Chaperone-Mediated Autophagy in the Liver:
Good or Bad?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
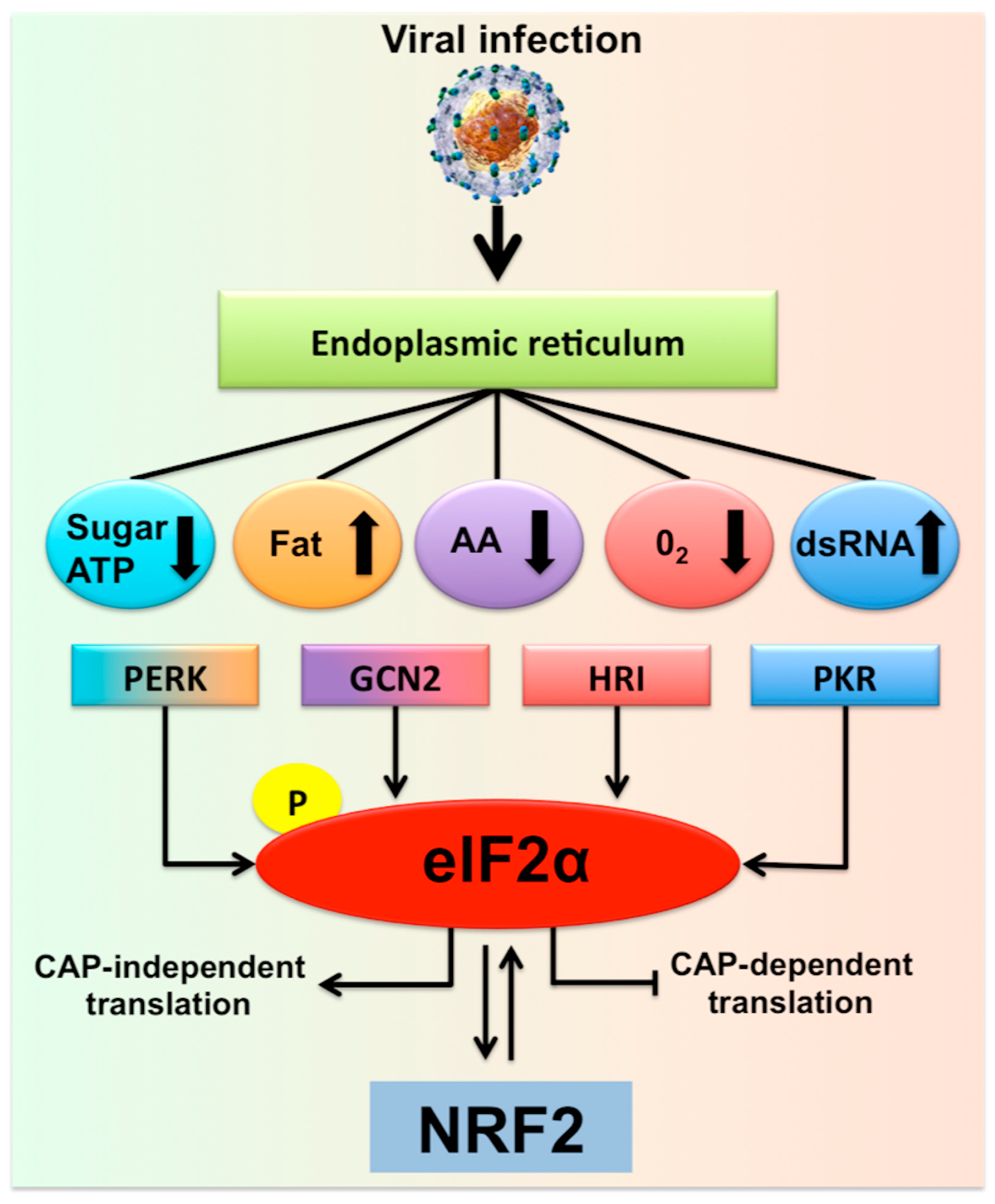
2. Hepatocytes Experience Multifaceted Stress Responses during Chronic HCV Infection
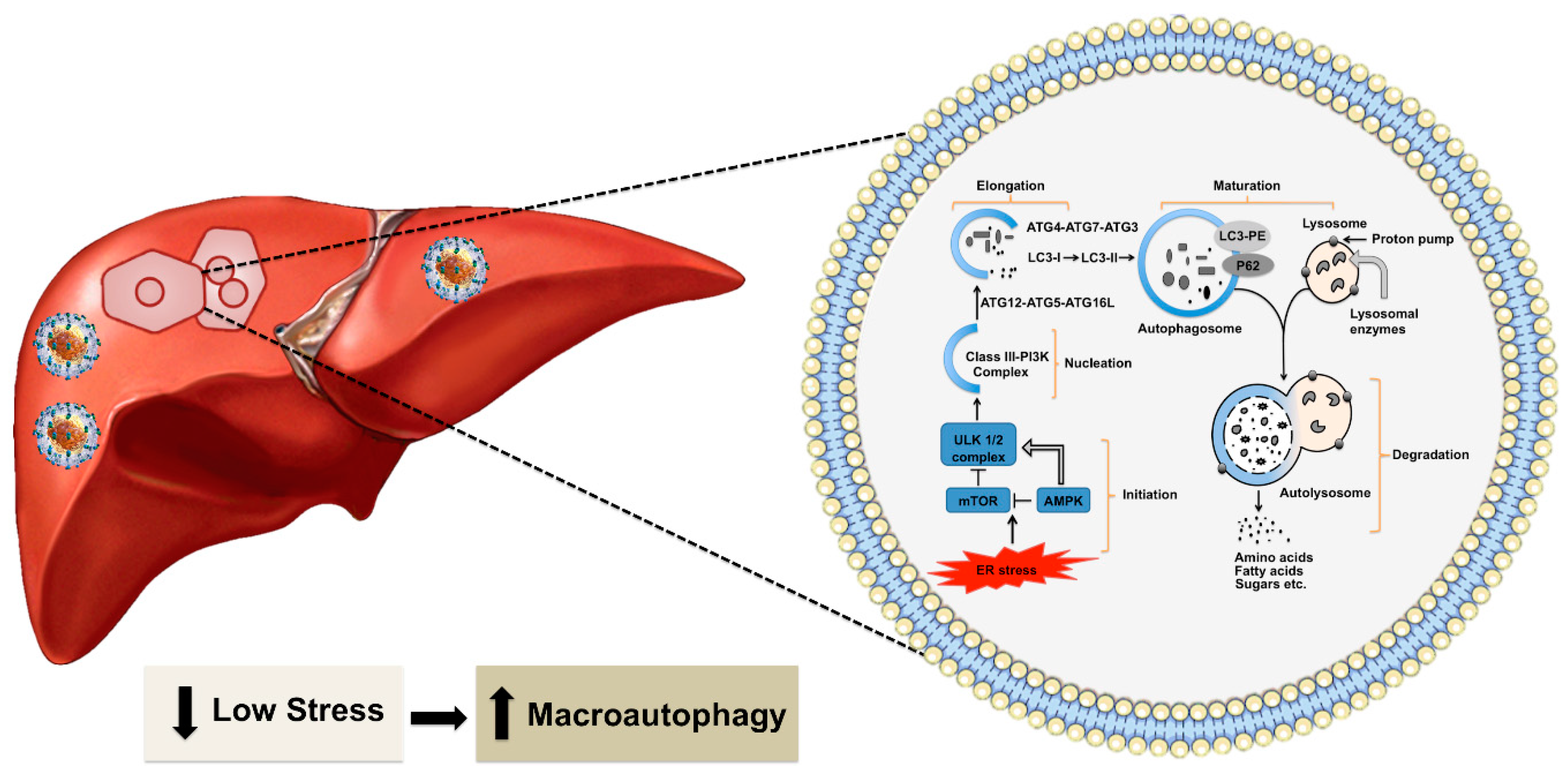
3. Excessive Hepatic Stress Modulates Autophagy Compensation
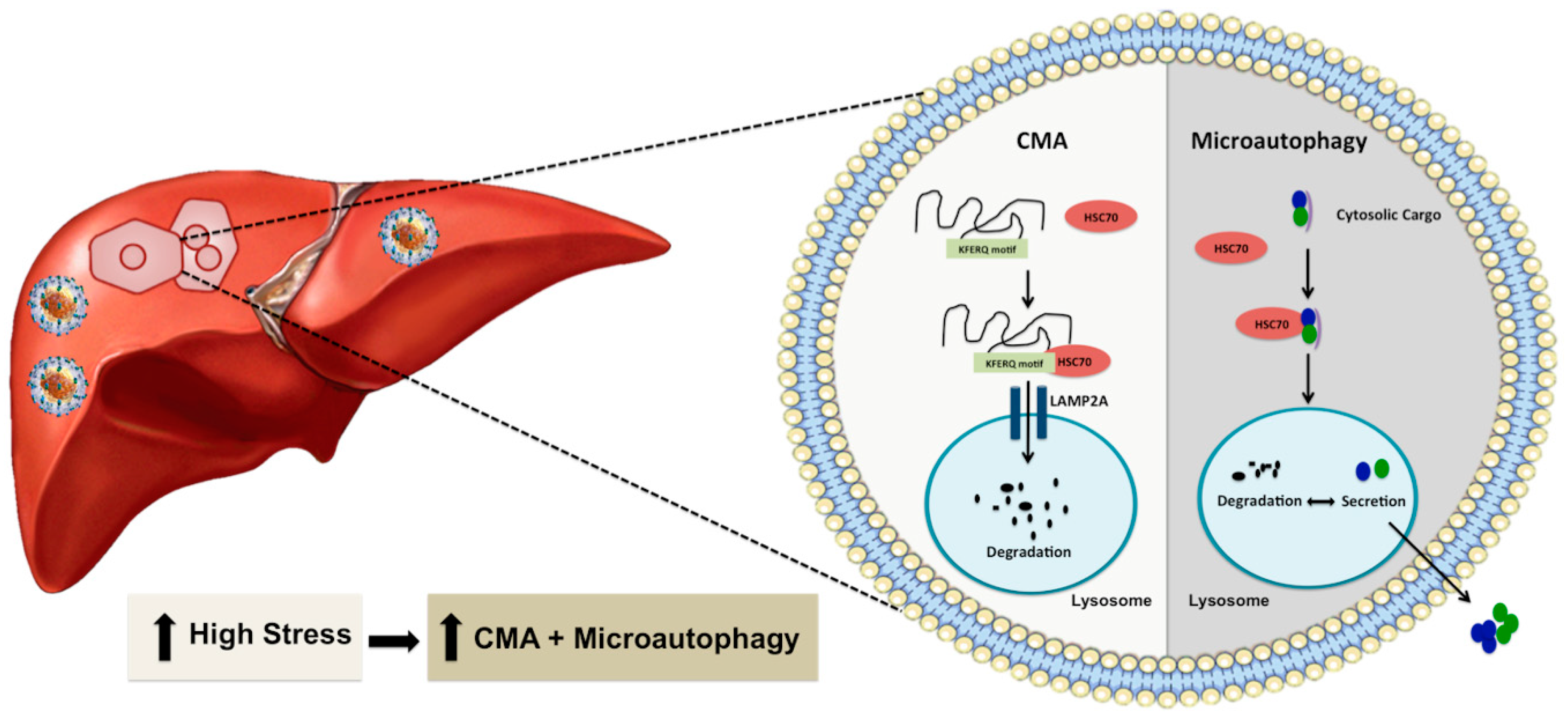
4. Chaperone-Mediated Autophagy Promotes Hepatocyte Survival under Excessive Stress
5. The Mechanism of Stress-Induced CMA
5.1. Multifaceted Stress Promotes Transcription of Hsc70 and Lamp2a through Nrf2 Activation
5.2. Oxidative Stress Promotes Nrf2-Mediated Lamp2a Activation
5.3. Regulation of CMA by Direct Phosphorylation of Lamp2a in A Non-Viral ER Stress Model
5.4. The MTOR Axis Modulates CMA through Autolysosome Lysosome Reformation
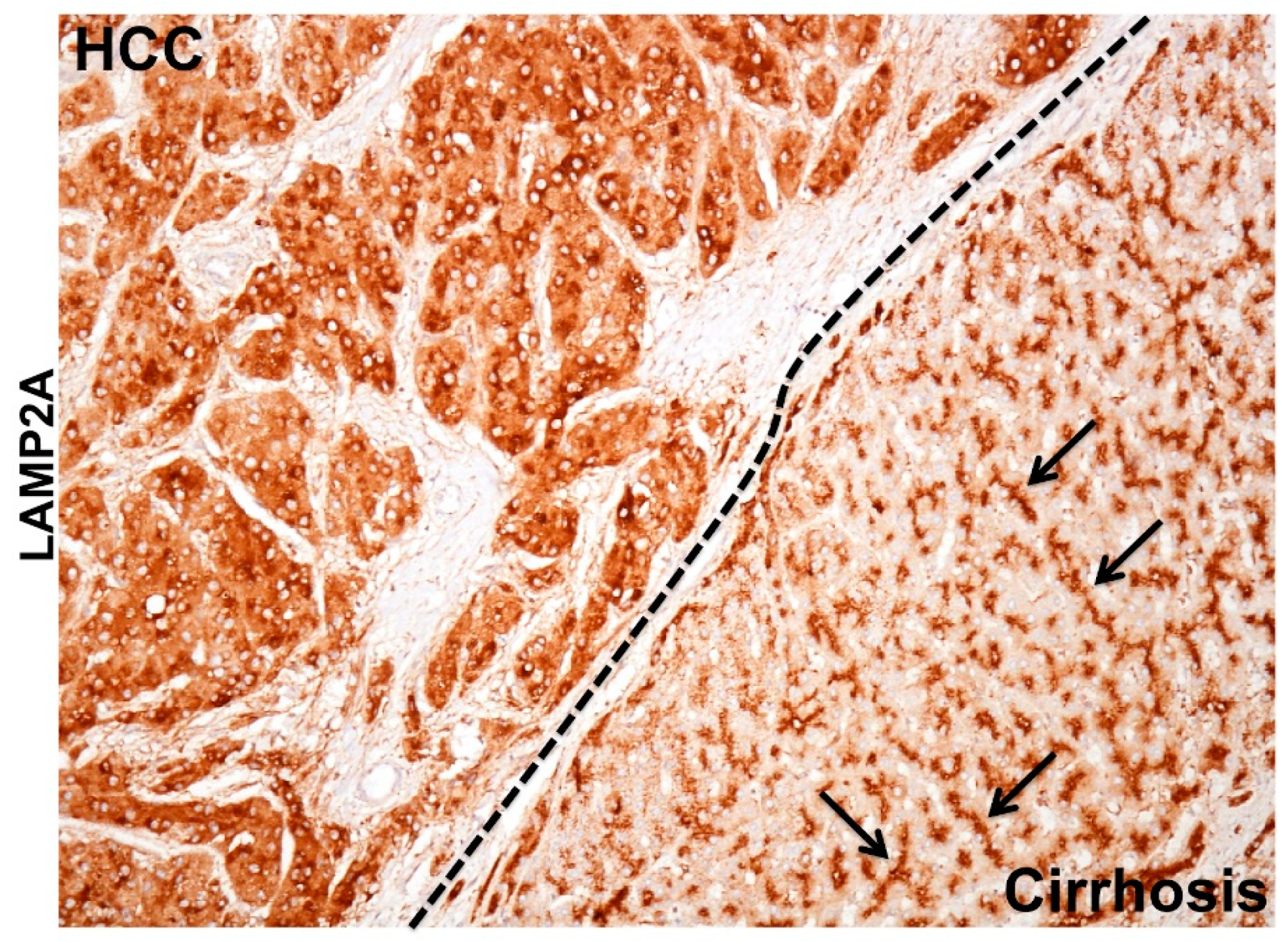
6. The Pathological Implication of CMA Activation in HCV-Induced Liver Disease
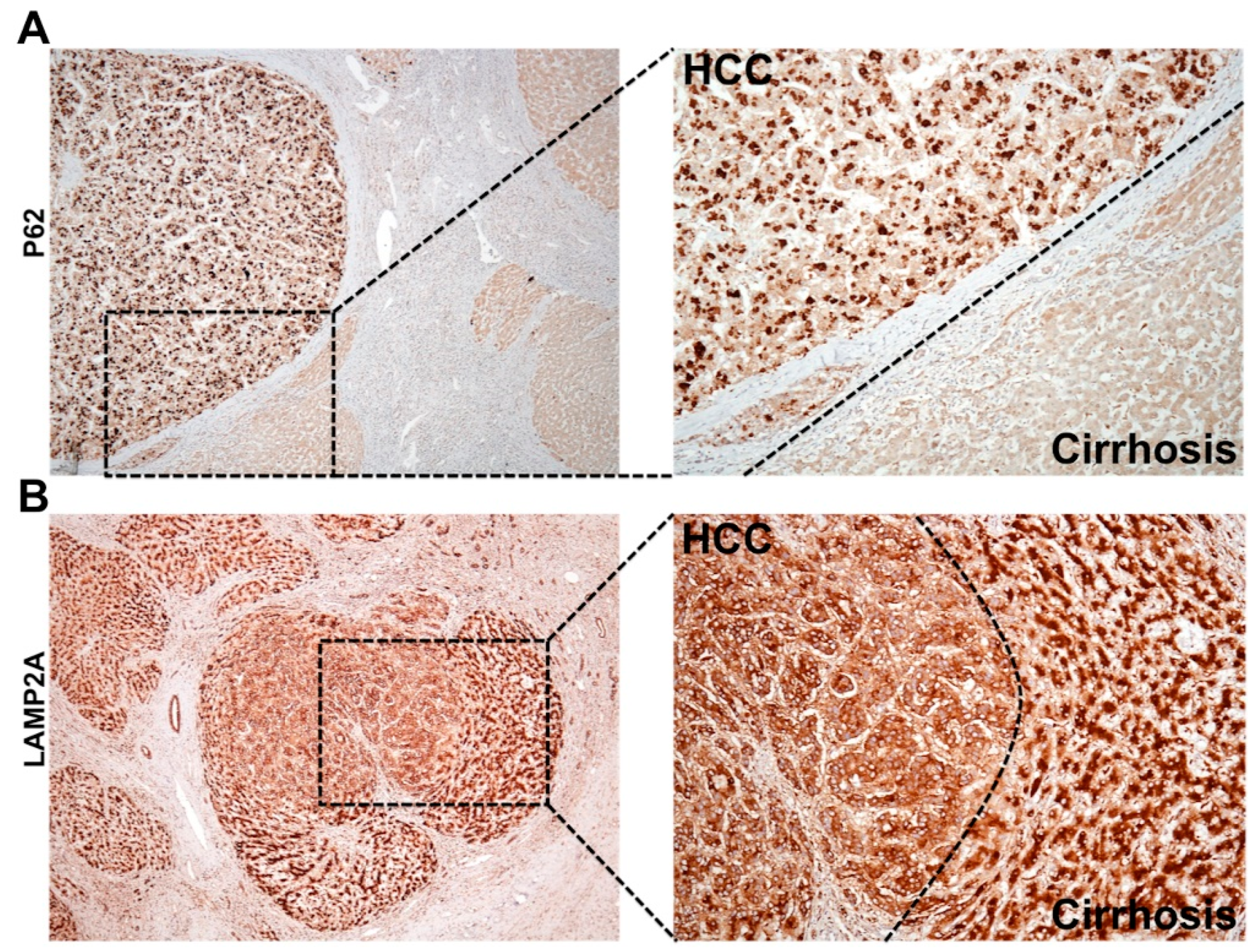
6.1. Cellular Adaptive Response to HCV Infection Promotes Cm-Associated Autophagy Compensation Favoring HCC Growth in the Cirrhotic Liver
6.2. Autophagy Inhibition Activates Oncogenic Signaling
6.3. Hepatic Adaptive Response to HCV-Associated Stress Degrades Tumor Suppressors
6.4. CMA Targets Interferon-Alpha Receptor Chain-1 (Ifnar1) Degradation
6.5. CMA Dampens Innate Hepatic Immunity by Preventing the P53-Ifnar1 Feedback Loop
7. Treatment of Liver Cirrhosis and Cancer Targeting Autophagy
8. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- White, D.L.; Thrift, A.P.; Kanwal, F.; Davila, J.; El-Serag, H.B. Incidence of Hepatocellular Carcinoma in All 50 United States, From 2000 Through 2012. Gastroenterology 2017, 152, 812–820. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Card, T.R.; Aithal, G.P.; Fleming, K.M.; Card, T.; Aithal, G. Risk of hepatocellular carcinoma among individuals with different aetiologies of cirrhosis: A population-based cohort study. Aliment. Pharmacol. Ther. 2017, 45, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef]
- Lombardi, A.; Mondelli, M.U. ESCMID Study Group for Viral Hepatitis (ESGVH) Hepatitis C: Is eradication possible? Liver Int. 2019, 39, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Lopatin, U. Drugs in the Pipeline for HBV. Clin. Liver Dis. 2019, 23, 535–555. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Urban, S.; Protzer, U. Towards curative therapy of chronic viral hepatitis. Zeitschrift für Gastroenterologie 2019, 57, 61–73. [Google Scholar] [CrossRef]
- Ward, J.W.; Hinman, A.R. What Is Needed to Eliminate Hepatitis B Virus and Hepatitis C Virus as Global Health Threats. Gastroenterology 2019, 156, 297–310. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease - A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Patel, Y.A.; Gifford, E.J.; Glass, L.M.; McNeil, R.; Turner, M.J.; Han, B.; Provenzale, D.; Choi, S.S.; Moylan, C.A.; Hunt, C.M. Risk factors for biopsy-proven advanced non-alcoholic fatty liver disease in the Veterans Health Administration. Aliment. Pharmacol. Ther. 2018, 47, 268–278. [Google Scholar] [CrossRef]
- Barrera, F.; George, J. The Role of Diet and Nutritional Intervention for the Management of Patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Sievenpiper, J.L.; De Souza, R.J.; I Cozma, A.; Mirrahimi, A.; Carleton, A.J.; Ha, V.; Di Buono, M.; Jenkins, A.L.; A Leiter, L.; et al. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2014, 68, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Barle, H.; Nyberg, B.; Essén, P.; Andersson, K.; McNurlan, M.A.; Wernerman, J.; Garlick, P.J. The synthesis rates of total liver protein and plasma albumin determined simultaneouslyin vivoin humans. Hepatology 1997, 25, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.L. synthesis of all plasma protein fractions except gamma globulins by the liver: The use of zone electrophoresis and lysine-epsi-c14 to define the plasma proteins synthesized by the isolated perfused liver. J. Exp. Med. 1954, 99, 125–132. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef]
