Roles of the Hepatic Endocannabinoid and Apelin Systems in the Pathogenesis of Liver Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of the Endogenous Cannabinoid System
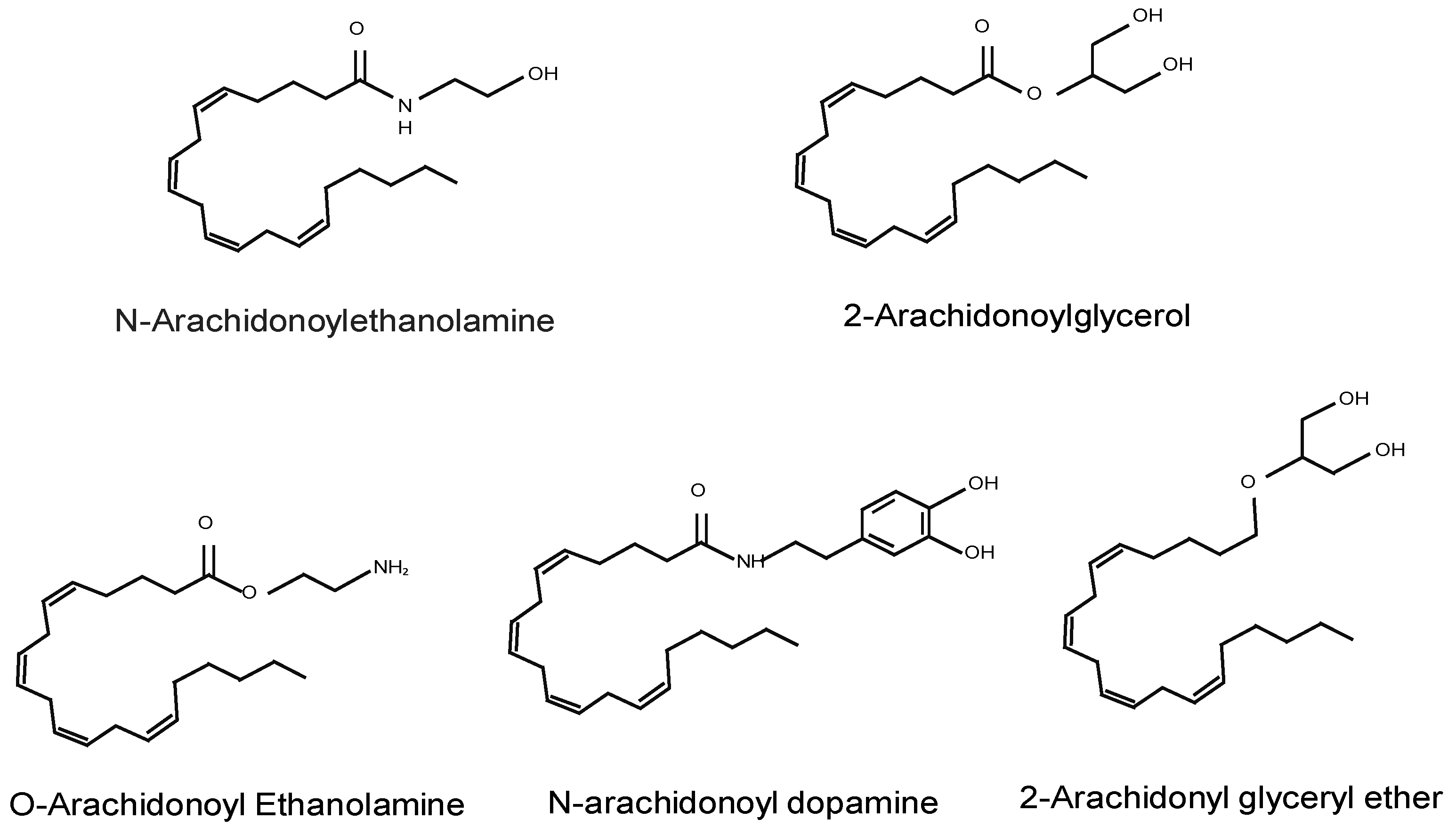
2.1. Endocannabinoids
2.2. EC Canonical Receptors
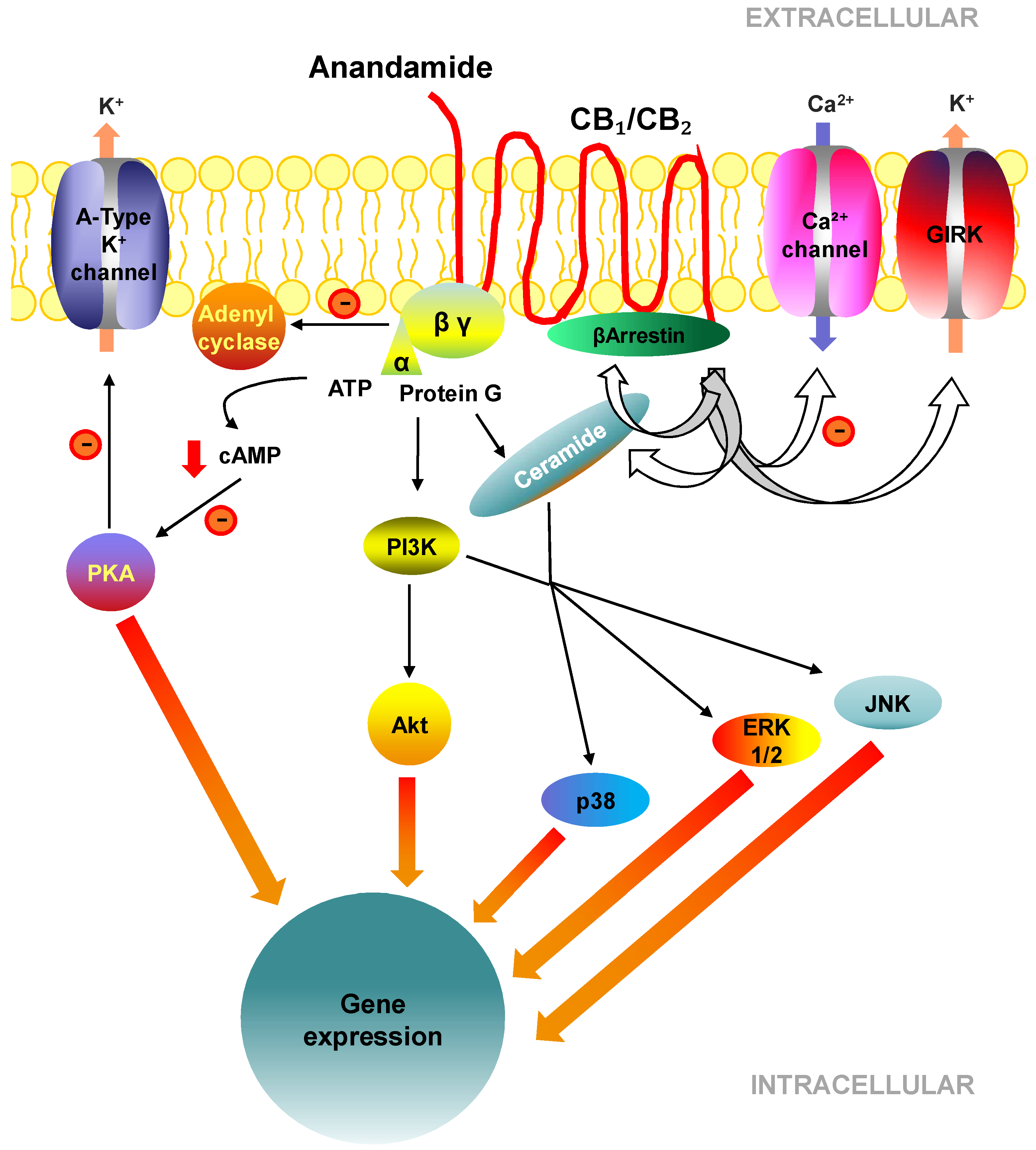
2.3. Cell Signalling Pathways Activated by CB1 and CB2
3. The Endocannabinoid System (ECS) in Health and Disease
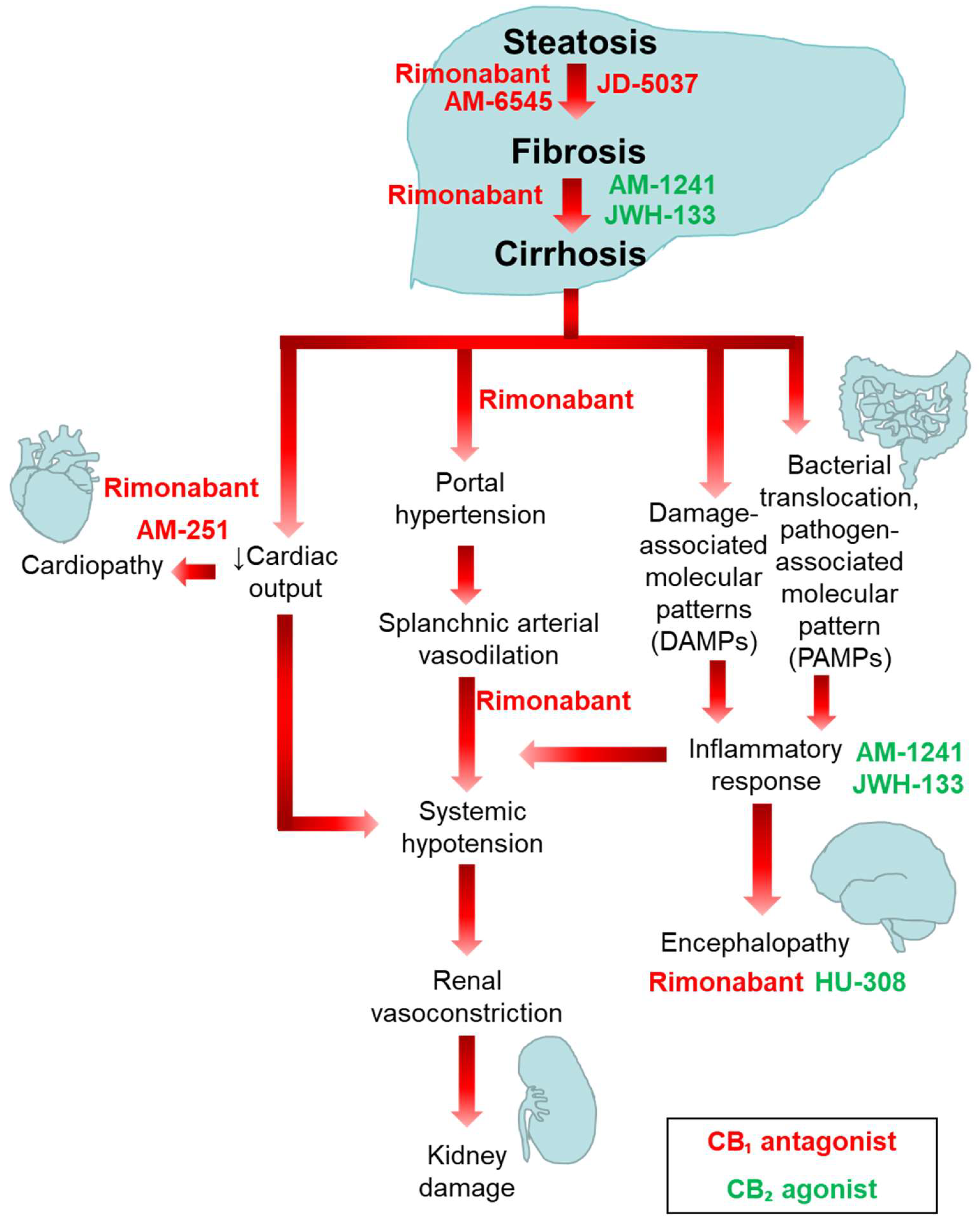
4. Involvement of the ECS in the Pathogenesis of Liver Fibrosis
5. Overview of the Apelin System
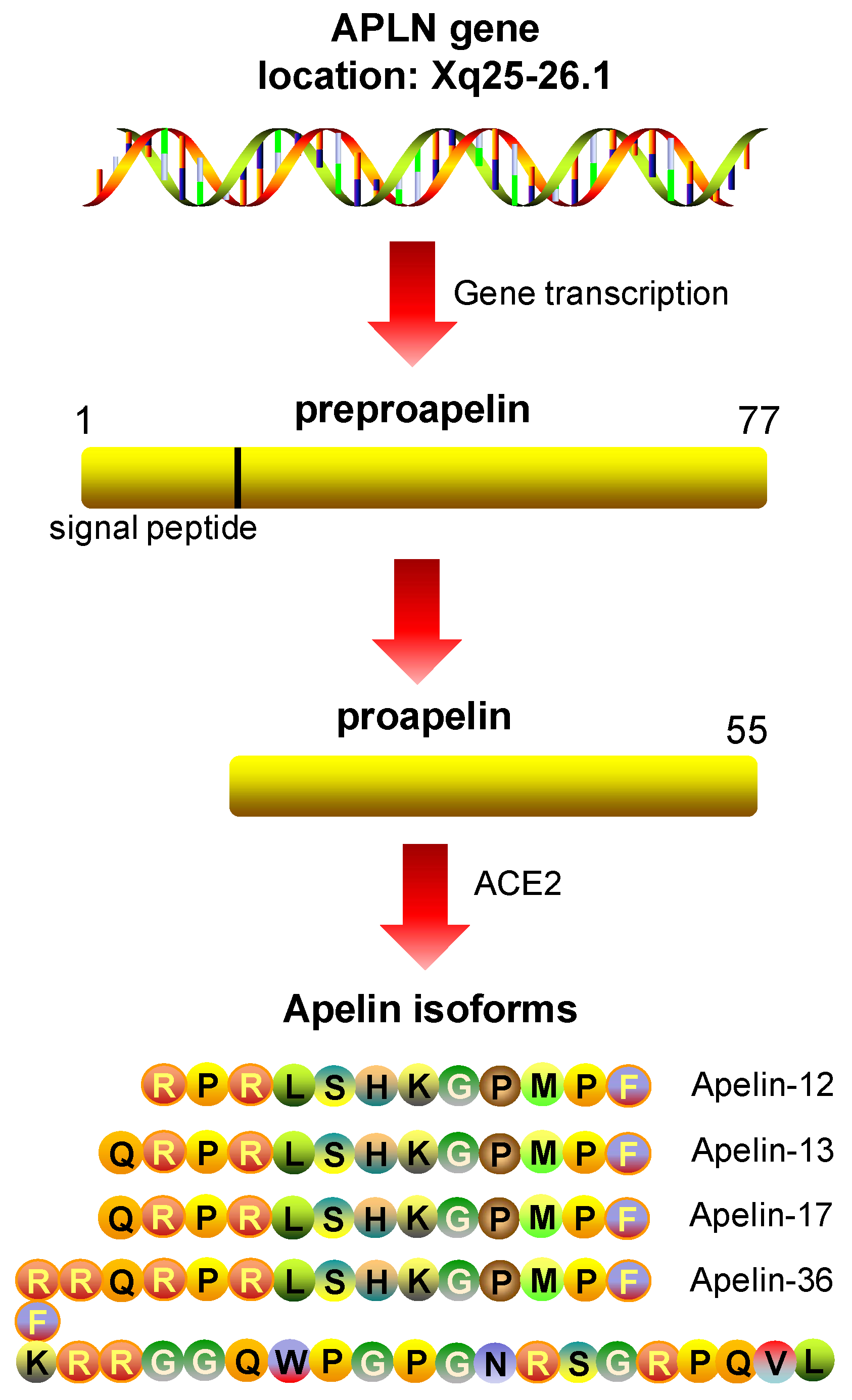
5.1. Apelin: Peptide Isoforms
5.2. APJ, the Apelin Receptor
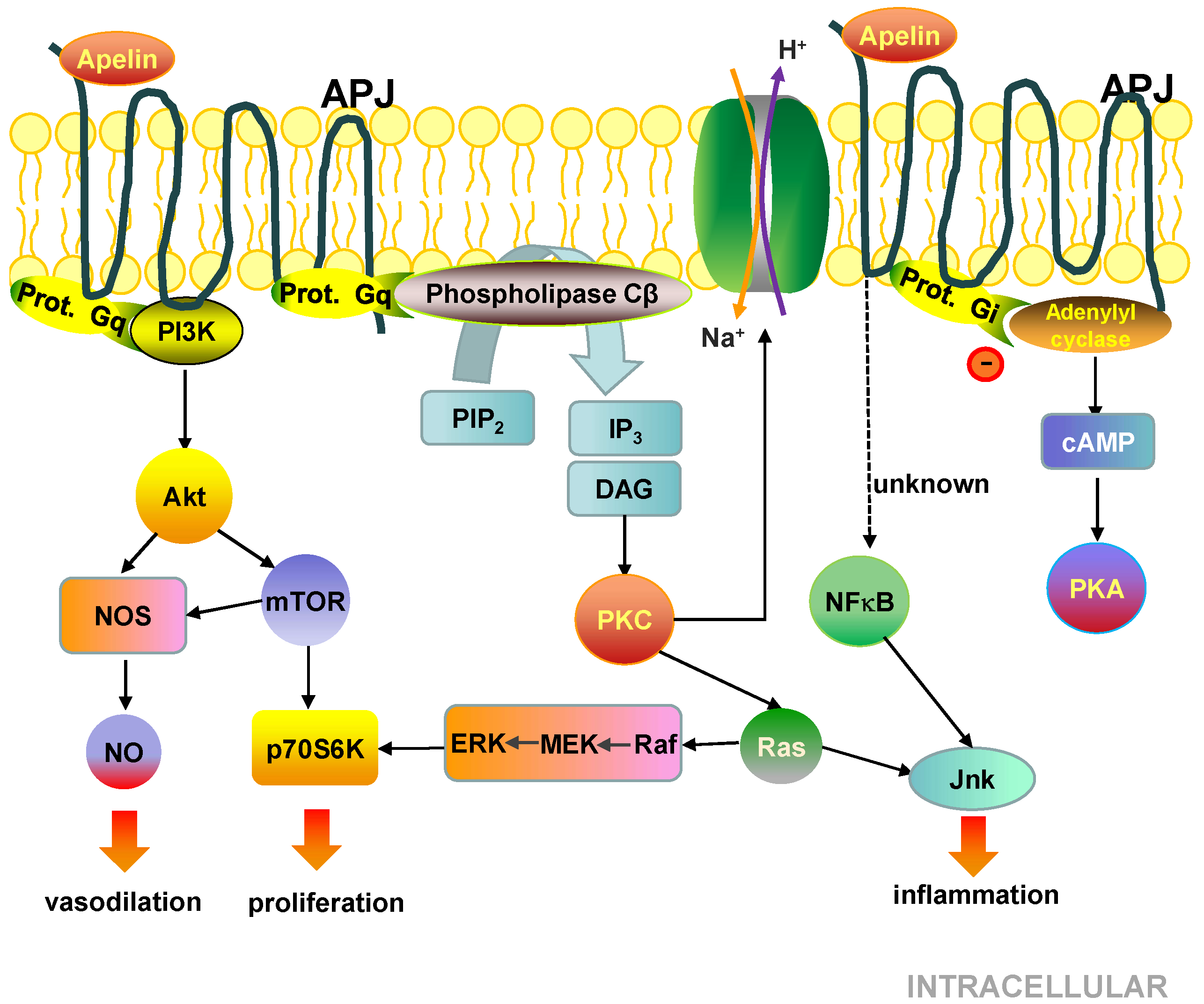
5.3. Cell Signalling Pathways Activated by the APJ Receptor
6. The Apelin System in Health and Disease
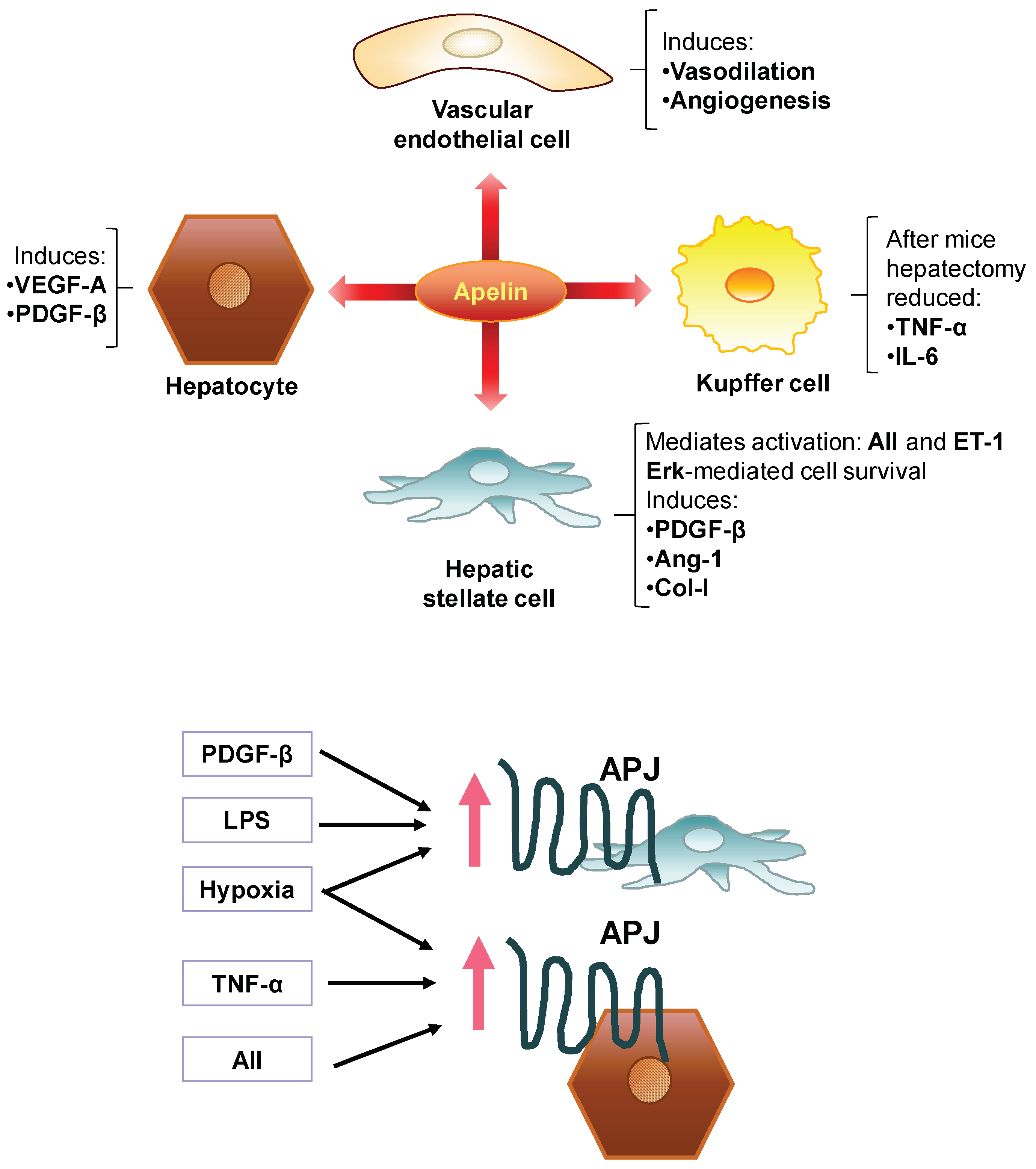
7. Involvement of the Apelin System in the Pathogenesis of Liver Fibrosis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Levene, A.P.; Goldin, R.D. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 2012, 61, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef]
- Rout, G.; Nayak, B.; Patel, A.H.; Gunjan, D.; Singh, V.; Kedia, S.; Shalimar. Therapy with oral directly acting agents in hepatitis C infection is associated with reduction in fibrosis and increase in hepatic steatosis on transient elastography. J. Clin. Exp. Hepatol. 2019, 9, 207–214. [Google Scholar] [CrossRef]
- Grgurevic, I.; Bozin, T.; Madir, A. Hepatitis C is now curable, but what happens with cirrhosis and portal hypertension afterwards? Clin. Exp. Hepatol. 2017, 3, 181–186. [Google Scholar] [CrossRef]
- Arthur, M.J. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology 2002, 122, 1525–1528. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Luquero, A.; Parra-Robert, M.; Mora, A.; Ribera, J.; Edelman, E.R.; Jimenez, W. Graphene-dendrimer nanostars for targeted macrophage overexpression of metalloproteinase 9 and hepatic fibrosis precision therapy. Nano Lett. 2018, 18, 5839–5845. [Google Scholar] [CrossRef]
- Oro, D.; Yudina, T.; Fernandez-Varo, G.; Casals, E.; Reichenbach, V.; Casals, G.; Gonzalez de la Presa, B.; Sandalinas, S.; Carvajal, S.; Puntes, V.; et al. Cerium oxide nanoparticles reduce steatosis, portal hypertension and display anti-inflammatory properties in rats with liver fibrosis. J. Hepatol. 2016, 64, 691–698. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Fernandez-Varo, G.; Munoz-Luque, J.; Ros, J.; Arroyo, V.; Rodes, J.; Friedman, S.L.; Carmeliet, P.; Jimenez, W.; Morales-Ruiz, M. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology 2007, 46, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Bru, P.; Bataller, R.; Fernandez-Varo, G.; Moreno, M.; Ramalho, L.N.; Colmenero, J.; Mari, M.; Claria, J.; Jimenez, W.; Arroyo, V.; et al. Bradykinin attenuates hepatocellular damage and fibrosis in rats with chronic liver injury. Gastroenterology 2007, 133, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Titos, E.; Claria, J.; Planaguma, A.; Lopez-Parra, M.; Villamor, N.; Parrizas, M.; Carrio, A.; Miquel, R.; Jimenez, W.; Arroyo, V.; et al. Inhibition of 5-lipoxygenase induces cell growth arrest and apoptosis in rat Kupffer cells: Implications for liver fibrosis. FASEB J. 2003, 17, 1745–1747. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, V.; Fernandez-Varo, G.; Casals, G.; Oro, D.; Ros, J.; Melgar-Lesmes, P.; Weiskirchen, R.; Morales-Ruiz, M.; Jimenez, W. Adenoviral dominant-negative soluble PDGFRbeta improves hepatic collagen, systemic hemodynamics, and portal pressure in fibrotic rats. J. Hepatol. 2012, 57, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.K.; Sarker, K.P.; Abeyama, K.; Kawahara, K.; Iino, S.; Otsubo, Y.; Saigo, K.; Izumi, H.; Hashiguchi, T.; Yamakuchi, M.; et al. Membrane cholesterol but not putative receptors mediates anandamide-induced hepatocyte apoptosis. Hepatology 2003, 38, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rodriguez, C.M.; Romero, J.; Petros, T.J.; Bradshaw, H.; Gasalla, J.M.; Gutierrez, M.L.; Lledo, J.L.; Santander, C.; Fernandez, T.P.; Tomas, E.; et al. Circulating endogenous cannabinoid anandamide and portal, systemic and renal hemodynamics in cirrhosis. Liver Int. 2004, 24, 477–483. [Google Scholar] [CrossRef]
- Principe, A.; Melgar-Lesmes, P.; Fernandez-Varo, G.; del Arbol, L.R.; Ros, J.; Morales-Ruiz, M.; Bernardi, M.; Arroyo, V.; Jimenez, W. The hepatic apelin system: A new therapeutic target for liver disease. Hepatology 2008, 48, 1193–1201. [Google Scholar] [CrossRef]
- Julien, B.; Grenard, P.; Teixeira-Clerc, F.; Van Nhieu, J.T.; Li, L.; Karsak, M.; Zimmer, A.; Mallat, A.; Lotersztajn, S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755. [Google Scholar] [CrossRef]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Batkai, S.; Harvey-White, J.; Mackie, K.; Offertaler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef]
- Velasco, G.; Sanchez, C.; Guzman, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Poleszak, E.; Wosko, S.; Slawinska, K.; Szopa, A.; Wrobel, A.; Serefko, A. Cannabinoids in depressive disorders. Life Sci. 2018, 213, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Schmole, A.C.; Lundt, R.; Toporowski, G.; Hansen, J.N.; Beins, E.; Halle, A.; Zimmer, A. Cannabinoid receptor 2-deficiency ameliorates disease symptoms in a mouse model with Alzheimer’s Disease-like pathology. J. Alzheimers Dis. 2018, 64, 379–392. [Google Scholar] [CrossRef]
- Lamontagne, D.; Lepicier, P.; Lagneux, C.; Bouchard, J.F. The endogenous cardiac cannabinoid system: A new protective mechanism against myocardial ischemia. Arch. Mal. Coeur. Vaiss. 2006, 99, 242–246. [Google Scholar]
