C. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via antiinflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J. Cereb. Blood Flow Metab. 2003, 23, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.G.; Zhang, R.L.; Lu, M.; Adams, J.; Elliott, P.J.; Chopp, M. Postischemic (6-Hour) treatment with recombinant human tissue plasminogen activator and proteasome inhibitor PS-519 reduces infarction in a rat model of embolic focal cerebral ischemia. Stroke 2001, 32, 2926–2931. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.; Wang, Y.; Lei, H.; Su, H.; Zeng, J.; Pei, Z.; Huang, R. Inhibition of immunoproteasome reduces infarction volume and attenuates inflammatory reaction in a rat model of ischemic stroke. Cell Death Dis. 2015, 6, e1626. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.K.; Andriola, I.F.; Kampinga, H.H.; Merry, D.E. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2002, 11, 515–523. [Google Scholar] [CrossRef]
- Buchan, A.M.; Li, H.; Blackburn, B. Neuroprotection achieved with a novel proteasome inhibitor which blocks NF-kappaB activation. Neuroreport 2000, 11, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.J.; Zollner, T.M.; Boehncke, W.H. Proteasome inhibition: A new anti-inflammatory strategy. J. Mol. Med. (Berl.) 2003, 81, 235–245. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liew, H.-K.; Hu, W.-F.; Lin, P.B.-C.; Wang, P.-K.; Tsai, A.P.-Y.; Pang, C.-Y.; Chen, T.-Y. Over-Activated Proteasome Mediates Neuroinflammation on Acute Intracerebral Hemorrhage in Rats. Cells 2019, 8, 1326. https://doi.org/10.3390/cells8111326
Liew H-K, Hu W-F, Lin PB-C, Wang P-K, Tsai AP-Y, Pang C-Y, Chen T-Y. Over-Activated Proteasome Mediates Neuroinflammation on Acute Intracerebral Hemorrhage in Rats. Cells. 2019; 8(11):1326. https://doi.org/10.3390/cells8111326
Chicago/Turabian StyleLiew, Hock-Kean, Wei-Fen Hu, Peter Bor-Chian Lin, Po-Kai Wang, Andy Po-Yi Tsai, Cheng-Yoong Pang, and Tsung-Ying Chen. 2019. "Over-Activated Proteasome Mediates Neuroinflammation on Acute Intracerebral Hemorrhage in Rats" Cells 8, no. 11: 1326. https://doi.org/10.3390/cells8111326
APA StyleLiew, H.-K., Hu, W.-F., Lin, P. B.-C., Wang, P.-K., Tsai, A. P.-Y., Pang, C.-Y., & Chen, T.-Y. (2019). Over-Activated Proteasome Mediates Neuroinflammation on Acute Intracerebral Hemorrhage in Rats. Cells, 8(11), 1326. https://doi.org/10.3390/cells8111326