- Brey, I.R.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Dicks, N.; Gutierrez, K.; Michalak, M.; Bordignon, V.; Agellon, L.B. Endoplasmic Reticulum Stress, Genome Damage, and Cancer. Front. Oncol. 2015, 5, 11. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Boil. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. Human Cytomegalovirus Infection Maintains mTOR Activity and Its Perinuclear Localization during Amino Acid Deprivation. J. Virol. 2011, 85, 9369–9376. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, H.H.; Horetsky, R.L.; Kimball, S.R.; Murthi, A.; Jeffereson, L.S.; Hooper, A.K. Retrograde nuclear accumulation of cytoplamsic tRNA in rat hepatoma cells in response to aminoa cid deprivation. Proc. Natl. Acad. Sci. USA 2007, 104, 8845–8850. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Guo, S.; Gao, J.; Zhong, M.; yan, G.; Wu, W.; Chao, Y.; Jiang, Y. General control nonderepressible 2 (GCN2) kinase inhibits target of rapamycin complex 1 in response to amino acid starvation in Saccharomyces cerevisiae. J. Biol. Chem. 2017, 292, 2660–2669. [Google Scholar] [CrossRef]
- Selitsky, S.R.; Baran-Gale, J.; Honda, M. Small tRNA-derived RNAs are increased and more abundant than microRNAs in chronic hepatitis B and C. Sci. Rep. 2015, 5, 7675. [Google Scholar] [CrossRef]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M. Temporal proteome and lipidome profiles reveals hepatitis C virus-associated reprogramming of hepatocellular metabolisms and bioenergetics. PLOS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Cooosing between glycolysis and oxidative phosphorylation: A tumor dilema? Biochica Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Balsa, E.; Soustel, M.; Thomas, A. ER and nutritional stress promotes assembly of respitatory chain supercomplexes through PERK-eIF2alpha axis. Mol. Cell 2019, 74, 877–890. [Google Scholar] [CrossRef]
- Hofmann, S.; Krajewski, M.; Scherer, C. Complex lipid metabolic remodelling is required for efficient hepatitis C virus replication. BBA-Mol. Cell Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef]
- Stoeck, I.K.; Lee, J.Y.; tabata, K. Hepatitis C virus replication depends on endosomal cholestrol homeostasis. J Virol. 2018, 92, e01196. [Google Scholar]
- Naito, H.; Yoshikawa-Bando, Y.; Yuan, Y.; Hashimoto, S.; Kitamori, K.; Yatsuya, H.; Nakajima, T. High-fat and high-cholesterol diet decreases phosphorylated inositol-requiring kinase-1 and inhibits autophagy process in rat liver. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tanaka, N.; Hu, X.; Kimura, T.; Lu, Y.; Jia, F.; Sato, Y.; Nakayama, J.; Moriya, K.; Koike, K.; et al. A high-cholesterol diet promotes steatohepatis and liver tumorigenesis in HVC core gene transgenic mice. Arch Toxicol. 2019, 93, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Ocampo, W.A.; Navas, M.-C.; Faber, K.N.; Daemen, T.; Moshage, H. The cellular stress response in hepatitis C virus infection: A balancing act to promote viral persistence and host cell survival. Virus Res. 2019, 263, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.H.; Petryshyn, R.; London, I.M. Characterization of double stranded RNA-activated kinase that phosphorylates alpha-subunit of eukaryotic intiatiation factor 2 (eIF2A) in reticulocytes lysates. Proc. Natl. Acad. Sci. USA 1980, 77, 832–836. [Google Scholar] [CrossRef]
- Meurs, E. Molecular cloning and characterization of the human double-stranded RNA activated protein kianse induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Mitchell, J.K.; Midkiff, B.R.; Israelow, B.; Evans, M.J.; Lanford, R.E.; Walker, C.M.; Lemon, S.M.; McGivern, D.R. Hepatitis C virus indirectly disrupt DNA damage-indued p53 response by activating protein kinase R. MBio 2017, 8, e00121-17. [Google Scholar] [CrossRef]
- Wang, S.C.; lai, K.R.; Li, C.Y. The paradoxial effects of different hepatitis C viral loads on host DNA damage and repair abilities. PLOS ONE 2017, 12, e0164281. [Google Scholar]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Boil. 2005, 6, 79–87. [Google Scholar] [CrossRef]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Asp. Med. 2009, 30, 191–296. [Google Scholar] [CrossRef]