- Bazwinsky-Wutschke, I.; Zipprich, A.; Dehghani, F. Endocannabinoid system in hepatic glucose metabolism, fatty liver disease, and cirrhosis. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Perez-Gomez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; Garcia-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martin-Villar, E.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer. Inst. 2015, 107, djv077. [Google Scholar] [CrossRef]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the endocannabinoid system as a potential anticancer strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef]
- Mechoulam, R.; Hanus, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef]
- Lu, H.C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating endocannabinoids: From whence do they come and where are they going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A.; et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar] [CrossRef]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef]
- Nakane, S.; Oka, S.; Arai, S.; Waku, K.; Ishima, Y.; Tokumura, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: Occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch. Biochem. Biophys. 2002, 402, 51–58. [Google Scholar] [CrossRef]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allara, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef]
- Baram, L.; Peled, E.; Berman, P.; Yellin, B.; Besser, E.; Benami, M.; Louria-Hayon, I.; Lewitus, G.M.; Meiri, D. The heterogeneity and complexity of Cannabis extracts as antitumor agents. Oncotarget 2019, 10, 4091–4106. [Google Scholar] [CrossRef] [PubMed]
- Kozak, K.R.; Rowlinson, S.W.; Marnett, L.J. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J. Biol. Chem. 2000, 275, 33744–33749. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, E.; Chahinian, H.; Sanchez, P.; Fantini, J. The insertion and transport of anandamide in synthetic lipid membranes are both cholesterol-dependent. PLoS ONE 2009, 4, e4989. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Vu, H.K.; Larsson, N.; Groblewski, T.; Hjorth, S.; Elebring, T.; Sjogren, S.; Greasley, P.J. Identification and characterisation of a novel splice variant of the human CB1 receptor. FEBS Lett. 2005, 579, 259–264. [Google Scholar] [CrossRef]
- Shire, D.; Carillon, C.; Kaghad, M.; Calandra, B.; Rinaldi-Carmona, M.; Le Fur, G.; Caput, D.; Ferrara, P. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J. Biol. Chem. 1995, 270, 3726–3731. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Wolfson, M.L.; Valchi, P.; Aisemberg, J.; Franchi, A.M. Endocannabinoid system and pregnancy. Reproduction 2016, 152, R191–R200. [Google Scholar] [CrossRef]
- Sun, X.; Dey, S.K. Aspects of endocannabinoid signaling in periimplantation biology. Mol. Cell Endocrinol. 2008, 286, S3–S11. [Google Scholar] [CrossRef]
- Zimmer, A.; Zimmer, A.M.; Hohmann, A.G.; Herkenham, M.; Bonner, T.I. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 5780–5785. [Google Scholar] [CrossRef]
- Li, Y.; Kim, J. CB2 Cannabinoid receptor knockout in mice impairs contextual long-term memory and enhances spatial working memory. Neural Plast 2016, 2016, 9817089. [Google Scholar] [CrossRef]
- Ravinet Trillou, C.; Delgorge, C.; Menet, C.; Arnone, M.; Soubrie, P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 640–648. [Google Scholar] [CrossRef]
- Mallat, A.; Lotersztajn, S. Endocannabinoids and liver disease. I. Endocannabinoids and their receptors in the liver. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 294, G9–G12. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D.; Giuffrida, A.; Calignano, A.; Rodriguez de Fonseca, F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol. Sci. 2000, 21, 218–224. [Google Scholar] [CrossRef]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease--successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 receptor signaling and bias. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Ozaita, A.; Puighermanal, E.; Maldonado, R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J. Neurochem. 2007, 102, 1105–1114. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Casado, V.; Canela, E. The endocannabinoid system as a target in cancer diseases: Are we there yet? Front. Pharmacol. 2019, 10, 339. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467. [Google Scholar] [CrossRef]
- Howlett, A.C.; Fleming, R.M. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol. Pharmacol. 1984, 26, 532–538. [Google Scholar]
- Mlost, J.; Wasik, A.; Starowicz, K. Role of endocannabinoid system in dopamine signalling within the reward circuits affected by chronic pain. Pharmacol. Res. 2019, 143, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tian, Y.; Zheng, R.; Li, L.; Qiu, F. Endocannabinoid system and the expression of endogenous ceramides in human hepatocellular carcinoma. Oncol. Lett. 2019, 18, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Kirilly, E.; Gonda, X.; Bagdy, G. CB1 receptor antagonists: New discoveries leading to new perspectives. Acta Physiol. 2012, 205, 41–60. [Google Scholar] [CrossRef]
- Ranganathan, M.; D’Souza, D.C. The acute effects of cannabinoids on memory in humans: A review. Psychopharmacology 2006, 188, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Blazquez, P.; Rodriguez-Munoz, M.; Vicente-Sanchez, A.; Garzon, J. Cannabinoid receptors couple to NMDA receptors to reduce the production of NO and the mobilization of zinc induced by glutamate. Antioxid Redox Signal. 2013, 19, 1766–1782. [Google Scholar] [CrossRef] [PubMed]