- Breusing, N.; Grune, T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Boil. Chem. 2008, 389, 203–209. [Google Scholar] [CrossRef]
- Ding, Q.; Dimayuga, E.; Keller, J.N. Proteasome Regulation of Oxidative Stress in Aging and Age-Related Diseases of the CNS. Antioxid. Redox Signal. 2006, 8, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Farout, L.; Friguet, B. Proteasome Function in Aging and Oxidative Stress: Implications in Protein Maintenance Failure. Antioxid. Redox Signal. 2006, 8, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses 2016, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Jewell, J.L.; Russell, R.C.; Guan, K.-L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Boil. 2013, 14, 133–139. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Boil. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Tazawa, H.; Kuroda, S.; Hasel, J.; Kagawa, S.; Fujiwara, T. Impact of autophagy in oncolytic adenoviral therapy of cancer. Int. J. Mol. Sci. 2017, 18, 1479. [Google Scholar] [CrossRef]
- Ito, H.; Aoki, H.; Kühnel, F.; Kondo, Y.; Kubicka, S.; Wirth, T.; Iwado, E.; Iwamaru, A.; Fujiwara, K.; Hess, K.R.; et al. Autophagic Cell Death of Malignant Glioma Cells Induced by a Conditionally Replicating Adenovirus. J. Natl. Cancer Inst. 2006, 98, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. Oncolytic adenovirus-induced autophagy: Tumor-suppressive effect and molecular basis. Acta Med. Okayama 2013, 67, 333–342. [Google Scholar] [PubMed]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of atg5 and fadd in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Sanyal, S. Manipulation of autophagy by (+) RNA viruses. Semin Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Rautou, P.-E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bieche, I.; Lebrec, D.; et al. Changes in Autophagic Response in Patients with Chronic Hepatitis C Virus Infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia–Monzon, C.; et al. Autophagy Protects Cells From HCV-Induced Defects in Lipid Metabolism. Gastroenterology 2012, 142, 644–653.e3. [Google Scholar] [CrossRef]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef]
- Falcon, V.; Acosta-Rivero, N.; Chinea, G.; Gavilondo, J.; De La Rosa, M.-C.; Menéndez, I.; Dueñas-Carrera, S.; Viña, A.; García, W.; Gra, B.; et al. Ultrastructural evidences of HCV infection in hepatocytes of chronically HCV-infected patients. Biochem. Biophys. Res. Commun. 2003, 305, 1085–1090. [Google Scholar] [CrossRef]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, R.; Chandra, P.K.; Ferraris, P.; Kurt, R.; Song, K.; Garry, R.F.; Reiss, K.; Coe, I.R.; Furihata, T.; Balart, L.A.; et al. Persistent hepatitis C virus infection impairs ribavirin antiviral activity through clathrin-mediated trafficking of equilibrative nucleoside transporter 1. J. Virol. 2015, 89, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Gündüz, F.; Aboulnasr, F.M.; Chandra, P.K.; Hazari, S.; Poat, B.; Baker, D.P.; A Balart, L.; Dash, S. Free fatty acids induce ER stress and block antiviral activity of interferon alpha against hepatitis C virus in cell culture. Virol. J. 2012, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Kurt, R.; Chandra, P.K.; Aboulnasr, F.; Panigrahi, R.; Ferraris, P.; Aydin, Y.; Reiss, K.; Wu, T.; Balart, L.A.; Dash, S. Chaperone-Mediated Autophagy Targets IFNAR1 for Lysosomal Degradation in Free Fatty Acid Treated HCV Cell Culture. PLOS ONE 2015, 10, e0125962. [Google Scholar] [CrossRef]
- Asselah, T.; Bieche, I.; Mansouri, A.; Laurendeau, I.; Feldmann, G.; Bedossa, P.; Paradis, V.; Lebrec, D.; Guichard, C.; Ogier-Denis, E.; et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef]
- Chandra, P.K.; Gündüz, F.; Hazari, S.; Kurt, R.; Panigrahi, R.; Poat, B.; Bruce, D.; Cohen, A.J.; Behorquez, H.E.; Carmody, I.; et al. Impaired Expression of Type I and Type II Interferon Receptors in HCV-Associated Chronic Liver Disease and Liver Cirrhosis. PLOS ONE 2014, 9, e108616. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Wang, H.-C.; Huang, W.; Lai, M.-D.; Su, I.-J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef]
- Baiceanu, A.; Mesdom, P.; Lagouge, M.; Foufelle, F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat. Rev. Endocrinol. 2016, 12, 710–722. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Balasus, D.; Way, M.; Fusilli, C.; Mazza, T.; Morgan, M.Y.; Cervello, M.; Giannitrapani, L.; Soresi, M.; Agliastro, R.; Vinciguerra, M.; et al. The association of variants in PNPLA3 and GRP78 and the risk of developing hepatocellular carcinoma in an Italian population. Oncotarget 2016, 7, 86791–86802. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhuang, L.; Szatmary, P.; Wen, L.; Sun, H.; Lu, Y.; Xu, Q.; Chen, X. Upregulation of Heat Shock Proteins (HSPA12A, HSP90B1, HSPA4, HSPA5 and HSPA6) in Tumour Tissues Is Associated with Poor Outcomes from HBV-Related Early-Stage Hepatocellular Carcinoma. Int. J. Med Sci. 2015, 12, 256–263. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic Reticulum Stress in Hepatic Stellate Cells Promotes Liver Fibrosis via PERK-Mediated Degradation of HNRNPA1 and Up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193. [Google Scholar] [CrossRef]
- Wang, L.; Ou, J.-H.J. Regulation of Autophagy by Hepatitis C Virus for Its Replication. DNA Cell Boil. 2018, 37, 287–290. [Google Scholar] [CrossRef]
- Gual, P.; Gilgenkrantz, H.; Lotersztajn, S. Autophagy in chronic liver diseases: The two faces of Janus. Am. J. Physiol. Physiol. 2017, 312, C263–C273. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.-S.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; A Gewirtz, D.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gélinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lan, S.H.; Wu, S.R. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology 2018, 68, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, S.B.; Wang, L.Y. Autophagy-dependent generation of Axin2+ cancer stem-like cells promotes hepatocarcinogenesis in liver cirrhosis. Oncogene 2017, 36, 6725–6737. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Li, H.-Y.; Liu, J. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Mol. BioSyst. 2016, 12, 2605–2612. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K. p62 upregulated during preneoplasia, induces Hepatocellular Carcinoma by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef]
- Bao, L.; Chandra, P.K.; Moroz, K.; Zhang, X.; Thung, S.N.; Wu, T.; Dash, S. Impaired autophagy response in human hepatocellular carcinoma. Exp. Mol. Pathol. 2014, 96, 149–154. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive Activation of Chaperone-mediated Autophagy in Cells with Impaired Macroautophagy. Mol. Boil. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Grünvogel, O.; Colasanti, O.; Lee, J.-Y.; Klöss, V.; Belouzard, S.; Reustle, A.; Esser-Nobis, K.; Hesebeck-Brinckmann, J.; Mutz, P.; Hoffmann, K.; et al. Secretion of Hepatitis C Virus Replication Intermediates Reduces Activation of Toll-Like Receptor 3 in Hepatocytes. Gastroenterology 2018, 154, 2237–2251. [Google Scholar] [CrossRef] [PubMed]
- Bainton, D.F. The discovery of lysosomes. J. Cell Boil. 1981, 91, 66s–76s. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Essner, E.; Novikoff, A.B. Localization of Acid Phosphatase Activity in Hepatic Lysosomes by Means of Electron Microscopy. J. Cell Boil. 1961, 9, 773–784. [Google Scholar] [CrossRef]
- Straus, W. Isolation and biochemical properties of droplets from the cells of rat kidney. J. Boil. Chem. 1954, 207, 745–755. [Google Scholar]
- Del Pino, V.H.L.; LaRusso, N.F. Dissociation of bile flow and biliary lipid secretion from biliary lysosomal enzyme output in experimental cholestasis. J. Lipid Res. 1981, 22, 229–235. [Google Scholar]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Boil. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Lamming, D.W.; Bar-Peled, L. Lysosome: The metabolic signaling hub. Traffic 2019, 20, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Altered degradation of proteins microinjected into senescent human fibroblasts. J. Boil. Chem. 1982, 257, 14624–14627. [Google Scholar]