- Khaspekov, L.G.; Brenz Verca, M.S.; Frumkina, L.E.; Hermann, H.; Marsicano, G.; Lutz, B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 2004, 19, 1691–1698. [Google Scholar] [CrossRef]
- Parolaro, D.; Realini, N.; Vigano, D.; Guidali, C.; Rubino, T. The endocannabinoid system and psychiatric disorders. Exp. Neurol. 2010, 224, 3–14. [Google Scholar] [CrossRef]
- Scotter, E.L.; Abood, M.E.; Glass, M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol. 2010, 160, 480–498. [Google Scholar] [CrossRef]
- Donvito, G.; Nass, S.R.; Wilkerson, J.L.; Curry, Z.A.; Schurman, L.D.; Kinsey, S.G.; Lichtman, A.H. The endogenous cannabinoid system: A budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacology 2018, 43, 52–79. [Google Scholar] [CrossRef]
- Salamone, J.D.; McLaughlin, P.J.; Sink, K.; Makriyannis, A.; Parker, L.A. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: Effects on food intake, food-reinforced behavior and food aversions. Physiol. Behav. 2007, 91, 383–388. [Google Scholar] [CrossRef]
- Smoum, R.; Baraghithy, S.; Chourasia, M.; Breuer, A.; Mussai, N.; Attar-Namdar, M.; Kogan, N.M.; Raphael, B.; Bolognini, D.; Cascio, M.G.; et al. CB2 cannabinoid receptor agonist enantiomers HU-433 and HU-308: An inverse relationship between binding affinity and biological potency. Proc. Natl. Acad. Sci. USA 2015, 112, 8774–8779. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Dubray, C.; Dualé, C. FAAH inhibitors in the limelight, but regrettably. Int. J. Clin. Pharmacol. Ther. 2016, 54, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, S.; Wang, Q.; Hu, W.; Wang, D.; Li, X.; Su, T.; Qin, X.; Zhang, X.; Ma, K.; et al. Effects of cannabinoid receptor type 2 on endogenous myocardial regeneration by activating cardiac progenitor cells in mouse infarcted heart. Sci. China Life Sci. 2014, 57, 201–208. [Google Scholar] [CrossRef]
- Ward, S.J.; Castelli, F.; Reichenbach, Z.W.; Tuma, R.F. Surprising outcomes in cannabinoid CB1/CB2 receptor double knockout mice in two models of ischemia. Life Sci. 2018, 195, 1–5. [Google Scholar] [CrossRef]
- Kunos, G.; Tam, J. The case for peripheral CB(1) receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. Br. J. Pharmacol. 2011, 163, 1423–1431. [Google Scholar] [CrossRef]
- Morales, P.; Hernandez-Folgado, L.; Goya, P.; Jagerovic, N. Cannabinoid receptor 2 (CB2) agonists and antagonists: A patent update. Expert Opin. Ther. Pat. 2016, 26, 843–856. [Google Scholar] [CrossRef]
- Izzo, A.A.; Sharkey, K.A. Cannabinoids and the gut: New developments and emerging concepts. Pharmacol. Ther. 2010, 126, 21–38. [Google Scholar] [CrossRef]
- Velasco, G.; Sanchez, C.; Guzman, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; DuBois, R.N. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008, 68, 6468–6476. [Google Scholar] [CrossRef]
- Orellana-Serradell, O.; Poblete, C.E.; Sanchez, C.; Castellon, E.A.; Gallegos, I.; Huidobro, C.; Llanos, M.N.; Contreras, H.R. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol. Rep. 2015, 33, 1599–1608. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Schwabe, R.F. Endocannabinoids and liver disease. II. Endocannabinoids in the pathogenesis and treatment of liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G357–G362. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Stoll, M.; Millonig, G.; Agaimy, A.; Wissniowski, T.; Schneider, V.; Mueller, S.; Brenneisen, R.; Seitz, H.K.; Ocker, M.; et al. Cannabinoid receptor type I modulates alcohol-induced liver fibrosis. Mol. Med. 2011, 17, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, P.; Viola, A.; Piscitelli, F.; Giannone, F.; Berzigotti, A.; Cescon, M.; Domenicali, M.; Petrosino, S.; Giampalma, E.; Riili, A.; et al. Circulating and hepatic endocannabinoids and endocannabinoid-related molecules in patients with cirrhosis. Liver Int. 2010, 30, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Patsenker, E.; Sachse, P.; Chicca, A.; Gachet, M.S.; Schneider, V.; Mattsson, J.; Lanz, C.; Worni, M.; de Gottardi, A.; Semmo, M.; et al. Elevated levels of endocannabinoids in chronic hepatitis C may modulate cellular immune response and hepatic stellate cell activation. Int. J. Mol. Sci. 2015, 16, 7057–7076. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Wojtalla, A.; Schlosser, M.; Zimmer, A.; Singer, M.V. Fatty acid amide hydrolase but not monoacyl glycerol lipase controls cell death induced by the endocannabinoid 2-arachidonoyl glycerol in hepatic cell populations. Biochem. Biophys. Res. Commun. 2013, 437, 48–54. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Seki, E.; Osawa, Y.; Uchinami, H.; Cravatt, B.F.; Schwabe, R.F. Fatty acid amide hydrolase determines anandamide-induced cell death in the liver. J. Biol. Chem. 2006, 281, 10431–10438. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Uchinami, H.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. Anandamide induces necrosis in primary hepatic stellate cells. Hepatology 2005, 41, 1085–1095. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Wojtalla, A.; Schlosser, M.; Schildberg, F.A.; Knolle, P.A.; Nusing, R.M.; Zimmer, A.; Strassburg, C.P.; Singer, M.V. Cyclooxygenase-2 contributes to the selective induction of cell death by the endocannabinoid 2-arachidonoyl glycerol in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2016, 470, 678–684. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J.; Qiu, Y. Oleoylethanolamide, an endogenous PPAR-alpha ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef]
- Batkai, S.; Mukhopadhyay, P.; Harvey-White, J.; Kechrid, R.; Pacher, P.; Kunos, G. Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in vivo in cirrhotic rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1689–H1695. [Google Scholar] [CrossRef]
- Dai, E.; Zhang, L.; Ye, L.; Wan, S.; Feng, L.; Qi, Q.; Yao, F.; Li, Z. Hepatic expression of cannabinoid receptors CB1 and CB2 correlate with fibrogenesis in patients with chronic hepatitis B. Int. J. Infect. Dis 2017, 59, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Teixeira-Clerc, F.; Belot, M.P.; Manin, S.; Deveaux, V.; Cadoudal, T.; Chobert, M.N.; Louvet, A.; Zimmer, A.; Tordjmann, T.; Mallat, A.; et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology 2010, 52, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Hezode, C.; Zafrani, E.S.; Roudot-Thoraval, F.; Costentin, C.; Hessami, A.; Bouvier-Alias, M.; Medkour, F.; Pawlostky, J.M.; Lotersztajn, S.; Mallat, A. Daily cannabis use: A novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology 2008, 134, 432–439. [Google Scholar] [CrossRef]
- Dibba, P.; Li, A.A.; Cholankeril, G.; Iqbal, U.; Gadiparthi, C.; Khan, M.A.; Kim, D.; Ahmed, A. The role of cannabinoids in the setting of cirrhosis. Medicines 2018, 5. [Google Scholar] [CrossRef]
- Batkai, S.; Jarai, Z.; Wagner, J.A.; Goparaju, S.K.; Varga, K.; Liu, J.; Wang, L.; Mirshahi, F.; Khanolkar, A.D.; Makriyannis, A.; et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat. Med. 2001, 7, 827–832. [Google Scholar] [CrossRef]
- Ros, J.; Claria, J.; To-Figueras, J.; Planaguma, A.; Cejudo-Martin, P.; Fernandez-Varo, G.; Martin-Ruiz, R.; Arroyo, V.; Rivera, F.; Rodes, J.; et al. Endogenous cannabinoids: A new system involved in the homeostasis of arterial pressure in experimental cirrhosis in the rat. Gastroenterology 2002, 122, 85–93. [Google Scholar] [CrossRef]
- Domenicali, M.; Caraceni, P.; Giannone, F.; Pertosa, A.M.; Principe, A.; Zambruni, A.; Trevisani, F.; Croci, T.; Bernardi, M. Cannabinoid type 1 receptor antagonism delays ascites formation in rats with cirrhosis. Gastroenterology 2009, 137, 341–349. [Google Scholar] [CrossRef]
- Giannone, F.A.; Baldassarre, M.; Domenicali, M.; Zaccherini, G.; Trevisani, F.; Bernardi, M.; Caraceni, P. Reversal of liver fibrosis by the antagonism of endocannabinoid CB1 receptor in a rat model of CCl(4)-induced advanced cirrhosis. Lab. Invest. 2012, 92, 384–395. [Google Scholar] [CrossRef]
- Caraceni, P.; Pertosa, A.M.; Giannone, F.; Domenicali, M.; Grattagliano, I.; Principe, A.; Mastroleo, C.; Perrelli, M.G.; Cutrin, J.; Trevisani, F.; et al. Antagonism of the cannabinoid CB-1 receptor protects rat liver against ischaemia-reperfusion injury complicated by endotoxaemia. Gut 2009, 58, 1135–1143. [Google Scholar] [CrossRef]
- Liu, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Wagner, J.A.; Cravatt, B.F.; Gao, B.; Kunos, G. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J. Biol. Chem. 2003, 278, 45034–45039. [Google Scholar] [CrossRef]
- Domenicali, M.; Ros, J.; Fernandez-Varo, G.; Cejudo-Martin, P.; Crespo, M.; Morales-Ruiz, M.; Briones, A.M.; Campistol, J.M.; Arroyo, V.; Vila, E.; et al. Increased anandamide induced relaxation in mesenteric arteries of cirrhotic rats: Role of cannabinoid and vanilloid receptors. Gut 2005, 54, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Deutsch, D.G. Unique pathway for anandamide synthesis and liver regeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6339–6340. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Picardi, P.; Pallottini, V.; Martini, C.; Petrosino, S.; Proto, M.C.; Vitale, M.; Laezza, C.; Gazzerro, P.; Di Marzo, V.; et al. Anandamide drives cell cycle progression through CB1 receptors in a rat model of synchronized liver regeneration. J. Cell Physiol. 2015, 230, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, V.; Ros, J.; Fernandez-Varo, G.; Casals, G.; Melgar-Lesmes, P.; Campos, T.; Makriyannis, A.; Morales-Ruiz, M.; Jimenez, W. Prevention of fibrosis progression in CCl4-treated rats: Role of the hepatic endocannabinoid and apelin systems. J. Pharmacol. Exp. Ther. 2012, 340, 629–637. [Google Scholar] [CrossRef]
- Horvath, B.; Magid, L.; Mukhopadhyay, P.; Batkai, S.; Rajesh, M.; Park, O.; Tanchian, G.; Gao, R.Y.; Goodfellow, C.E.; Glass, M.; et al. A new cannabinoid CB2 receptor agonist HU-910 attenuates oxidative stress, inflammation and cell death associated with hepatic ischaemia/reperfusion injury. Br. J. Pharmacol. 2012, 165, 2462–2478. [Google Scholar] [CrossRef]
- Munoz-Luque, J.; Ros, J.; Fernandez-Varo, G.; Tugues, S.; Morales-Ruiz, M.; Alvarez, C.E.; Friedman, S.L.; Arroyo, V.; Jimenez, W. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J. Pharmacol. Exp. Ther. 2008, 324, 475–483. [Google Scholar] [CrossRef]
- Guillot, A.; Hamdaoui, N.; Bizy, A.; Zoltani, K.; Souktani, R.; Zafrani, E.S.; Mallat, A.; Lotersztajn, S.; Lafdil, F. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology 2014, 59, 296–306. [Google Scholar] [CrossRef]
- Avraham, Y.; Israeli, E.; Gabbay, E.; Okun, A.; Zolotarev, O.; Silberman, I.; Ganzburg, V.; Dagon, Y.; Magen, I.; Vorobia, L.; et al. Endocannabinoids affect neurological and cognitive function in thioacetamide-induced hepatic encephalopathy in mice. Neurobiol. Dis. 2006, 21, 237–245. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- De Mota, N.; Reaux-Le Goazigo, A.; El Messari, S.; Chartrel, N.; Roesch, D.; Dujardin, C.; Kordon, C.; Vaudry, H.; Moos, F.; Llorens-Cortes, C. Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc. Natl. Acad. Sci. USA 2004, 101, 10464–10469. [Google Scholar] [CrossRef]