- Dice, J.F.; Chiang, H.L.; Spencer, E.P.; Backer, J.M. Regulation of catabolism of microinjected ribonuclease A. Identification of residues 7-11 as the essential pentapeptide. J. Boil. Chem. 1986, 261, 6853–6859. [Google Scholar]
- Dice, J.F.; Backer, J.M.; Chiang, H.-L.; Goff, S.A. Lysosomal degradation of microinjected proteins. Biochem. Soc. Trans. 1987, 15, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Boil. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Chiang, H.; Plant, C.P.; Dice, J. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Lysosomes, a meeting point of proteins, chaperones, and proteases. J. Mol. Med. 1998, 76, 6–12. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Männ, L.; Bonten, E.J.; D’Azzo, A.; Dice, J.F. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003, 22, 47–59. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Boil. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Boil. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Cuervo, A.M. Methods to study chaperone-mediated autophagy. Methods 2015, 75, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Yabu, T.; Imamura, S.; Mohammed, M.S.; Touhata, K.; Minami, T.; Terayama, M.; Yamashita, M. Differential gene expression of HSC70/HSP70 in yellowtail cells in response to chaperone-mediated autophagy. FEBS J. 2011, 278, 673–685. [Google Scholar] [CrossRef]
- Mukherjee, A.; Patel, B.; Koga, H.; Cuervo, A.M.; Jenny, A. Selective endosomal microautophagy is starvation-inducible in Drosophila. Autophagy 2016, 12, 1984–1999. [Google Scholar] [CrossRef]
- Eisermann, D.J.; Wenzel, U.; Fitzenberger, E. Inhibition of chaperone-mediated autophagy prevents glucotoxicity in the Caenorhabditis elegans mev-1 mutant by activation of the proteasome. Biochem. Biophys. Res. Commun. 2017, 484, 171–175. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Boil. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Boil. 2019, 17, e3000301. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar]
- Ferreira, J.V.; Fôfo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girão, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef]
- Rout, A.K.; Strub, M.-P.; Piszczek, G.; Tjandra, N. Structure of Transmembrane Domain of Lysosome-associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-mediated Autophagy. J. Boil. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Sridhar, S.; Kaushik, S.; Kiffin, R.; Cuervo, A.M. Identification of regulators of chaperone-mediated autophagy. Mol. Cell 2010, 39, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholí, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Inpanathan, S.; Botelho, R.J. The Lysosome Signaling Platform: Adapting With the Times. Front. Cell Dev. Boil. 2019, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Genet. 2018, 16, 341–354. [Google Scholar] [CrossRef]
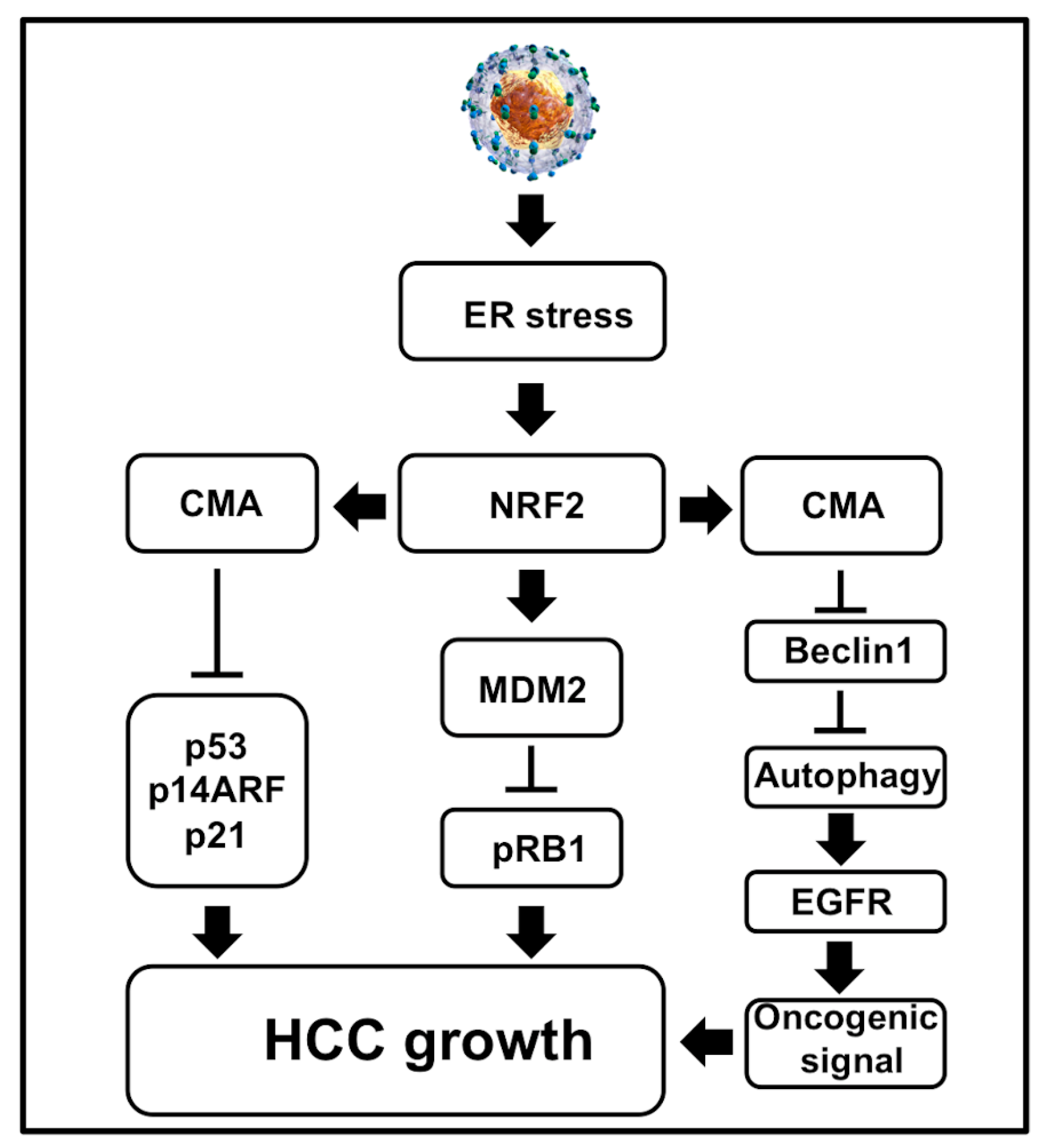
- Dash, S.; Aydin, Y.; Wu, T. Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: A novel mechanism for host-microbe survival and HCC development in liver cirrhosis. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Aydin, Y.; Stephens, C.M.; Chava, S.; Heidari, Z.; Panigrahi, R.; Williams, D.D.; Wiltz, K.; Bell, A.; Wilson, W.; Reiss, K.; et al. Chaperone-Mediated Autophagy Promotes Beclin1 Degradation in Persistently Infected Hepatitis C Virus Cell Culture. Am. J. Pathol. 2018, 188, 2339–2355. [Google Scholar] [CrossRef]
- Singh, V.; Finke-Isami, J.; Hopper-Chidlaw, A.C.; Schwerk, P.; Thompson, A.; Tedin, K. Salmonella Co-opts Host Cell Chaperone-mediated Autophagy for Intracellular Growth. J. Biol. Chem. 2017, 292, 1847–1864. [Google Scholar] [CrossRef]