- Szokodi, I.; Tavi, P.; Foldes, G.; Voutilainen-Myllyla, S.; Ilves, M.; Tokola, H.; Pikkarainen, S.; Piuhola, J.; Rysa, J.; Toth, M.; et al. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ. Res. 2002, 91, 434–440. [Google Scholar] [CrossRef]
- Ishida, J.; Hashimoto, T.; Hashimoto, Y.; Nishiwaki, S.; Iguchi, T.; Harada, S.; Sugaya, T.; Matsuzaki, H.; Yamamoto, R.; Shiota, N.; et al. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J. Biol. Chem. 2004, 279, 26274–26279. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, Y.; Fujii, T.; Kamimura, Y.; Kawashima, K. The endogenous, immunologically active peptide apelin inhibits lymphocytic cholinergic activity during immunological responses. J. Neuroimmunol. 2003, 144, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Dray, C.; Sakar, Y.; Vinel, C.; Daviaud, D.; Masri, B.; Garrigues, L.; Wanecq, E.; Galvani, S.; Negre-Salvayre, A.; Barak, L.S.; et al. The intestinal glucose-apelin cycle controls carbohydrate absorption in mice. Gastroenterology 2013, 144, 771–780. [Google Scholar] [CrossRef]
- Daviaud, D.; Boucher, J.; Gesta, S.; Dray, C.; Guigne, C.; Quilliot, D.; Ayav, A.; Ziegler, O.; Carpene, C.; Saulnier-Blache, J.S.; et al. TNFalpha up-regulates apelin expression in human and mouse adipose tissue. FASEB J. 2006, 20, 1528–1530. [Google Scholar] [CrossRef]
- Scott, I.C.; Masri, B.; D’Amico, L.A.; Jin, S.W.; Jungblut, B.; Wehman, A.M.; Baier, H.; Audigier, Y.; Stainier, D.Y. The G protein-coupled receptor agtrl1b regulates early development of myocardial progenitors. Dev. Cell 2007, 12, 403–413. [Google Scholar] [CrossRef]
- Kawamata, Y.; Habata, Y.; Fukusumi, S.; Hosoya, M.; Fujii, R.; Hinuma, S.; Nishizawa, N.; Kitada, C.; Onda, H.; Nishimura, O.; et al. Molecular properties of apelin: Tissue distribution and receptor binding. Biochim. Biophys Acta 2001, 1538, 162–171. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Casals, G.; Pauta, M.; Ros, J.; Reichenbach, V.; Bataller, R.; Morales-Ruiz, M.; Jimenez, W. Apelin mediates the induction of profibrogenic genes in human hepatic stellate cells. Endocrinology 2010, 151, 5306–5314. [Google Scholar] [CrossRef]
- Tatemoto, K.; Takayama, K.; Zou, M.X.; Kumaki, I.; Zhang, W.; Kumano, K.; Fujimiya, M. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul. Pept. 2001, 99, 87–92. [Google Scholar] [CrossRef]
- Masri, B.; Knibiehler, B.; Audigier, Y. Apelin signalling: A promising pathway from cloning to pharmacology. Cell Signal. 2005, 17, 415–426. [Google Scholar] [CrossRef]
- Hosoya, M.; Kawamata, Y.; Fukusumi, S.; Fujii, R.; Habata, Y.; Hinuma, S.; Kitada, C.; Honda, S.; Kurokawa, T.; Onda, H.; et al. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J. Biol. Chem. 2000, 275, 21061–21067. [Google Scholar] [CrossRef]
- Akcilar, R.; Turgut, S.; Caner, V.; Akcilar, A.; Ayada, C.; Elmas, L.; Ozcan, T.O. The effects of apelin treatment on a rat model of type 2 diabetes. Adv. Med. Sci. 2015, 60, 94–100. [Google Scholar] [CrossRef]
- Casals, G.; Fernandez-Varo, G.; Melgar-Lesmes, P.; Marfa, S.; Reichenbach, V.; Morales-Ruiz, M.; Jimenez, W. Factors involved in extracellular matrix turnover in human derived cardiomyocytes. Cell Physiol. Biochem. 2013, 32, 1125–1136. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Pauta, M.; Reichenbach, V.; Casals, G.; Ros, J.; Bataller, R.; Morales-Ruiz, M.; Jimenez, W. Hypoxia and proinflammatory factors upregulate apelin receptor expression in human stellate cells and hepatocytes. Gut 2011, 60, 1404–1411. [Google Scholar] [CrossRef]
- Eyries, M.; Siegfried, G.; Ciumas, M.; Montagne, K.; Agrapart, M.; Lebrin, F.; Soubrier, F. Hypoxia-induced apelin expression regulates endothelial cell proliferation and regenerative angiogenesis. Circ. Res. 2008, 103, 432–440. [Google Scholar] [CrossRef]
- Glassford, A.J.; Yue, P.; Sheikh, A.Y.; Chun, H.J.; Zarafshar, S.; Chan, D.A.; Reaven, G.M.; Quertermous, T.; Tsao, P.S. HIF-1 regulates hypoxia- and insulin-induced expression of apelin in adipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1590–E1596. [Google Scholar] [CrossRef]
- Mazzucotelli, A.; Ribet, C.; Castan-Laurell, I.; Daviaud, D.; Guigne, C.; Langin, D.; Valet, P. The transcriptional co-activator PGC-1alpha up regulates apelin in human and mouse adipocytes. Regul. Pept. 2008, 150, 33–37. [Google Scholar] [CrossRef]
- Zhang, L.; Li, F.; Su, X.; Li, Y.; Wang, Y.; Fang, R.; Guo, Y.; Jin, T.; Shan, H.; Zhao, X.; et al. Melatonin prevents lung injury by regulating apelin 13 to improve mitochondrial dysfunction. Exp. Mol. Med. 2019, 51, 73. [Google Scholar] [CrossRef]
- Wei, L.; Hou, X.; Tatemoto, K. Regulation of apelin mRNA expression by insulin and glucocorticoids in mouse 3T3-L1 adipocytes. Regul. Pept. 2005, 132, 27–32. [Google Scholar] [CrossRef]
- Kralisch, S.; Lossner, U.; Bluher, M.; Paschke, R.; Stumvoll, M.; Fasshauer, M. Growth hormone induces apelin mRNA expression and secretion in mouse 3T3-L1 adipocytes. Regul. Pept. 2007, 139, 84–89. [Google Scholar] [CrossRef]
- Jiang, H.; Ye, X.P.; Yang, Z.Y.; Zhan, M.; Wang, H.N.; Cao, H.M.; Xie, H.J.; Pan, C.M.; Song, H.D.; Zhao, S.X. Aldosterone directly affects apelin expression and secretion in adipocytes. J. Mol. Endocrinol. 2013, 51, 37–48. [Google Scholar] [CrossRef]
- Llorens-Cortes, C.; Moos, F. Opposite potentiality of hypothalamic coexpressed neuropeptides, apelin and vasopressin in maintaining body-fluid homeostasis. Prog. Brain Res. 2008, 170, 559–570. [Google Scholar] [CrossRef]
- Rastellini, C.; Han, S.; Bhatia, V.; Cao, Y.; Liu, K.; Gao, X.; Ko, T.C.; Greeley, G.H., Jr.; Falzon, M. Induction of chronic pancreatitis by pancreatic duct ligation activates BMP2, apelin, and PTHrP expression in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 309, G554–G565. [Google Scholar] [CrossRef][Green Version]
- Han, S.; Englander, E.W.; Gomez, G.A.; Aronson, J.F.; Rastellini, C.; Garofalo, R.P.; Kolli, D.; Quertermous, T.; Kundu, R.; Greeley, G.H., Jr. Pancreatitis activates pancreatic apelin-APJ axis in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2013, 305, G139–G150. [Google Scholar] [CrossRef]
- Kasai, A.; Shintani, N.; Kato, H.; Matsuda, S.; Gomi, F.; Haba, R.; Hashimoto, H.; Kakuda, M.; Tano, Y.; Baba, A. Retardation of retinal vascular development in apelin-deficient mice. Arterioscler Thromb Vasc. Biol. 2008, 28, 1717–1722. [Google Scholar] [CrossRef]
- Cox, C.M.; D’Agostino, S.L.; Miller, M.K.; Heimark, R.L.; Krieg, P.A. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev. Biol. 2006, 296, 177–189. [Google Scholar] [CrossRef]
- Sato, T.; Kadowaki, A.; Suzuki, T.; Ito, H.; Watanabe, H.; Imai, Y.; Kuba, K. Loss of apelin augments angiotensin II-induced cardiac dysfunction and pathological remodeling. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Siddiquee, K.; Hampton, J.; McAnally, D.; May, L.; Smith, L. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br. J. Pharmacol. 2013, 168, 1104–1117. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Wang, W.; Jin, H.Y.; Chen, X.; Cheng, Y.W.; Xu, Y.L.; Song, B.; Penninger, J.M.; Oudit, G.Y.; Zhong, J.C. Apelin is a negative regulator of angiotensin II-mediated adverse myocardial remodeling and dysfunction. Hypertension 2017, 70, 1165–1175. [Google Scholar] [CrossRef]
- Falcao-Pires, I.; Goncalves, N.; Henriques-Coelho, T.; Moreira-Goncalves, D.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F. Apelin decreases myocardial injury and improves right ventricular function in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H2007–H2014. [Google Scholar] [CrossRef]
- Hou, J.; Wang, L.; Long, H.; Wu, H.; Wu, Q.; Zhong, T.; Chen, X.; Zhou, C.; Guo, T.; Wang, T. Hypoxia preconditioning promotes cardiac stem cell survival and cardiogenic differentiation in vitro involving activation of the HIF-1alpha/apelin/APJ axis. Stem Cell Res. Ther. 2017, 8, 215. [Google Scholar] [CrossRef]
- Dray, C.; Debard, C.; Jager, J.; Disse, E.; Daviaud, D.; Martin, P.; Attane, C.; Wanecq, E.; Guigne, C.; Bost, F.; et al. Apelin and APJ regulation in adipose tissue and skeletal muscle of type 2 diabetic mice and humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1161–E1169. [Google Scholar] [CrossRef]
- O’Carroll, A.M.; Don, A.L.; Lolait, S.J. APJ receptor mRNA expression in the rat hypothalamic paraventricular nucleus: Regulation by stress and glucocorticoids. J. Neuroendocrinol. 2003, 15, 1095–1101. [Google Scholar] [CrossRef]
- Deng, C.; Chen, H.; Yang, N.; Feng, Y.; Hsueh, A.J. Apela regulates fluid homeostasis by binding to the APJ receptor to activate Gi signaling. J. Biol. Chem. 2015, 290, 18261–18268. [Google Scholar] [CrossRef]
- Yang, P.; Read, C.; Kuc, R.E.; Buonincontri, G.; Southwood, M.; Torella, R.; Upton, P.D.; Crosby, A.; Sawiak, S.J.; Carpenter, T.A.; et al. Elabela/toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 2017, 135, 1160–1173. [Google Scholar] [CrossRef]
- Ho, L.; Tan, S.Y.; Wee, S.; Wu, Y.; Tan, S.J.; Ramakrishna, N.B.; Chng, S.C.; Nama, S.; Szczerbinska, I.; Chan, Y.S.; et al. ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell 2015, 17, 435–447. [Google Scholar] [CrossRef]
- Langelaan, D.N.; Reddy, T.; Banks, A.W.; Dellaire, G.; Dupre, D.J.; Rainey, J.K. Structural features of the apelin receptor N-terminal tail and first transmembrane segment implicated in ligand binding and receptor trafficking. Biochim. Biophys. Acta 2013, 1828, 1471–1483. [Google Scholar] [CrossRef]
- Liu, C.; Su, T.; Li, F.; Li, L.; Qin, X.; Pan, W.; Feng, F.; Chen, F.; Liao, D.; Chen, L. PI3K/Akt signaling transduction pathway is involved in rat vascular smooth muscle cell proliferation induced by apelin-13. Acta Biochim. Biophys. Sin. 2010, 42, 396–402. [Google Scholar] [CrossRef]
- Chaves-Almagro, C.; Castan-Laurell, I.; Dray, C.; Knauf, C.; Valet, P.; Masri, B. Apelin receptors: From signaling to antidiabetic strategy. Eur. J. Pharmacol. 2015, 763, 149–159. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Bai, B.; Du, H.; Liu, Y.; Liu, H. Heterodimerization of human apelin and kappa opioid receptors: Roles in signal transduction. Cell Signal. 2012, 24, 991–1001. [Google Scholar] [CrossRef]
- Masri, B.; Morin, N.; Cornu, M.; Knibiehler, B.; Audigier, Y. Apelin (65-77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. FASEB J. 2004, 18, 1909–1911. [Google Scholar] [CrossRef]
- Berry, M.F.; Pirolli, T.J.; Jayasankar, V.; Burdick, J.; Morine, K.J.; Gardner, T.J.; Woo, Y.J. Apelin has in vivo inotropic effects on normal and failing hearts. Circulation 2004, 110, II187–II193. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, X.; Liang, G.X.; Cui, R.R.; Liu, Y.; Wu, S.S.; Liang, Q.H.; Liu, G.Y.; Jiang, Y.; Liao, X.B.; et al. Apelin-APJ induces ICAM-1, VCAM-1 and MCP-1 expression via NF-kappaB/JNK signal pathway in human umbilical vein endothelial cells. Amino Acids 2012, 43, 2125–2136. [Google Scholar] [CrossRef]
- Kim, J.D.; Kang, Y.; Kim, J.; Papangeli, I.; Kang, H.; Wu, J.; Park, H.; Nadelmann, E.; Rockson, S.G.; Chun, H.J.; et al. Essential role of Apelin signaling during lymphatic development in zebrafish. Arterioscler Thromb Vasc. Biol. 2014, 34, 338–345. [Google Scholar] [CrossRef]
- Mughal, A.; O’Rourke, S.T. Vascular effects of apelin: Mechanisms and therapeutic potential. Pharmacol. Ther. 2018, 190, 139–147. [Google Scholar] [CrossRef]