- Jones-Jamtgaard, K.N.; Wozniak, A.L.; Koga, H.; Ralston, R.; Weinman, S.A. Hepatitis C virus infection increases autophagosome stability by suppressing lysosomal fusion through an Arl8b-dependent mechanism. J. Biol. Chem. 2019, 294, 14257–14266. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Boil. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Dhar, D. Liver carcinogenesis: From naughty chemicals to soothing fat and the surprising role of NRF2. Carcinogenesis 2016, 37, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M. Potential role of p62 in tumor development. Autophagy 2011, 7, 1088–1090. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Boil. 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Singh, R.; Czaja, M.J. Compensatory mechanisms and the type of injury determine the fate of cells with impaired macroautophagy. Autophagy 2008, 4, 516–518. [Google Scholar] [CrossRef]
- Wang, K.; Wei, Y.; Liu, W.; Liu, L.; Guo, Z.; Fan, C.; Wang, L.; Hu, J.; Li, B. Mechanical stress-dependent autophagy component release via extracellular nanovesicles in tumor cells. ACS Nano 2019, 13, 4589–4602. [Google Scholar] [CrossRef]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprograming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; I Rojo, A.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Boil. 2019, 29, 635–646. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef]
- Christen, V.; Treves, S.; Duong, F.H.T.; Heim, M.H. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology 2007, 46, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J. Gastroenterol. 2004, 10, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Shi, Y.-H.; Zhou, J.; Qiu, S.-J.; Xu, Y.; Dai, Z.; Shi, G.-M.; Wang, X.-Y.; Ke, A.-W.; Wu, B.; et al. Association of Autophagy Defect with a Malignant Phenotype and Poor Prognosis of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Fu, X.-T.; Shi, Y.-H.; Zhou, J.; Peng, Y.-F.; Liu, W.-R.; Shi, G.-M.; Gao, Q.; Wang, X.-Y.; Song, K.; et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Levine, B.; Liu, R.; Dong, X.; Zhong, Q. Beclin orthologs: Integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Boil. 2015, 25, 533–544. [Google Scholar] [CrossRef]
- Hill, S.M.; Wrobel, L.; Rubinsztein, D.C. Post-translational modification of Beclin1 provide multiple strategies of autophagy regulation. Cell Death Differ. 2019, 234, 12562–12568. [Google Scholar]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation - at the Center of Autophagy Regulation. Front. Cell Dev. Boil. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-H.; Ding, Z.-B.; Zhou, J.; Qiu, S.-J.; Fan, J. Prognostic significance of Beclin 1-dependent apoptotic activity in hepatocellular carcinoma. Autophagy 2009, 5, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.M.; Wang, G.L.; LChen, L. The expression of beclin-1, an autophagy gene in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Exp. Med. 2011, 208, 275–284. [Google Scholar] [CrossRef]
- Ni, H.-M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.-X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Saito, T. p62/Sqtm1 promotes malignancy of HCV positive hepatocellular carcinoma carcinogenesis through Nrf2-dependnet metabolic reprograming. Nature Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.; Nouri, K.; Van Rensburg, H.J.J.; Maritan, S.M.; Wu, L.; Hao, Y.; Montminy, T.; Yu, J.; Khanal, P.; Mulligan, L.M.; et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2019, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, J.J.; Di Guglielmo, G.M.; Dahan, S.; Dominguez, M.; Posner, B.I. Spatial and Temporal Regulation of Receptor Tyrosine Kinase Activation and Intracellular Signal Transduction. Annu. Rev. Biochem. 2016, 85, 573–597. [Google Scholar] [CrossRef] [PubMed]
- Abella, J.V.; Park, M. Breakdown of endocytosis in the oncogenic activation of receptor tyrosine kinases. Am. J. Physiol. Metab. 2009, 296, E973–E984. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Di Fiore, P.P.; Sigismund, S. Endocytic control of signaling at the plasma membrane. Curr. Opin. Cell Boil. 2016, 39, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, Y.; Mills, G.B.; Yarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signaling decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signaling. Exp Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Komposh, K.; Sibilia, M. EGFR signaling in Liver disease. Int. J. Mol. Sci. 2016, 17, 30. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Kira, S.; Nakanishi, T.; Suemori, S.; Kitamoto, M.; Watanabe, Y.; Kajiyama, G. Expression of transforming growth factor α and epidermal growth factor receptor in human hepatocellular carcinoma. Liver 1997, 17, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Daveau, M.; Scotte, M.; Francois, A.; Coulouarn, C.; Ros, G.; Tallet, Y.; Hiron, M.; Hellot, M.-F.; Salier, J.-P. Hepatocyte growth factor, transforming growth factor α, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol. Carcinog. 2003, 36, 130–141. [Google Scholar] [CrossRef]