- Xiao, Z.Y.; Wang, B.; Fu, W.; Jin, X.; You, Y.; Tian, S.W.; Kuang, X. The hippocampus is a critical site mediating antidepressant-like activity of apelin-13 in rats. Neuroscience 2018, 375, 1–9. [Google Scholar] [CrossRef]
- Huang, Z.; Luo, X.; Liu, M.; Chen, L. Function and regulation of apelin/APJ system in digestive physiology and pathology. J. Cell Physiol. 2019, 234, 7796–7810. [Google Scholar] [CrossRef]
- Ureche, C.; Tapoi, L.; Volovat, S.; Voroneanu, L.; Kanbay, M.; Covic, A. Cardioprotective apelin effects and the cardiac-renal axis: Review of existing science and potential therapeutic applications of synthetic and native regulated apelin. J. Hum. Hypertens 2019, 33, 429–435. [Google Scholar] [CrossRef]
- Yin, J.; Wang, Y.; Chang, J.; Li, B.; Zhang, J.; Liu, Y.; Lai, S.; Jiang, Y.; Li, H.; Zeng, X. Apelin inhibited epithelial-mesenchymal transition of podocytes in diabetic mice through downregulating immunoproteasome subunits beta5i. Cell Death Dis. 2018, 9, 1031. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, D.; Yang, R.; Xia, W.; Qian, K.; Shi, Z.; Brown, R.; Zhou, H.; Xi, Y.; Shi, L.; et al. Hepatic and cardiac beneficial effects of a long-acting Fc-apelin fusion protein in diet-induced obese mice. Diabetes Metab. Res. Rev. 2018, 34, e2997. [Google Scholar] [CrossRef] [PubMed]
- Renaud-Gabardos, E.; Tatin, F.; Hantelys, F.; Lebas, B.; Calise, D.; Kunduzova, O.; Masri, B.; Pujol, F.; Sicard, P.; Valet, P.; et al. Therapeutic benefit and gene network regulation by combined gene transfer of apelin, FGF2, and SERCA2a into ischemic heart. Mol. Ther. 2018, 26, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Tatin, F.; Renaud-Gabardos, E.; Godet, A.C.; Hantelys, F.; Pujol, F.; Morfoisse, F.; Calise, D.; Viars, F.; Valet, P.; Masri, B.; et al. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Han, B.; Yu, T.; Zong, Z. Effect of apelin on the cardiac hemodynamics in hypertensive rats with heart failure. Int. J. Mol. Med. 2014, 34, 756–764. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhong, J.C.; Yu, X.Y.; Huang, Y.; Yung, L.M.; Lau, C.W.; Lin, S.G. Apelin modulates aortic vascular tone via endothelial nitric oxide synthase phosphorylation pathway in diabetic mice. Cardiovasc. Res. 2007, 74, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D.; Alam, S.; Carter, G.; Pedersen, C.M.; Lee, K.M.; Hubbard, T.J.; Veitch, S.; Jeong, H.; White, A.; Cruden, N.L.; et al. Sustained cardiovascular actions of APJ agonism during renin-angiotensin system activation and in patients with heart failure. Circ. Heart Fail. 2013, 6, 482–491. [Google Scholar] [CrossRef]
- Foldes, G.; Horkay, F.; Szokodi, I.; Vuolteenaho, O.; Ilves, M.; Lindstedt, K.A.; Mayranpaa, M.; Sarman, B.; Seres, L.; Skoumal, R.; et al. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure. Biochem. Biophys. Res. Commun. 2003, 308, 480–485. [Google Scholar] [CrossRef]
- Chong, K.S.; Gardner, R.S.; Morton, J.J.; Ashley, E.A.; McDonagh, T.A. Plasma concentrations of the novel peptide apelin are decreased in patients with chronic heart failure. Eur. J. Heart Fail. 2006, 8, 355–360. [Google Scholar] [CrossRef]
- Japp, A.G.; Cruden, N.L.; Barnes, G.; van Gemeren, N.; Mathews, J.; Adamson, J.; Johnston, N.R.; Denvir, M.A.; Megson, I.L.; Flapan, A.D.; et al. Acute cardiovascular effects of apelin in humans: Potential role in patients with chronic heart failure. Circulation 2010, 121, 1818–1827. [Google Scholar] [CrossRef]
- Perea, R.J.; Morales-Ruiz, M.; Ortiz-Perez, J.T.; Bosch, X.; Andreu, D.; Borras, R.; Acosta, J.; Penela, D.; Prat-Gonzalez, S.; de Caralt, T.M.; et al. Utility of galectin-3 in predicting post-infarct remodeling after acute myocardial infarction based on extracellular volume fraction mapping. Int. J. Cardiol. 2016, 223, 458–464. [Google Scholar] [CrossRef]
- Miettinen, K.H.; Magga, J.; Vuolteenaho, O.; Vanninen, E.J.; Punnonen, K.R.; Ylitalo, K.; Tuomainen, P.; Peuhkurinen, K.J. Utility of plasma apelin and other indices of cardiac dysfunction in the clinical assessment of patients with dilated cardiomyopathy. Regul. Pept. 2007, 140, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Sans-Rosello, J.; Casals, G.; Rossello, X.; Gonzalez de la Presa, B.; Vila, M.; Duran-Cambra, A.; Morales-Ruiz, M.; Ferrero-Gregori, A.; Jimenez, W.; Sionis, A. Prognostic value of plasma apelin concentrations at admission in patients with ST-segment elevation acute myocardial infarction. Clin. Biochem. 2017, 50, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Hampton, J.; Khan, S.; Zadory, D.; Gleaves, L.; Vaughan, D.E.; Smith, L.H. Apelin protects against angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J. Hypertens 2011, 29, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Kobayashi, N.; Takeshima, H.; Koguchi, W.; Ishimitsu, T. Effects of olmesartan on Apelin/APJ and Akt/endothelial nitric oxide synthase pathway in Dahl rats with end-stage heart failure. J. Cardiovasc. Pharmacol. 2010, 55, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Okumura, Y.; Oka, T.; Toiyama, K.; Ozawa, S.; Itoi, T.; Hamaoka, K. The role of apelin on the alleviative effect of Angiotensin receptor blocker in unilateral ureteral obstruction-induced renal fibrosis. Nephron Extra 2012, 2, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Diao, Z.L.; Zhang, D.L.; Zheng, J.F.; Zhang, Q.D.; Ding, J.X.; Liu, W.H. The regulatory peptide apelin: A novel inhibitor of renal interstitial fibrosis. Amino Acids 2014, 46, 2693–2704. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Sekiguchi, A.; Fujiwara, C.; Uchiyama, A.; Uehara, A.; Ogino, S.; Torii, R.; Ishikawa, O.; Motegi, S.I. Inhibitory regulation of skin fibrosis in systemic sclerosis by Apelin/APJ signaling. Arthritis Rheumatol. 2018, 70, 1661–1672. [Google Scholar] [CrossRef]