- Buckley, A.F.; Burgart, L.J.; Sahai, V.; Kakar, S. Epidermal Growth Factor Receptor Expression and Gene Copy Number in Conventional Hepatocellular Carcinoma. Am. J. Clin. Pathol. 2008, 129, 245–251. [Google Scholar] [CrossRef]
- Divella, R.; Daniele, A.; Gadaleta, C.; Tufaro, A.; Venneri, M.T.; Paradiso, A.; Quaranta, M. Circulating transforming growth factor-β and epidermal growth factor receptor as related to virus infection in liver carcinogenesis. Anticancer Res. 2012, 32, 141–145. [Google Scholar]
- Tornesello, M.L.; Annunziata, C.; Tornesello, A.L.; Buonaguro, L.; Buonaguro, F.M. Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers 2018, 10, 213. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med. Virol. 2013, 23, 213–220. [Google Scholar] [CrossRef]
- Zaika, A.I.; Wei, J.; Noto, J.M.; Peek, R.M. Microbial Regulation of p53 Tumor Suppressor. PLOS Pathog. 2015, 11, e1005099. [Google Scholar] [CrossRef]
- Li, N.; Xie, C.; Lu, N.-H. p53, a potential predictor of helicobacter pylori infection associated gastric carcinogenesis. Oncotarget 2016, 7, 66276. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Marcelli, E.; Casini, N.; Rizzo, V.; Giordono, A. Emerging role of RB family: New defense mechanisms against tumor progression. J. cell Physiol. 2013, 228, 525–535. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C Virus Induces E6AP-Dependent Degradation of the Retinoblastoma Protein. PLOS Pathog. 2007, 3, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Chatterjee, A.; Chandra, P.K.; Chava, S.; Chen, W.; Tandon, A.; Dash, A.; Chedid, M.; Moehlen, M.W.; Regenstein, F.; et al. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol. Commun. 2017, 1, 256–269. [Google Scholar] [CrossRef]
- Aydin, Y.; Chedid, M.; Chava, S.; Williams, D.D.; Liu, S.; Hagedorn, C.H.; Sumitran-Holgersson, S.; Reiss, K.; Moroz, K.; Lu, H.; et al. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci. Rep. 2017, 7, 9223. [Google Scholar] [CrossRef]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef]
- Han, S.Y.; Ko, A.; Kitano, H. Molecular chaperone HSP70 is necessary to prevent cellular senescence via lysosomal degradation of p14ARF. Cancer Res. 2016, 77, 343–354. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; A Donehower, L.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.; Varanasi, S.; Kirkwood, J.M. Interferons in the treatment of solid tumors. Cancer Treat. Res. 2005, 126, 207–241. [Google Scholar] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Tschurtschenthaler, M.; Wang, J.; Fricke, C.; Fritz, T.M.J.; Niederreiter, L.; Adolph, T.E.; Sarcevic, E.; Künzel, S.; Offner, F.A.; Kalinke, U.; et al. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 2014, 63, 1921–1931. [Google Scholar] [CrossRef]
- Guillot, B.; Portales, P.; Du Thanh, A.; Merlet, S.; Dereure, O.; Clot, J.; Corbeau, P. The expression of cytotoxic mediators is altered in mononuclear cells of patients with melanoma and increased by interferon-alpha treatment. Br. J. Dermatol. 2005, 152, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Bacher, N.; Raker, V.; Hofmann, C.; Graulich, E.; Schwenk, M.; Baumgrass, R.; Bopp, T.; Zechner, U.; Merten, L.; Becker, C.; et al. Interferon- Suppresses cAMP to Disarm Human Regulatory T Cells. Cancer Res. 2013, 73, 5647–5656. [Google Scholar] [CrossRef]
- Schiavoni, G.; Mattei, F.; Gabriele, L. Type I Interferons as Stimulators of DC-Mediated Cross-Priming: Impact on Anti-Tumor Response. Front. Immunol. 2013, 4, 483. [Google Scholar] [CrossRef]
- Spadaro, F.; Lapenta, C.; Donati, S.; Abalsamo, L.; Barnaba, V.; Belardelli, F.; Santini, S.M.; Ferrantini, M. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood 2012, 119, 1407–1417. [Google Scholar] [CrossRef]
- Chen, H.-M.; Tanaka, N.; Mitani, Y.; Oda, E.; Nozawa, H.; Chen, J.-Z.; Yanai, H.; Negishi, H.; Choi, M.K.; Iwasaki, T.; et al. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci. 2009, 100, 449–456. [Google Scholar] [CrossRef]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: the biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Lee, H.-C.; Narayanan, S.; Park, S.-J.; Seong, S.-Y.; Hahn, Y.S. Transcriptional Regulation of IFN-λ Genes in Hepatitis C Virus-infected Hepatocytes via IRF-3·IRF-7·NF-κB Complex*. J. Boil. Chem. 2014, 289, 5310–5319. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Tamura, T.; Taniguchi, T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 2008, 99, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Miciak, J.; Bunz, F. Long story short: p53 mediates innate immunity. Biochim Biophys Acta 2016, 1865, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Aaronson, S.A.; Muñoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef]
- Layese, R.; Bourcier, V.; Cagnot, C.; Zucman, D.; Sutton, A.; Roudot-Thoraval, F.; Audureau, E.; Nahon, P.; Marcellin, P.; Guyader, D.; et al. Incidence of Hepatocellular Carcinoma After Direct Antiviral Therapy for HCV in Patients With Cirrhosis Included in Surveillance Programs. Gastroenterology 2018, 155, 1436–1450. [Google Scholar]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Holmes, J.A.; Dai, C.-Y.; Huang, C.-F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef]
- Hamdane, N.; Jühling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Suarez, A.A.R.; et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329. [Google Scholar] [CrossRef]
- Amaddeo, G.; Nguyen, C.T.; Maille, P.; Mule, S.; Luciani, A.; Machou, C.; Rodrigues, A.; Regnault, H.; Mallat, A.; Laurent, A.; et al. Intra-hepatic immune changes after hepatitis c virus eradication by direct-acting antiviral therapy. Liver Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.; Bosch, J.; Burroughs, A.K. Future treatments of cirrhosis. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 571–581. [Google Scholar] [CrossRef]
- Borzio, M.; Salerno, F.; Piantoni, L.; Cazzaniga, M.; Angeli, P.; Bissoli, F.; Boccia, S.; Colloredo-Mels, G.; Corigliano, P.; Fornaciari, G.; et al. Bacterial infection in patients with advanced cirrhosis: A multicentre prospective study. Dig. Liver Dis. 2001, 33, 41–48. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpedek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dzikl, L.; Diehl, J.A.; Majsterek, I. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, F.; Altucci, L.; Cobellis, G. Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy. Cancers 2019, 11, 1465. [Google Scholar] [CrossRef]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef]
- Bruch, J.; Xu, H.; Rösler, T.W.; De Andrade, A.; Kuhn, P.; Lichtenthaler, S.F.; Arzberger, T.; Winklhofer, K.F.; Müller, U.; Höglinger, G.U. PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol. Med. 2017, 9, 371–384. [Google Scholar] [CrossRef]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.-Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef]
- Panieri, E.; Soso, L. Potential application of Nrf2 inhibitors in cancer therapy. Oxidative Med. Cell. Longev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, S.; Aydin, Y.; Moroz, K.
Chaperone-Mediated Autophagy in the Liver:
Good or Bad? Cells 2019, 8, 1308.
https://doi.org/10.3390/cells8111308
Dash S, Aydin Y, Moroz K.
Chaperone-Mediated Autophagy in the Liver:
Good or Bad? Cells. 2019; 8(11):1308.
https://doi.org/10.3390/cells8111308
Dash, Srikanta, Yucel Aydin, and Krzysztof Moroz.
2019. "Chaperone-Mediated Autophagy in the Liver:
Good or Bad?" Cells 8, no. 11: 1308.
https://doi.org/10.3390/cells8111308
Dash, S., Aydin, Y., & Moroz, K.
(2019). Chaperone-Mediated Autophagy in the Liver:
Good or Bad? Cells, 8(11), 1308.
https://doi.org/10.3390/cells8111308