- Wang, L.Y.; Diao, Z.L.; Zheng, J.F.; Wu, Y.R.; Zhang, Q.D.; Liu, W.H. Apelin attenuates TGF-beta1-induced epithelial to mesenchymal transition via activation of PKC-epsilon in human renal tubular epithelial cells. Peptides 2017, 96, 44–52. [Google Scholar] [CrossRef]
- Wang, Y.; Song, J.; Bian, H.; Bo, J.; Lv, S.; Pan, W.; Lv, X. Apelin promotes hepatic fibrosis through ERK signaling in LX-2 cells. Mol. Cell Biochem. 2019, 460, 205–215. [Google Scholar] [CrossRef]
- Yokomori, H.; Oda, M.; Yoshimura, K.; Machida, S.; Kaneko, F.; Hibi, T. Overexpression of apelin receptor (APJ/AGTRL1) on hepatic stellate cells and sinusoidal angiogenesis in human cirrhotic liver. J. Gastroenterol. 2011, 46, 222–231. [Google Scholar] [CrossRef]
- Van Steenkiste, C.; Ribera, J.; Geerts, A.; Pauta, M.; Tugues, S.; Casteleyn, C.; Libbrecht, L.; Olievier, K.; Schroyen, B.; Reynaert, H.; et al. Inhibition of placental growth factor activity reduces the severity of fibrosis, inflammation, and portal hypertension in cirrhotic mice. Hepatology 2011, 53, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; You, Z.; Yu, H.; Zhou, L.; Zhao, H.; Yan, X.; Li, D.; Wang, B.; Zhu, L.; Xu, Y.; et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat. Mater. 2017, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, J.; Xiao, W.; Long, J.; Zhang, H. The STAT3 inhibitor S3I-201 suppresses fibrogenesis and angiogenesis in liver fibrosis. Lab. Investig. 2018, 98, 1600–1613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Hao, M.; Jin, H.; Yao, Z.; Lian, N.; Wu, L.; Shao, J.; Chen, A.; Zheng, S. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br. J. Pharmacol. 2017, 174, 409–423. [Google Scholar] [CrossRef]
- Pauta, M.; Ribera, J.; Melgar-Lesmes, P.; Casals, G.; Rodriguez-Vita, J.; Reichenbach, V.; Fernandez-Varo, G.; Morales-Romero, B.; Bataller, R.; Michelena, J.; et al. Overexpression of angiopoietin-2 in rats and patients with liver fibrosis. Therapeutic consequences of its inhibition. Liver Int. 2015, 35, 1383–1392. [Google Scholar] [CrossRef]
- Bocca, C.; Novo, E.; Miglietta, A.; Parola, M. Angiogenesis and fibrogenesis in chronic liver diseases. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 477–488. [Google Scholar] [CrossRef]
- Taura, K.; De Minicis, S.; Seki, E.; Hatano, E.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Miura, K.; Ikai, I.; Uemoto, S.; et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology 2008, 135, 1729–1738. [Google Scholar] [CrossRef]
- Rosmorduc, O.; Housset, C. Hypoxia: A link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin. Liver Dis. 2010, 30, 258–270. [Google Scholar] [CrossRef]
- Cannito, S.; Paternostro, C.; Busletta, C.; Bocca, C.; Colombatto, S.; Miglietta, A.; Novo, E.; Parola, M. Hypoxia, hypoxia-inducible factors and fibrogenesis in chronic liver diseases. Histol. Histopathol. 2014, 29, 33–44. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Tugues, S.; Ros, J.; Fernandez-Varo, G.; Morales-Ruiz, M.; Rodes, J.; Jimenez, W. Vascular endothelial growth factor and angiopoietin-2 play a major role in the pathogenesis of vascular leakage in cirrhotic rats. Gut 2009, 58, 285–292. [Google Scholar] [CrossRef]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [PubMed]
- Ercin, C.N.; Dogru, T.; Tapan, S.; Kara, M.; Haymana, C.; Karadurmus, N.; Karslioglu, Y.; Acikel, C. Plasma apelin levels in subjects with nonalcoholic fatty liver disease. Metabolism 2010, 59, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.L.; Choi, E.; Jang, Y.O.; Cho, Y.Z.; Kang, Y.S.; Baik, S.K.; Kwon, S.O.; Kim, M.Y. Clinical implications of the serum apelin level on portal hypertension and prognosis of liver cirrhosis. Gut Liver 2016, 10, 109–116. [Google Scholar] [CrossRef]
- Yasuzaki, H.; Yoshida, S.; Hashimoto, T.; Shibata, W.; Inamori, M.; Toya, Y.; Tamura, K.; Maeda, S.; Umemura, S. Involvement of the apelin receptor APJ in Fas-induced liver injury. Liver Int. 2013, 33, 118–126. [Google Scholar] [CrossRef]
- Yoshiya, S.; Shirabe, K.; Imai, D.; Toshima, T.; Yamashita, Y.; Ikegami, T.; Okano, S.; Yoshizumi, T.; Kawanaka, H.; Maehara, Y. Blockade of the apelin-APJ system promotes mouse liver regeneration by activating Kupffer cells after partial hepatectomy. J. Gastroenterol. 2015, 50, 573–582. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, R.; Wang, W.; Xu, F.; Xi, Y.; Brown, R.A.; Zhang, H.; Shi, L.; Zhu, D.; Gong, D.W. Fc-apelin fusion protein attenuates lipopolysaccharide-induced liver injury in mice. Sci. Rep. 2018, 8, 11428. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melgar-Lesmes, P.; Perramon, M.; Jiménez, W. Roles of the Hepatic Endocannabinoid and Apelin Systems in the Pathogenesis of Liver Fibrosis. Cells 2019, 8, 1311. https://doi.org/10.3390/cells8111311
Melgar-Lesmes P, Perramon M, Jiménez W. Roles of the Hepatic Endocannabinoid and Apelin Systems in the Pathogenesis of Liver Fibrosis. Cells. 2019; 8(11):1311. https://doi.org/10.3390/cells8111311
Chicago/Turabian StyleMelgar-Lesmes, Pedro, Meritxell Perramon, and Wladimiro Jiménez. 2019. "Roles of the Hepatic Endocannabinoid and Apelin Systems in the Pathogenesis of Liver Fibrosis" Cells 8, no. 11: 1311. https://doi.org/10.3390/cells8111311
APA StyleMelgar-Lesmes, P., Perramon, M., & Jiménez, W. (2019). Roles of the Hepatic Endocannabinoid and Apelin Systems in the Pathogenesis of Liver Fibrosis. Cells, 8(11), 1311. https://doi.org/10.3390/cells8111311